

Abstract
Laboratory testing is of great value when evaluating a patient with a suspected autoimmune disease. The results can confirm a diagnosis, estimate disease severity, aid in assessing prognosis and are useful to follow disease activity. Components of the laboratory exam include complete blood count with differential, comprehensive metabolic panel, inflammatory markers, autoantibodies, and flow cytometry. This chapter discusses these components and includes a discussion about organ-specific immunologic diseases where immunological laboratory testing is employed. Comprehensive laboratory evaluation of a suspected autoimmune illness in conjunction with a thorough clinical evaluation provides a better understanding of a patient's immunologic disease.
Keywords: autoimmune, disease, laboratory, inflammatory markers, evaluation, rheumatic, serologies, flow cytometry, HLA, organ-specific
I. Introduction
Autoimmunity involves the loss of normal immune homeostasis such that the organism produces an abnormal response to its own self tissue. The hallmark of autoimmune diseases generally involves the presence of self-reactive T cells, autoantibodies and inflammation. An area of intense research is determining why the immune system turns against its host. Over the past decade, research has greatly advanced our understanding of autoimmunity and the scientific findings from these investigations are translating new clinical laboratory studies of patients to aid in diagnoses.
Examining patients for potential autoimmune diseases is fraught with difficulty because not one laboratory test establishes such a diagnosis. Typically, multiple laboratory tests are needed and include basic studies like a complete blood count, comprehensive metabolic panel, acute phase reactants, immunologic studies, serologies, flow cytometry, cytokine analysis, and HLA typing. Although some tests may be non-specific, such as the erythrocyte sedimentation rate (ESR), they are useful to assess disease activity. These tests can be useful in the diagnosis and management of patients with autoimmune diseases and help in providing a prognosis, or indicate the severity of organ involvement or damage.
1. Initial laboratory evaluation
Inflammatory diseases will cause abnormalities in routine laboratory studies. Characteristic findings can include a normochromic, normocytic anemia indicating the chronicity or severity of disease. Common hematologic parameters also include an elevated or decreased platelet count and/or white blood cell count. Leukopenia and thrombocytopenia are common in patients with systemic lupus erythematosus (SLE).
Testing will find aberrations in serum levels of specific organ enzymes or abnormalities in metabolic processes that are reflected in the comprehensive metabolic panel. For example, autoimmune hepatitis can be manifested by elevations of transaminases, bilirubin, and serum proteins. One should be aware that these abnormalities can also be associated with drug toxicity.
Coagulation studies such as a prolongation of the activated partial thromboplastin time (aPTT) and/or the prothrombin time (PT) that does not correct with mixing studies suggests an inhibitor of the clotting process is present as seen in the antiphospholipid syndrome. Hypercalcemia can be observed in approximately 30% of patients with sarcoidosis. An increase in muscle enzymes, [creatinine kinase, alanine transaminase (ALT), and aspartate aminotransferase (AST)] can be seen in autoimmune inflammatory myopathies (dermatomyositis, polymyositis, and inclusion body myositis). Serum protein levels are helpful to screen for abnormal elevations of immunoglobulin.
The urinalysis is commonly used to assess renal injury (glomerulonephritis, interstitial nephritis) and will show proteinuria, hematuria or active sediment (white blood cell casts or red blood cell casts). Many other illnesses such as diabetic nephropathy, poorly controlled hypertension, or infections will test similarly but when autoimmune disease is suspect, the common laboratory evaluation will serve as an initial red flag to pursue further testing.1
2. Inflammatory markers
Serum proteins that are produced in response to inflammation can be referred to as inflammatory markers. These proteins are mainly produced by the liver in response to stress and can also be called acute phase reactants. Pro-inflammatory cytokines such as IL-1, IL-6, and TNF-alpha induce synthesis of some acute phase reactants that include CRP, fibrinogen and haptoglobin. Other proteins, like albumin, are not sensitive to inflammatory cytokines for increased synthesis; instead chronic stress (inflammation) results in a lower synthesis rate with resultant decreased serum concentrations. The inflammatory markers are not diagnostic of inflammation, but reflect abnormalities that are seen in autoimmune diseases, infections, malignancies and other illnesses.
Erythrocyte sedimentation rate (ESR)
The ESR is the measure of the quantity of red blood cells (RBC) that precipitate in a tube in a defined time and is based upon serum protein concentrations and RBC interactions with these proteins. Inflammation causes an increase in the ESR. Multiple factors influence the ESR and include patient's age, gender, RBC morphology, hemoglobin concentration, and serum levels of immunoglobulin. The sample must be handled appropriately and processed within a few hours to assure test accuracy. While the ESR is not a diagnostic test, it can be used to monitor disease activity and treatment response and signal that inflammatory or infectious stress is present. For example, in rheumatoid arthritis, the ESR correlates well with disease activity; however normalization of ESR often lags behind successful treatment that causes resolution of the inflammatory state.2, 3
C-reactive protein (CRP)
C-reactive protein (CRP/CRP-high sensitivity) was discovered and named for its reactivity to the C polysaccharide in the cell wall of S. pneumoniae. CRP, an innate immune protein, helps opsonize pathogens for phagocytosis and activates the complement system. CRP production is under the control of IL-1, IL-6, and TNF-alpha. Changes in serum CRP concentration change more quickly than ESR and therefore CRP may be a better reflection of current inflammation. Unlike the ESR, CRP is a fairly stable serum protein whose measurement is not time-sensitive and is not affected by other serum components. The magnitude of inflammation directly relates to the concentration of CRP. Levels < 0.2 mg/dl are considered normal, while those >1.0 mg/dL are suggestive off inflammation and/or infection. More recently, the use of high sensitivity CRP has been utilized. This test may better quantify lower levels of inflammation and has been important in evaluating cardiac disease and other inflammatory states.2, 3
Ferritin
Serum ferritin is a storage protein for iron and its synthesis is regulated by intracellular iron, cytokines (TNF-alpha, IL-1, and IL-6), products of oxidative stress, and growth factors. Elevated levels can indicate acute or chronic sepsis, inflammation or malignancy. Diseases such as adult Still's disease, systemic-onset juvenile idiopathic arthritis, hemophagocytic lymphohistiocytosis and iron overload diseases, including hemochromatosis or hemosiderosis, should be considered with elevated ferritin levels.4, 5
Less common indicators of inflammatory states include:
Ceruloplasmin
the major copper containing protein in the blood that plays a role in iron metabolism and is increased in acute and chronic inflammatory states, pregnancy, lymphoma, rheumatoid arthritis and Alzheimer's disease.7
Fibrinogen
a hemostatic coagulation factor produced in response to tissue injury. Fibrinogen synthesis is controlled at the transcription level and is increased in the presence of inflammation and stress that is mediated by IL-6.
Haptoglobin
is produced in response to tissue injury. Increased levels of haptoglobin can be seen during inflammation, malignancy, surgery, trauma, peptic ulcer disease and ulcerative colitis. Decreased levels may indicate chronic liver disease or anemia.
Albumin
a serum protein synthesized by the liver that aids body tissues in maintaining oncotic pressure necessary for proper body fluid distribution. The average amount of albumin in the plasma is approximately 300 to 400 grams, and about 15 grams is produced by the liver per day. While the rate of synthesis can double in situations of rapid albumin loss as seen in glomerulonephritis or inflammatory bowel disease, serum levels will decline.
3. Autoantibodies and Immunologic Studies
The presence of an autoantibody in a patient does not assure a diagnosis of an autoimmune disease. Rather, a positive serologic test in the company of appropriate signs and symptoms helps to support a diagnosis. Serologic testing is flawed by the presence of autoantibodies in healthy individuals and other patients with non-autoimmune diseases and imperfect testing systems. Historically, many different methods were used to test for the presence an autoantibody. Today, testing is principally done with enzyme immunosorbent assays (EIA) because of cost saving measures with mechanization.
Enzyme-linked immunosorbent assay (ELISA)
ELISA is an immunometric method for detecting and measuring specific antibodies. The basic components of this laboratory method include a substrate where an antigen is fixed (typically a 96 well micro-well plate), patient's sera, washing solutions and a detection method where an enzyme is linked to an antibody that detects the antigen. In a typical double-antibody sandwich ELISA, an antibody that is attached to the bottom of a well provides both antigen capture and immune specificity, while another antibody linked to an enzyme provides detection and acts as an amplification factor. This allows for accurate and sensitive detection of the antigen of interest. However, performance is largely dependent on antibody quantity, kit manufacturer, and operator skill and experience. ELISA permits measurement of only one antigen at a time for a given aliquot of sample. Furthermore, ELISA has a limited dynamic range (that is, the range over which there is a linear relationship between antigen concentration and absorbance reading) – the range is narrow relative to the range for other technologies such as multiplex assays.8
Rheumatoid factor (RF) and Anti-cyclic citrullinated peptide antibody (CCP)
RF is an autoantibody that reacts to the Fc portion of polyclonal IgG; but they can be any class of immunoglobulin. Most assays detect the IgM rheumatoid factor. RF is helpful when evaluating patients who may have rheumatoid arthritis as the sensitivity is ∼70% with a specificity of ∼70%. Rheumatoid factor is absent in ∼15% of patients with rheumatoid arthritis. However, ∼15% of the healthy population may have a low titer RF. Rheumatoid factor positive patients are more likely to have progressive, erosive arthritis with loss of joint mobility and also have extraarticular manifestations including rheumatoid nodules, vasculitis, Felty's syndrome and secondary Sjögren's syndrome. In addition, the presence of RF is seen in other autoimmune disorders including Sjogren's syndrome, SLE, cryoglobulinemia, in pulmonary diseases such as interstitial fibrosis and silicosis and various infectious diseases.
Recently, a new biomarker for RA has been described, autoantibodies to cyclic citrullinated peptide (CCP). Inflammation activates the enzyme peptidylarginine deiminase which incorporates citrulline into certain proteins. In RA, autoantibodies are formed against the citrullinated protein (anti-CCP). The presence of serum anti-CCP antibodies are ∼95% specific for the diagnosis of RA, with sensitivity similar to rheumatoid factor. Testing for both anti-CCP and RF is beneficial when excluding the diagnosis of RA rather than testing for either antibody alone. In early undifferentiated disease, anti-CCP positive patients tend to go on to have more severe, erosive and aggressive disease. Anti-CCP can also be present in other disease states such as some children with JIA, psoriatic arthritis, lupus, Sjögren's syndrome, inflammatory myopathies and active tuberculosis.2, 3
Anti-nuclear antibody (ANA)
Autoantibodies to nuclear antigens are a diverse group of antibodies that react against nuclear, nucleolar, or perinuclear antigens. These antigens represent cellular components such as nucleic acid, histone, chromatin, nuclear and ribonuclear proteins. Classically, the ANA hallmarks the serologic diagnosis of SLE, but finding an ANA is common to most other autoimmune diseases. Methods used for detection utilize immunofluorescence testing of the patient's serum, at various dilutions, using a cell substrate. Typically, screening patient's serum for the detection of an ANA with ELISA provides high sensitivity but lacks specificity. Results are reported as either the dilution of serum that tests positive or the degree of positivity measured by the testing procedure. Historically, HEp2 cells (a human laryngeal epithelioma cancer cell line) have been used as the cell substrate because the result offers the advantage of detecting a nuclear fluorescent pattern. The fluorescent patterns (homogenous, diffuse, speckled, peripheral and rim) suggest clinical associations with certain autoimmune diseases. However, because of the time and expense for testing with HEp2 cells, the assay procedures are largely done by ELISA methods.2, 3
Immunofluorescence is particularly useful as an initial screening test for those individuals suspected of having an autoimmune disease – SLE, Sjögren's syndrome, RA, mixed connective tissue disease (MCTD), scleroderma, polymyositis/dermatomyositis (PM/DM). However, one must use caution when interpreting ANA as this autoantibody is found in nonrheumatic diseases such as Hashimoto's thyroiditis, Graves' disease, autoimmune hepatitis, primary autoimmune cholangitis, primary pulmonary hypertension, and in various infections and malignancies. Furthermore, the presence of low titer ANA occurs more frequently in elderly populations.
Table 1 details the sensitivity and specificity of the various antinuclear antibodies in several autoimmune diseases. Values are reported as approximate percentages as seen in several published reviews.
Table 1.
| ANA | dsDNA | Histone | Nucleoprotein | Sm (ribonuclear) | |
|---|---|---|---|---|---|
| SLE | |||||
| Sensitivity | >95% | 70% | ∼50% | 60% | 25% |
| Specificity | 60 | 95 | 50 | Medium | 99 |
| RA | |||||
| Sensitivity | 45 | 1 | Low | 25 | 1 |
| Specificity | 60 | Low | |||
| Scleroderma | |||||
| Sensitivity | 60 | <1 | <1 | <1 | <1 |
| Specificity | 50 | ||||
| PM/DM | |||||
| Sensitivity | 60 | <1 | <1 | <1 | <1 |
| Specificity | 60 | ||||
| Sjögren's | |||||
| Sensitivity | 50 | <5 | Low | Medium | <5 |
| Specificity | 50 | Low | Medium |
Anti-double stranded DNA (anti-dsDNA)
Autoantibodies to double stranded DNA are an important marker used in the diagnosis and monitoring of SLE. Antibodies to dsDNA are highly specific for SLE. However, some patients with other rheumatic diseases or chronic active hepatitis may have mildly or moderately elevated serum titers. Previously, anti-dsDNA was typically measured using radioimmunoassay (particularly the Farr assay). The more common current tests employ an immunofluorescence assay (IFA) or ELISA. The IFA utilizes a target antigen Crithidia luciliae, a flagellated protozoa containing a dsDNA-containing small organelle called a kinetoplast. The antibodies to dsDNA are detected semiquantitatively by demonstrating IgG bound to the kinetoplast. In contrast, with ELISA testing, the dsDNA is bound to the solid phase of the microwell plate. The serum is incubated and then the bound IgG is detected.2, 3
Anti-extractable nuclear antigen (anti-ENA)
The extractable nuclear antigens consist of the Smith (Sm) antigen, ribonuclear protein (RNP) or U1RNP, anti-SSA (Ro) and anti-SSB (La). They are called extractable because they are readily soluble or extractable in neutral buffers. The Sm antigen is highly specific for SLE, but it is found only in ∼25% of SLE patients. The U1RNP antigen is seen in patients with SLE plus systemic sclerosis and in patients with mixed connective tissue disease. The SSA (Ro) and SSB (La) nuclear antigens are often found together in those patients with Sjögren's syndrome. Anti-SSA and anti-SSB are also seen in some subsets of SLE patients. This group includes those patients with subcutaneous lupus erythematosus (SCLE) (prominent photosensitive rashes and sometimes vasculitis), but without severe renal disease. The presence of anti-SSA and anti-SSB is associated with neonatal lupus, in which transplacental transfer of these antibodies (maternal IgG) can cause transient photosensitive rash and/or congenital heart block.2, 3
Anti-signal recognition particle (anti-SRP), anti-JO-1, anti-Mi2, anti-PM/Scl
Anti-SRP, anti-JO-1, anti-Mi-2 and anti-PM/Scl are termed myositis specific antibodies because of the high specificity to the autoimmune inflammatory myopathies (IIM). Anti-SRP antibodies are directed toward an RNA-protein complex consisting of 6 proteins and a 300-nucleotide RNA molecule (7SL RNA). Patients with this antibody have a distinct type of IIM that is characterized by acute onset of muscle weakness, a muscle biopsy that lacks inflammation and the patient shows a poor response to therapy. Anti-JO-1 autoantibodies are the most common autoantibody found in the group of inflammatory myopathies called the anti-synthetase syndrome. Anti-JO-1 autoantibodies are directed against histidyl-tRNA synthetase (an enzyme that attaches histidine to growing polypeptide chains). Other less well understood antisynthetase autoantibodies include anti-PL12, anti-EJ, anti-OJ, anti-PL7, and anti-KS. These antibodies are reportedly more common in PM than DM and are rare in children. Antisynthetase syndrome patients have disease characteristic that are very different than anti-SRP patients and often present with muscle weakness, interstitial lung disease, arthritis and fevers. The anti-JO-1 response appears to be self-antigen driven, with isotype switching and affinity maturation. Anti-Mi2 antibodies recognize a major protein of a nuclear complex formed by about 7 proteins involved in transcription. Autoantibodies to Mi2 are specific for dermatomyositis and associated with acute onset, a better prognosis and good response to therapy. Anti-PM/Scl is an autoantibody to the nucleolar granular component. This is often seen with myositis with scleroderma overlap. Detection of such autoantibodies is done mostly through commercial blot assays utilizing immunoblotting or dot immunoblotting methodologies, in addition to traditional ELISA.8
Antineutrophil cytoplasmic antibody (ANCA) [myeloperoxidase (MPO), proteinase-3 (PR3)
Antineutrophil cytoplasmic antibodies (ANCA) react with cytoplasmic granules of neutrophils. Initial ANCA testing screens sera for the presence of ANCA and two general immunofluorescent staining patterns are observed – cytoplasmic (cANCA) and perinuclear (pANCA). The immunofluorescence pattern is helpful to distinguish various ANCA associated vasculitis syndromes. cANCA is most often seen in Wegener's Granulomatosis (WG), microscopic polyangiitis (MPA) and Churg-Strauss syndrome. pANCA patterns were initially described in microscopic polyangiitis (MPA), but pANCA has now been observed in a variety of diseases including other types of vasculitis, inflammatory bowel disease (IBD), SLE, RA, juvenile idiopathic arthritis (JIA). Antigenic determinants of the ANCA that are important to detect in vasculitis are proteinase 3 (PR3) and myeloperoxidase (MPO). Vasculitic states that test positive are named PR3-ANCA positive or MPO-ANCA positive. The presence of PR3 or MPO can help the clinician to determine the type of vasculitis and activity of disease.
Antibodies to PR3 or MPO are predictive of the various vasculitis syndromes. cANCA plus PR3 have increased positive predictive value (PPV) for ANCA-associated vasculitis, particularly WG. pANCA plus MPO has an increased PPV for MPA and less often for Churg-Strauss. ANCA plus MPO more than PR3 often leads toward a diagnosis of Churg-Straus. With increased disease activity, there is a greater likelihood that ANCA will be positive. ANCA titers may normalize with treatment although persistent ANCA positivity or rising ANCA does not reliably predict disease exacerbation or flare. Therefore, one should not use ANCA titers to determine treatment efficacy. Furthermore, one should also be wary of ANCA positivity as this can be seen in other disease states that include infection, drug use (eg. thyroid medication, particularly PTU), and other autoimmune disease. If the suspicion for vasculitis remains, tissue biopsy should be considered.2, 3, 4, 9
Complement
The complement cascade is a complex, tightly regulated series of proteolytic enzymes, regulatory proteins and cell surface receptors that mediate and augment both complement, humoral and cellular immune response. The classical pathway is initiated by immune complexes binding to C1q and involves C4 and C2. The alternative pathway involves factors B, D, and properdin. The mannose-binding lectin pathway, classical and alternative pathways all involve cleavage of C3. This release product induces the formation of the terminal membrane attack complex (C5-C9).
Individual components, such as C3, C4 and factor B, are measured by nephelometry and ELISA. Plasma total hemolytic complement assay (CH50) utilizes a functional assay to assess the functional integrity of the classical pathway. To measure these values, diluted serum is added to sheep antibody-coated erythrocytes and the subsequent value is the reciprocal of the highest dilution able to lyse 50% of the red blood cells. CH50 is a useful screening tool to detect deficiencies of the classical pathway.
Serum levels of complement components can serve as markers of disease activity. In immune complex deposition disease, serum complement proteins are consumed and serum levels decrease. Immune complex disease results from the deposition of antigen-antibody complexes in involved organ tissues. In immune complex glomerulonephritis in SLE, decreased C3 and C4 indicate increased consumption and indicate disease activity. In contrast, increases of C3 and C4 indicate inflammatory disorders as these proteins are also acute phase reactants. Hypocomplementemia is generally not specific for any disease and may be secondary to nonrheumatic diseases such as subacute bacterial endocarditis or poststreptococal glomerulonephritis. If C4 is low compared to C3, it may indicate the presence of cryoglobulins or the genotype C4 null allele. If CH50 is low or undetectable, it may indicate a deficiency of one or more complement components. Genetic/congenital deficiencies of early complement components (C1 to C4) may increase the risk for development of immune-complex diseases. For example, congenital C1q deficiency, although rare in the general population, is associated with individuals who develop lupus.2, 3, 4, 8
Immunoglobulins (quantitative and qualitative)
Measuring total quantitative immunoglobulin (Ig) levels are a key component to any immunologic evaluation. Ig levels reflect B cell function (humoral production and T cell interaction) and serum Ig levels aid in disease detection. Quantitative measurements of serum immunoglobulins, mainly IgG, IgA and IgM are measured via nephelometry. Table 3 lists diseases that are associated with increased or decreased serum Ig levels.
Table 3.
Differential diagnosis for immunoglobulin levels
| Immunoglobulin | Increased | Decreased |
|---|---|---|
| Ig G | Infection, inflammation, hyperimmunization, IgG multiple myeloma, liver disease, rheumatic fever, systemic rheumatic disease | Agammaglobulinemia, amyloidosis, leukemia, myeloma, preeclampsia |
| IgM | Early HIV infection, infectious mononucleosis, lymphoma, macroglobulinemia, myeloma, rheumatoid arthritis | Rarely agammaglobulinemai, amyloidosis, leukemia, myeloma |
| IgA | Chronic infections (especially of gastrointestinal tract), inflammatory bowel disease, myeloma, rheumatic fever | Agammaglobulinemia, hereditary IgA deficiency, myeloma or protein-losing enteropathy |
Simple qualitative measurements of serum immunoglobulins reflect an individual's ability to mount a humoral immune response. Titers to tetanus, Haemophilus influenza type B (HiB), and pneumococcus can easily be tested to evaluate the quality of the immune response. These levels assess the function of B cells and also detect defects that may indicate immunodeficiency. To assess antibody production, responses to protein and polysaccharide antigens should be evaluated. B-cell testing is done primarily by in vivo (vaccination) studies. Protein vaccinations, like tetanus toxoid, measure T-cell dependent responses. Polysaccharide vaccines, like Pneumovax, measure T-cell independent responses.
Testing of specific antibody titers (such as to influenza immunization) are reported relative to protective values. These values are based on epidemiologic data regarding protection in larger populations. For randomly acquired antibody levels, an initial comparison to protective values can be used to decide if a proper immune response was achieved. A four-fold increase in titers to protein vaccination indicates a normal response. A two-fold increase in titers to a polysaccharide antigen indicates a normal response. Failure to mount an appropriate response to antigen is a clue to the physician to pursue B and T cell function further.2, 3, 4, 8
Cryoglobulins
Cryoglobulins are immunoglobulins that precipitate reversibly in cold temperatures. In disease states, these antibodies can bind with complement proteins and other peptides to form immune complexes and cause tissue damage. Three types of cryoglobulins exist. Type I cryoglobulins are monoclonal immunoglobulins often of the IgM isotype. Type II cryoglobulins are a mixture of polyclonal IgG and monoclonal IgM. Type III cryoglobulins are a combination of polyclonal IgG and polyclonal IgM.
At phlebotomy, whole blood is obtained in pre-warmed tubes without anti-coagulant and maintained at body temperature until coagulation occurs (about one hour). The sample is then centrifuged and the clot is removed. The remaining serum is placed at 4°C up to several days. The specimen is then examined daily to determine if proteins have precipitated. Once a precipitate is present, the sample is spun again and a cryocrit is measured in a calibrated tube. Confirmation of the cryocrit is seen if the precipitate redissolves when placed in a 37°C water bath.
Cryoglobulins are nonspecific indicators of disease states. Type I monoclonal cryoglobulins are associated with multiple myeloma, Waldenstrom's macroglobulinemia, and lymphoproliferative disorders. Type II and III cryoglobulins can bind complement, unlike type I, and are associated with Hepatitis C and small vessel vasculitis. The presence of multiple immunoglobulin components within the cryoglobulin is known as “mixed cryoglobulin.” Symptoms generally associated with cryoglobulins include purpura, ulcerations, Raynaud's phenomenon, arthralgias, proteinuria and renal failure. Cryoglobulins are rarely found in children.2, 3, 4, 8
Lupus Anticoagulant (LAC)/anti-cardiolipin (aCL)/antiphospholipid autoantibodies (aPL)
In autoimmune disease, serum antibodies that inhibit or prolong in vitro clinical laboratory coagulation tests are termed anti-phospholipid antibodies (aPL, also called anti-cardiolipin antibodies or lupus anticoagulant) because they are directed against phospholipids and phospholipid-binding proteins. The existence of these antibodies is associated with the anti-phospholipid antibody syndrome (APS). APS is suspected in individuals that have venous and/or arterial thromboses, recurrent fetal loss, or thrombocytopenia. APS can occur independent or with systemic rheumatic diseases. Anti-phospholipid antibodies can also be found in healthy individuals and in patients with various infections that do not have features of anti-phospholipid antibody syndrome. The term anticoagulant is paradoxical in that the presence of aPL are associated with thromboses in patients. The presence of aPL can be directly measured by enzyme linked assays. IgG anticardiolipin has a higher predictive value than IgM or IgA of a thrombosis. Beta-2 glycoprotein I has been identified as one of the major antigenic determinants of antiphospholipid antibodies.11, 12, 13, 14
4. Flow cytometry
Flow cytometry is a technique where particles or tagged cells flow through laser light so that populations of particle/cells can be counted and phenotyped using cell characteristics and surface proteins. Initial applications of flow cytometry pertained to the interest in certain cell populations, for example the numbers of lymphocytes in patients infected with human immunodeficiency virus (HIV). The number of T cells that are CD4 positive is an important gauge of severity of HIV infection. However, the methodology has greatly expanded its role such that cell cycle analysis, quantification of malignant cells and activation status of lymphocyte subpopulations can be determined. When evaluating a patient with a suspected immunodeficiency, flow cytometry is crucial to determine the quantitative number of immune cells (typically T, B and NK (natural killer) cells). Remember, flow cytometry testing reveals numbers of cells and does not indicate cellular function. Testing for cellular functioning involves other laboratory methods, such as quantitative immunoglobulin levels to indicate proper B cell function.15, 16
The markers commonly used to assess lymphocyte subsets by flow cytometry are as listed in Table 4.
Table 4.
| Marker | Cell type | Comment |
|---|---|---|
| CD3 | T cells | Expressed on all T cells |
| CD4 | T cell subset | Helper/inducer T cells |
| CD8 | T cell subset | Cytotoxic T cells: expressed by up to 1/3 of NK cells |
| CD19 or CD20 | B cells | |
| CD16 | NK cells | Some NK cells may not express CD16 |
| CD56 | NK cells | Expressed on majority of NK cells |
The flow cytometry device consists of an illumination source, an optical bench, fluidic system, electronic monitoring and computer. Cells that will flow through the cytometer are first prepared by tagging cell surface molecules with fluorescently labeled monoclonal antibodies. Illumination of the cell occurs by air-cooled lasers that provide a monochromatic light source (argon at 488 nm or blue, helium neon at 633 nm or red). The point of illumination occurs within the flow cell. The optical bench contains lenses that shape and focus the illumination beam. Nonfluorescent and fluorescent signals generated by the labeled cell are collected and measured by a detection system consisting of filters linked to a photodetector. Cells are injected into a moving fluid sheath to establish a focused single-file flow of cells that move through the analysis point. Differences in the magnitude of emission signal generated from each cell reflect biologic differences between the cells. Software collects data that analyzes cell subpopulations based on the presence or absence of labeled antibody binding. Data is then presented as fluorescence intensity versus cell number.8
5. Cytokine studies
Cytokines are molecules secreted by a variety of cells that function in cellular communication. Immunologists are keenly interested in cytokines, particularly those that influence immune function and inflammation. Commercial testing laboratories do not routinely assay most serum cytokine levels, as this testing is largely done in research laboratories. Testing is laborious because of the labile nature of these small molecules. After phlebotomy, the serum needs to be quickly removed from the cellular components and frozen as quickly as possible and testing should not be delayed. Laboratory methods commonly used to assay cytokine levels include flow cytometry and ELISA.15, 16
Cytokines that influence inflammation include IL-1, IL-6 and TNF-alpha. There is extensive evidence that these cytokines promote inflammation and therefore have become targets for therapy. Rheumatoid arthritis is the best example of an autoimmune illness where anti-TNF therapy has revolutionized the natural history of the disease. Targeting TNF with proteins (fusion produced or monoclonal antibodies) that antagonize TNF action results in dramatic improvement of disease activity. In fact, rheumatoid arthritis is the prototypic autoimmune disease where the efficacy of anti-cytokine therapy is best demonstrated. Currently, anti-TNF, anti-IL-1 and anti-IL-6 therapies have all proven to be effective in treating rheumatoid arthritis.
6. Major Histocompatibility Complex (MHC) (human leukocyte antigen (HLA))
Human leukocyte antigen (HLA) is synonymous with the major histocompatibility complex (MHC). MHC class I and II genes are the major genetic determinants of susceptibility to many autoimmune diseases. MHC class I molecules include HLA-A, -B, and –C. MHC class II molecules include HLA-DR, HLA-DQ, and HLA-DP. Detection of HLA type can be done routinely and can be assayed using several methods that include gel electrophoresis, polymerase chair reaction (PCR), ELISA, and newer methods employing high-throughput detection of nucleic acid. Many antigens of the MHC, especially of HLA class I and II, have been associated with rheumatic disorders. HLA-B27 is present in approximately 90% - 95% of white patients with ankylosing spondylitis and only 7% to 8% of the general population. HLA-DR1 and HLA-DR4 increase the risk of polyarticular juvenile idiopathic arthritis (JIA) in many populations. HLA-DR3 and HLA-DR2 are associated with lupus in Caucasian populations, while much of the risk attributable to MHC is associated with variation at HLA-DRB1 in patients with rheumatoid arthritis.3, 17, 18
II. Specific Immunologic Disease Entities
1. Immunologic Lung Disease
Sarcoidosis
Sarcoidosis is a systemic granulomatous disease characterized by noncaseating granulomas affecting multiple organ systems. The etiology of sarcoidosis is not known but believed to involve chronic inflammation, with a Th-1 cellular contribution, and because immunosuppressive therapy is beneficial. The organ systems most frequently involved, in decreasing order, include lungs, skin, sinus and upper respiratory tract, eye, musculoskeletal, abdominal, hematologic, salivary/parotid, cardiac and neurologic. Biopsy of the affected tissue is vital to diagnosis and histological findings should show noncaseating granulomas. Imaging studies, in particular, chest x-ray or chest CT, show bilateral hilar lymphadenopathy and or interstitial infiltrates. Chest CT reveals nodular infiltrates that tend to be distributed along the bronchoalveolar structures.
Laboratory investigations helpful in the diagnosis of sarcoidosis include a serum angiotensin converting enzyme (ACE) level and vitamin D levels. ACE levels are generally elevated, however, ACE levels lack disease specificity and therefore have limited diagnostic and therapeutic utility. 4 High serum levels of 1, 25(OH)2D3 vitamin D are commonly seen in granulomatous disease and are believed to induce hypercalcemia. IFN-gamma produced by Th1 cells is possibly a stimulus for 25(OH)2D3 synthesis by macrophages.
Churg-Strauss syndrome
Churg-Strauss syndrome (CSS) is a necrotizing vasculitis affecting small and medium blood vessels characterized by eosinophilic infiltration, eosinophilic granulomas, nasal polyps, allergic rhinitis, conductive hearing loss, eye disease (scleritis, episcleritis, uveitis), asthma, fleeting infiltrates, alveolar hemorrhage, segmental necrotizing glomerulonephritis, heart failure, and vasculitic neuropathy. Initial symptoms typically suggest a reactive airway process similar to asthma. Immunologic studies that aid in the diagnosis of CSS include a CBC that reveals a peripheral eosinophila and the serologic presence of an ANA and a p-ANCA directed against MPO. As is the case with any suspected vasculitis, a biopsy of the involved organ showing an inflammatory destruction of the blood vessels, with eosinophilic infiltrates and granuloma formation is vital for diagnosis. Characteristic of all inflammatory vasculitidies are significantly elevated ESR and CRP.
Wegener's granulomatosis (WG) and microscopic polyangiitis (MPA)
WG is characterized by systemic granulomatous vasculitic lesions of the upper and lower respiratory tract and the kidney. MPA is characterized by nongranulomatous vasculitic lesions of the lower respiratory tract, kidney, skin and nerve. Both of these diseases can be manifested by acute renal failure with the urinalysis showing RBCs, RBC casts, and proteinuria. Renal biopsy reveals a segmental necrotizing glomerulonephritis with a characteristic lack of immune complexes. WG can be a chronic, undiagnosed illness where a patient complains of chronic sinusitis that may cause nasal septal perforation. The auditory nerve can be inflamed causing a conductive or sensorineural hearing loss. Cartilage inflammation will cause subglottic stenosis and/or saddle-nose deformity; ocular inflammation leads to orbital pseudotumor, scleritis, episcleritis, or uveitis; and lung disease with nodules, infiltrates, and/or cavitary lesions and/or alveolar hemorrhage. Characteristic of vasculitis, the ESR and CRP will be elevated. Other laboratory evaluations for WG and MPA include a serologic finding of C-ANCA/PR3 generally associated with WG and P-ANCA/MPO with MPA. Important HLA associations with WG are HLA-DR4, DR13.
Goodpasture's syndrome
Antiglomerular basement membrane disease or Goodpasture's syndrome is characterized by pulmonary hemorrhage and/or glomerulonephritis. Pathognomonic to Goodpasture's syndrome are autoantibodies to the renal glomerular and lung alveolar basement membrane. The specific autoantigen is the 235 amino acid carboxy-terminal noncollagenous domain of type IV collagen. Urinalysis of affected individuals reveals proteinuria, dysmorphic RBCs, WBCs, and RBC cellular and granular casts. Diagnostic testing includes the detection of antiglomerular basement membrane antibodies. In cases where only the presence of diffuse alveolar hemorrhage occurs, antibodies may not be present. In those cases, the diagnosis is established by demonstrating linear immunofluorescence in lung tissue.
Other autoimmune diseases with pulmonary manifestations
Many autoimmune diseases have pulmonary complications. For example, extraarticular manifestations of rheumatoid arthritis include parenchymal lung disease with nodules and/or diffuse interstitial lung disease, obliterative bronchiolitis, or bronchiectasis. Pleural effusions and pleurisy can be bilateral in up to one-forth of cases of RA. Pleurisy is very common in SLE. Other pulmonary diseases in SLE include pneumonitis, pulmonary hemorrhage, and shrinking lung syndrome. These pulmonary processes may cause cough with hemoptysis, and dyspnea. The antiphospholipid syndrome's pulmonary manifestations consist of pulmonary thromboembolism and pulmonary hypertension. Scleroderma pulmonary manifestations of interstitial lung disease and pulmonary hypertension are the leading cause of death in systemic sclerosis. Lastly, idiopathic inflammatory myositis – polymyositis, dermatomyositis, and inclusion body myositis (PM/DM/IBM) - have varying lung disease, primarily that of interstitial lung disease.
The astute physician will be aware of the patient's symptoms so that they can pursue further investigations into the etiology of pulmonary disease. Remembering that chronic lung disease can be associated with spurious rheumatoid factor autoantibodies, one must test for serum autoantibodies that are specific to a disease state. Anti-CCP antibodies help to diagnose rheumatoid arthritis if accompanied by radiographic evidence demonstrating erosive disease and clinical evidence of joint involvement. High titer ANA with anti-DNA, anti-ENA autoantibodies are helpful to diagnose SLE, particularly if hypocomplementemia exists and the urine shows hematuria, proteinuria and cellular elements present. The presence of anti-phospholipid antibodies can be confirmed with a positive lupus anticoagulant test, or anti-cardiolipin antibody or beta-2 glycoprotein antibody. The diagnosis of scleroderma is assisted with anti-SCL-70 antibodies or anti-centromere antibodies. For suspected lung disease associated with a myopathy, elevations of CK, AST, ALT, aldolase in conjunction with the presence of a myositis-specific autoantibody are very helpful.8
2. Inflammatory or immune mediated renal disease
Amyloidosis
Amyloidosis describes a group of disorders characterized by the extracellular tissue deposition of a variety of low molecular weight proteins called amyloid. Currently, there are more than 25 types of amyloid. The most frequent is type AL, which is found in primary amyloidosis as well as in myeloma-associated amyloidosis. Type AA, is found in secondary amyloidosis, associated with chronic infections or inflammatory disease and some periodic fever syndromes, such as Familial Mediterranean fever. Deposition of AL and AA, occurs primarily in the kidneys (causing asymptomatic proteinuria, nephrotic syndrome, renal failure, and ESRD), heart (cardiomyopathy, systolic or diastolic dysfunction, heart block, angina or infarct) and liver (hepatomegaly) and gastrointestinal (gastroparesis, constipation, bacterial overgrowth, malabsorption and intestinal pseudo-obstruction).
Amyloidosis from chronic inflammation will reveal an elevated ESR and CRP – clues to suggest inflammation is present and amyloid should be considered as a cause of organ dysfunction. Testing serum levels of type A amyloid is possible in research laboratories. Diagnosis is confirmed by tissue or aspiration biopsy of the affected organ looking for birefringent material on Congo red stain.
Henoch-Schonlein purpura (HSP) and IgA nephropathy
Henoch-Schonlein purpura is an immune-mediated vasculitis associated with immunoglobulin A (IgA) deposition and is the most common form of systemic vasculitis in children. Signs and symptoms include palpable purpura, arthritis, arthralgias, abdominal pain and renal disease. Adults with HSP have presentations similar to children and are at increased risk for developing significant renal disease. Renal disease presents similarly to IgA nephropathy - characterized by deposition of IgA immune complexes causing glomerulonephritis.
Diagnosis by laboratory tests can include elevated levels of serum IgA, elevated ESR and CRP, a normochromic, normocytic anemia and a urinalysis showing RBCs or WBCs, cellular casts, and proteinuria. Renal biopsy discloses IgA deposition in the mesangium.
Systemic lupus erythematosus (SLE)
Systemic lupus erythematosus is an autoimmune disease that can affect most organs. Renal involvement occurs with a high incidence and clinical features include hematuria, proteinuria, nephrotic and/or nephritic syndrome, acute renal failure and chronic renal failure. Analysis of urine can show various casts – RBC, WBC, granular and waxy, in addition to oval fat bodies. In active disease, both the classic and alternative complement cascades are activated resulting in depressed serum levels of complement components (C3 and C4 are typically tested). Most patients with lupus nephritis will have high titers of anti-dsDNA auto-antibodies.19
Anti-glomerular basement membrane (GBM) disease
As described previously in the immunologic lung disease section of this chapter, antiglomerular basement membrane disease or Goodpasture's syndrome is characterized by anti-GBM autoantibodies. Patients presenting with renal involvement often have abrupt onset of oliguria or anuria, hematuria, and anemia. Renal biopsy can reveal crescents in more than 50% of the glomeruli on light microscopy (LM). Immunofluorescence (IF) demonstrates linear deposition of IgG along the glomerular capillaries and occasionally the distal tubules. Rarely, IgA or IgM may be present. Anti-GBM antibodies are also detected in the serum via IF or ELISA. Many patients are also found to test positive for ANCA, particularly p-ANCA/MPO.
Wegener's granulomatosis (WG) and microscopic polyangiitis (MPA)
As discussed previously, this small to medium sized vasculitis is able to cause inflammation in several tissues. Vasculitis often involves the kidney causing proteinuria, hematuria, cellular casts, hypoalbuminemia and renal failure. Serologic testing shows WG and MPA to be ANCA associated diseases with specificity of the ANCA to PR3 in WG and MPO in MPA.
Other autoimmune diseases with renal manifestations
In a similar fashion discussed previously where autoimmune diseases cause pulmonary disease, the same scenario is seen with autoimmune disease and renal involvement. Rheumatoid arthritis, anti-phospholipid antibody syndrome, scleroderma and Sjögren's syndrome all have known renal complications. Proteinuria may be the first clue that the kidney is involved from the result of chronic inflammation or immune complex deposition. Persistent inflammation seen in rheumatoid arthritis can cause a membranous nephropathy, the result of reactive amyloid deposition. One must also remember that therapies to treat inflammatory disease, as seen in RA, such as medications like gold or penicillamine can cause proteinuria. Vasculitis lesions may also occur in patients with severe RA. Therefore, a vigilant watch of renal function with frequent urinalysis is critically important in monitoring the autoimmune patient.20
3. Immunologic neuromuscular disease
Polymyositis (PM)/Dermatomyositis (DM)/Inclusion body myositis (IBM)
Polymyositis, dermatomyositis, and inclusion body myositis constitute the idiopathic inflammatory myopathies (IIM). While the etiology is not well defined, muscles become inflamed as the result of both humoral and cellular immune dysfunction causing a lymphocytic infiltration and muscle damage. These myopathies differ in their etiology, clinical presentation, and histology.
Polymyositis is defined by symmetric proximal muscle weakness, elevations in muscle enzymes, characteristic EMG findings and a muscle biopsy that shows inflammation. The illness is progressive with clinical symptoms that may include myalgias, dysphagia, and dyspnea. Dermatomyositis mimics polymyositis with the addition of skin rashes such as a heliotrope rash and Gottron's papules. The rate of malignancy is increased in patients with IIM, more so in DM patients and may precede, coincide with or postdate the diagnosis. Therefore, screening for malignancy is very important. Inclusion body myositis is the most common form of IIM in patients over the age of 50. Features that set IBM apart from other forms of myositis are asymmetry and distal involvement, particularly affecting the foot extensors and finger flexors. The disease tends to be indolent in its progression and resistant to therapy.
Diagnostic testing for myositis includes common laboratory tests (CBC, CMP), serologies, imaging studies and muscle biopsy. Serum muscle enzymes are elevated such as creatinine kinase (CK) and what are most often thought of as liver function tests - AST, ALT and lactate dehydrogenase (LDH). The AST, ALT and LDH actually reflect muscle disease as well as liver disease. CK levels usually rise 10 to 100 fold of normal and transaminases can increase to 10 fold. Myositis-specific antibodies (MSA) are found in approximately 50% of affected patients. These antibodies are listed in Table 2 and are helpful in predicting the future course and/or prognosis of disease. For example, anti-tRNA synthetase antibodies (eg. anti-JO-1 antibodies) are strong predictors of interstitial lung disease. Genetic risk factors for the development of myositis include the alleles HLA-DRB1 *0301 and HLA-DQA1 *0501. Muscle biopsy is helpful to distinguish PM, DM and IBM. PM typically demonstrates a lymphocytic infiltration seen mostly within the fascicles (endomysial inflammation), some fiber necrosis, degenerative and regenerative fibers. MHC class I antigens on fibers identified by immunohistochemistry can also be seen. In dermatomyositis, perifascicular atrophy is common. The main features of IBM include endomysial inflammation, vacuolization (“red-rimmed vacuoles” on Gomori trichrome stain) and loss of muscle fibers. Large, atrophic or angulated fibers are also present. 8, 21, 22
Table 2.
lists specific autoantibodies with their antigenic determinant and clinical disease associations.
| Autoantibody | Antigenic Determinant | Clinical Associations |
|---|---|---|
| Anti-dsDNA | Double stranded-DNA | High specificity for SLE |
| Often correlates with active, severe disease | ||
| Anti-Extractable | ||
| Nuclear Antigens | ||
| Anti-Sm | Smith | High specificity for SLE |
| Anti-RNP | proteins containing U1-RNA | MCTD, SLE, RA, Scleroderma, Sjögren's |
| Anti-SSA (Ro) | Ribonucleoproteins | Sjögren's syndrome, SLE (subacute cutaneous lupus), neonatal lupus |
| Anti-SSB (La) | Ribonucleoproteins | Sjögren's syndrome, SLE, neonatal SLE |
| Anti-centromere | Centromere/kinetochore region of chromosome | Limited scleroderma, pulmonary hypertension, primary biliary cirrhosis |
| Anti-Scl 70 | DNA topoisomerase I | Diffuse scleroderma |
| Anti-Jo-1 (anti-synthetase Abs) | Histidyl tRNA synthetase (other tRNA synthetases) | Inflammatory myopathies with interstitial lung disease, fever and arthritis |
| Anti-SRP | Antibody to signal recognition protein | Inflammatory myopathies with poor prognosis |
| Anti-PM/Scl | Antibody to nucleolar granular component | Polymyositis/scleroderma overlap syndrome |
| Anti-Mi-2 | Antibodies to a nucleolar antigen of unknown function | Dermatomyositis |
MCTD = mixed connective tissue disease
4. Hematologic disease
Autoimmune hemolytic anemia (AIHA)
AIHA is characterized by increased erythrocyte destruction and/or decreased red cell survival due to autoantibodies directed against self-antigens on red cells. Two major types of AIHA exist – warm and cold. AIHA due to the presence of warm agglutinins is almost always due to IgG antibodies that react with protein antigens on the erythrocyte surface at body temperature (37°C). The etiology of warm AIHA includes infections (often in children), autoimmune diseases (SLE), malignancies of the immune system (Non-Hodgkin's lymphoma, chronic lymphocytic leukemia), prior allogenic blood transfusion, and certain drugs (cephalosporins, hydralazine, isoniazid (INH), sulfonamides, tetracycline, triamterene). Signs and symptoms of mild warm AIHA include anemia, occasionally jaundice, and mild to moderate splenomegaly with severe cases also involving fever, tachypnea, tachycardia, angina or heart failure. Peripheral blood exam cam show leukopenia, neutropenia or thrombocytopenia but often patients have a normal platelet count, neutrophilia, and mild leukocytosis. Peripheral blood smear reveals polychromasia, spherocytosis, fragmented and nucleated erythrocytes and sometimes erythrophagocytosis by monocytes in severe cases. Reticulocytosis is commonly present. Serum haptoglobin levels are decreased and LDH increased. Diagnosis depends on the demonstration of immunoglobulin and/or complement proteins bound to the patient's erythrocytes via direct antiglobulin test (DAT).23, 24, 25
The other major form of AIHA is due to cold-reactive antibodies and includes the cold agglutinin syndrome (CAS) and paroxysmal cold hemoglobinuria (PCH). In cold reactive AIHA, the antibodies exhibit a greater affinity for erythrocytes at temperatures below 37°C and cause red blood cell membrane injury by activating complement. The majority of cold-reactive autoantibodies are cold agglutinins.26
CAS usually occurs in middle-aged and elderly persons with IgM the responsible immunoglobulin. Signs and symptoms are chronic anemia, dark urine, acrocyanosis, pallor and jaundice. Laboratory testing includes mild to moderate anemia with prominent autoagglutination, abnormal erythrocyte morphology, reticulocytosis, jaundice, hemoglobinuria, and erythroid hyperplasia in the bone marrow
PCH frequently occurs in children, often after an upper respiratory infection and IgG is typically the causative immunoglobulin. Acute attacks are often severe but the illness usually resolves spontaneously within a few days to several weeks. Signs and symptoms consist of fever, malaise, abdominal pain, dark colored urine, jaundice and pallor. Laboratory findings show anemia (often severe), reticulocytosis, abnormal RBC morphology, hemoglobinuria, usually erythroid hyperplasia, leukocytosis and a platelet count that is normal or slightly elevated.
Vascular thrombotic disorders
Antiphospholipid syndrome is a thrombophilic disease defined by two major components – the presence of at least one type of autoantibody (antiphospholipid antibody (aPL) as mentioned earlier in this chapter), which are directed against phospholipid-binding plasma proteins, and the occurrence of at least one of several clinical features (recurrent fetal loss, arterial thromboses, or thrombocytopenia). Primary antiphospholipid antibody syndrome is diagnosed in the absence of other underlying diseases and secondary if another illness (such as SLE) is identified. Therapy consists of anticoagulation and treating any coexisting illness that may give rise to antiphospholipid antibodies.
Thrombocytopenia (Idiopathic Thrombocytopenia, Post-transfusion purpura)
Immune or idiopathic thrombocytopenic purpura (ITP) is an acquired disorder without a clear etiology. Both acute and chronic forms occur. The pathogenesis is thought to occur through a combination of increased platelet destruction and inhibition of megakaryocyte platelet production by specific IgG autoantibodies. Clinical manifestations are variable, can be abrupt or acute, and often are insidious. Symptoms include bleeding ranging from petechiae and easy bruising to severe bleeding diathesis. Intracranial hemorrhage is quite rare. Children often exhibit symptoms after a viral or bacterial infection. Thrombocytopenia is noted on lab work. Bone marrow cellularity is normal with normal erythropoiesis and myelopoiesis. Megakaryocytes are present in normal to increased numbers. However, bone marrow examination is not usually required unless another disease is suspected or if the patient is greater than 60 years of age because of concern for myelodysplasia.
Post-transfusion purpura (PTP) is a rare condition that usually develops seven to ten days after a red blood cell transfusion. The syndrome is characterized by severe thrombocytopenia and bleeding caused by alloimmunization to human platelet specific antigens following a blood component transfusion. Patients are usually middle-aged multiparous females, although PTP has also been reported in males. Most cases occur with the formation of human platelet antigen – 1a antibodies. These antibodies destroy transfused HPA-1a+ cells and the negative recipient's cells by a poorly understood antiplatelet mechanism.27
Figure 1.
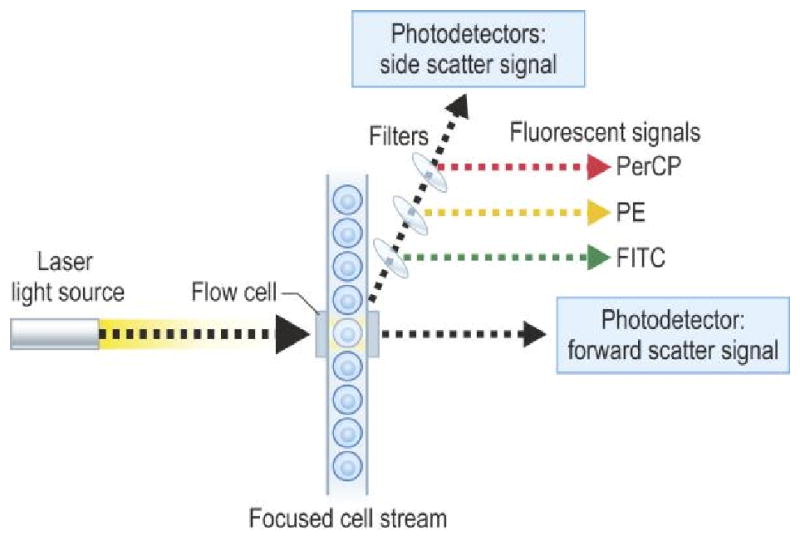
Figure of flow chamber and presentation of data of CD8/CD4 as dot plot (A) and contour plot (B).
from Rich RR, Fleischer TA, Shearer WT, Schreder HW, Frew AJ, Weyand CM. Clinical Immunology Principles and Practice, Third edition. Chapter 97, page 1436, Figure 97.1. Reprinted with permission.
Figure 2.
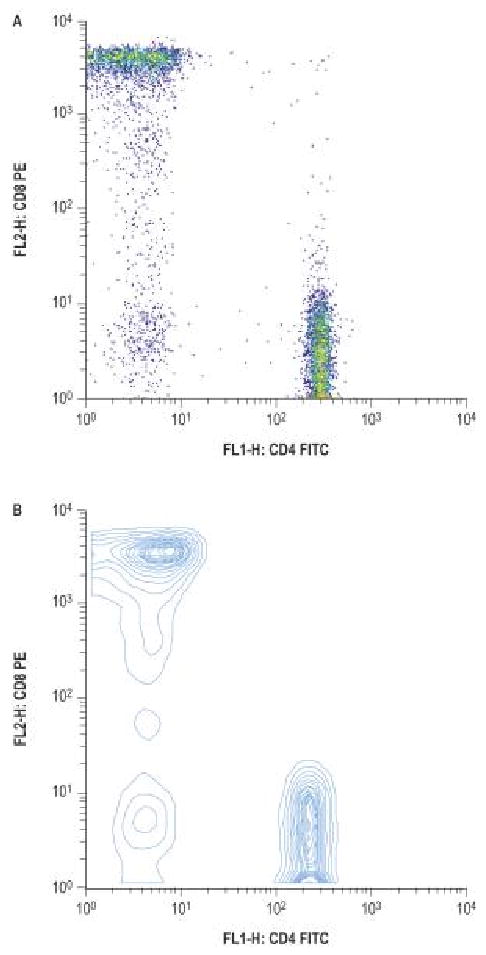
from Rich RR, Fleischer TA, Shearer WT, Schreder HW, Frew AJ, Weyand CM. Clinical Immunology Principles and Practice, Third edition. Chapter 97, page 1438, figure 97.4, A and B. Reprinted with permission.
Abbreviations
- ACE
angiotensin converting enzyme
- aCL
anti-cardiolipin
- ADB
anti-DNase B
- AIHA
autoimmune hemolytic anemia
- ALT
alanine transaminase
- ANCA
anti-neutrophil cytoplasmic antibody
- Anti-dsDNA
anti-double stranded DNA
- Anti-ENA
anti-extractable nuclear antigen
- Anti-GBM
anti-glomerular basement membrane
- aPTT
activated partial thromboplastin time
- aPL
anti-phospholipid
- AST
aspartate aminotransferase
- RBC
red blood cell
- CAS
cold agglutinin syndrome
- CBC
complete blood count
- CCP
cyclic citrullinated peptide
- CK
creatinine kinase
- CMP
complete metabolic panel
- CRP
c-reactive protein
- CSS
Churg-Strauss
- DM
dermatomyositis
- EIA
enzyme immunosorbent assays
- ELISA
Enzyme-linked immunosorbent assay
- ESR
erythrocyte sedimentation rate
- ESRD
end-stage renal disease
- GN
glomerulonephritis
- HiB
Haemophilus influenza type B
- HLA
human leukocyte antigen
- HSP
Henoch-Schonlein purpura
- IBM
inclusion body myositis
- IF
immunofluorescence
- Ig
immunoglobulin
- IIM
idiopathic inflammatory myopathy
- IL
interleukin
- INH
isoniazid
- ITP
idiopathic or immune thrombocytopenic purpura
- JIA
juvenile idiopathic arthritis
- LAC
lupus anticoagulant
- LDH
lactate dehydrogenase
- LM
light microscopy
- MHC
major histocompatibility complex
- MCTD
mixed connective tissue disease
- MPA
microscopic polyangiitis
- MPO
myeloperoxidase
- PCH
paroxysmal cold hemoglobinuria
- PCR
polymerase chain reaction
- PM
polymyositis
- PR3
proteinase-3
- PT
prothrombin time
- PTP
post-transfusion purpura
- RA
rheumatoid arthritis
- RF
rheumatoid factor
- RNP
ribonucleoprotein
- SLE
systemic lupus erythematosus
- Sm
Smith
- TNF
tumor necrosis factor
- WBC
white blood cell
- WG
Wegener's granulomatosis
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.McPherson RA, Pincus MR. Henry's Clinical Diagnosis and Management by Laboratory Methods. 21st. Philadelphia, Pa: WB Saunders; 2007. pp. 40–1. [Google Scholar]
- 2.Klippel JH, Stone JH, Crofford LJ, White PH. Primer on Rheumatic Diseases. Thirteenth. Chapter 2. Springer; 2008. pp. 15–20. [Google Scholar]
- 3.Lahita RG, Weinstein A. Educational Review Manual in Rheumatology. 4th. Chapter 1. New York, NY: Castle Connolly Graduate Medical Publishing, Ltd.; 2007. pp. 1–42. [Google Scholar]
- 4.Breda L, Nozzi M, de Sanctis S, Chiarelli F. Laboratory tests in the Diagnosis and follow-up of Pediatric Rheumatic Diseases: An Update. Sem Arth Rheum. 2009 doi: 10.1016/j.semarthrit.2008.12.001. Article in press. [DOI] [PubMed] [Google Scholar]
- 5.Koulaouzidis A, Cottier R, Bhat S, Said E, Linaker BD, Saeed AA. A ferritin level > 50 ug/L is frequently consistent with iron deficiency. Eur J Int Med. 2009;20:168–170. doi: 10.1016/j.ejim.2007.09.024. [DOI] [PubMed] [Google Scholar]
- 6.Therul I, Aigner E, Theuri M, Nairz M, Seifert M, Schroll A, et al. Regulation of iron homeostasis in anemia of chronic disease and iron deficiency anemia: diagnostic and therapeutic implications. Blood. 2009;113:5277–5286. doi: 10.1182/blood-2008-12-195651. [DOI] [PubMed] [Google Scholar]
- 7.Hellman NE, Gitlin JD. Ceruloplasmin Metabolism and Function. Ann Rev Nutr. 2002;22:439–458. doi: 10.1146/annurev.nutr.22.012502.114457. [DOI] [PubMed] [Google Scholar]
- 8.Rich RR, Fleischer TA, Shearer WT, Schreder HW, Frew AJ, Weyand CM. Clinical Immunology Principles and Practice. Third. Chapters 96, 97, and 98 [Google Scholar]
- 9.Miller FW. Arthritis and Allied Conditions – A textbook of Rheumatology. Lippcott, Williams, and Wilkins; Philadelphia: 2005. Inflammatory Myopathies: Polymyositis, Dermatomyositis, and Related Conditions; pp. 1593–1620. [Google Scholar]
- 10.Colglazier CL, Sutej PG. Laboratory testing in the rheumatic diseases: a practical review. South Med J. 2005 Feb;98(2):185–91. doi: 10.1097/01.SMJ.0000153572.22346.E9. [DOI] [PubMed] [Google Scholar]
- 11.Reymond JL, Fluxa VS, Maillard N. Enzyme assays. Chem Comm. 2009;1:34–46. doi: 10.1039/b813732c. [DOI] [PubMed] [Google Scholar]
- 12.Milos M, Herak D, Kuric L, Horvat I, Zadro R. Evaluation and performance characteristics of the coagulation system: ACL TOP analyzer – HemosIL reagents. Int J Lab Hema. 2009;31(1):26–35. doi: 10.1111/j.1751-553x.2007.00999.x. [DOI] [PubMed] [Google Scholar]
- 13.Tebo AE, Jaskowski TD, Hill HR, Branch DW. Clinical relevance of multiple antibody specific testing in antiphospholipid syndrome and recurrent pregnancy loss. Clin Exp Imm. 2008;154(3):332–338. doi: 10.1111/j.1365-2249.2008.03774.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tripodi A. Testing for lupus anticoagulants: all that a clinician should know. Lupus. 2008;18:291–298. doi: 10.1177/0961203308101436. [DOI] [PubMed] [Google Scholar]
- 15.Favaloro EJ, Wong CW. Laboratory Testing and Identification of Antiphospholipid Antibodies and the Antiphospholipid Syndrome: A Potpourri of Problems, a Compilation of Possible Solutions. Sem Thromb Hemost. 2008;34(4):389–410. doi: 10.1055/s-0028-1085482. [DOI] [PubMed] [Google Scholar]
- 16.Fleisher TA. Basic principles and clinical applications of flow cytometry. In: Rodgers G, Young NS, editors. Bethesda Handbook of Clinical Hematology. Lippincott Williams & Wilkins; Baltimore: 2005. pp. 415–424. [Google Scholar]
- 17.Rostaing LM, Tkaczuk J, Durand M, Peres C, Durand D, de Preval C, et al. Kinetics of intracytoplasmic Th1 and Th2 cytokine production assessed by flow cytometry following in vitro activation of peripheral blood mononuclear cells. Cytometry. 1999;35:318–328. doi: 10.1002/(sici)1097-0320(19990401)35:4<318::aid-cyto4>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 18.Hamdahl M, Justesen S, Lamberth K, Roder G, Neilsen M, Buus S. Peptide binding to HLA Class I molecules: Homogenous, high-throughput screening, and affinity assays. J Biomol Screen. 2009;14(2):173–180. doi: 10.1177/1087057108329453. [DOI] [PubMed] [Google Scholar]
- 19.Dunbar SA. Applications of Luminex xMAP technology for rapid, high-throughput multiplexed nucleic acid detection. Clin Chim Acta. 2006;363:71–82. doi: 10.1016/j.cccn.2005.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seshan SV, Jennette JC. Renal Disease in Systemic Lupus Erythematosus with Emphasis on Classification of Lupus Glomerulonephritis. Arch Pathol Lab Med. 2009;133:233–248. doi: 10.5858/133.2.233. [DOI] [PubMed] [Google Scholar]
- 21.Batal I, Domsic RT, Shafer A, Medsger TA, Kiss LP, Randhawa P, et al. Renal biopsy findings predicting outcome in scleroderma renal crisis. Hum Path. 2009;40:332–340. doi: 10.1016/j.humpath.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 22.Dalakis MC. Muscle biopsy findings in inflammatory myopathies. Rheum Dis Clin N Am. 2003;28:779–798. doi: 10.1016/s0889-857x(02)00030-3. [DOI] [PubMed] [Google Scholar]
- 23.Katz JD, Nayyar G. Introduction: Arthritis and Myositis. Ann of New York Acad of Sciences. 2009;1154:3–9. doi: 10.1111/j.1749-6632.2009.04380.x. [DOI] [PubMed] [Google Scholar]
- 24.Das SS, Nityanand S, Chaudhary R. Clinical and serological characterization of autoimmune hemolytic anemia in a tertiary care hospital in North India. Ann Hematol. 2009;88:727–732. doi: 10.1007/s00277-008-0674-6. [DOI] [PubMed] [Google Scholar]
- 25.Lin JS, Hao TC, Lyou JY, Chen YJ, Liu HM, Tzeng CH, et al. Clinical application of flow cytometric direct antiglobulin test. Transfusion. 2009;49(7):1335–1346. doi: 10.1111/j.1537-2995.2009.02130.x. [DOI] [PubMed] [Google Scholar]
- 26.Packman CH. Hemolytic anemia due to warm autoantibodies. Blood Reviews. 2008;22:17–31. doi: 10.1016/j.blre.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 27.Petz LD. Cold antibody autoimmune hemolytic anemias. Blood Rev. 2008;22:1–15. doi: 10.1016/j.blre.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 28.Shtalrid M, Shvidel L, Vorst E, Weinmann EE, Berrebi A, Sigler E. Post-Transfusion Purpura: A Challenging Diagnosis. IMAJ. 2006;8(10):672–674. [PubMed] [Google Scholar]