

Abstract
The analysis of damage products as biomarkers of inflammation has been hampered by a poor understanding of the chemical biology of inflammation, the lack of sensitive analytical methods, and a focus on single chemicals as surrogates for inflammation. To overcome these problems, we developed a general and sensitive liquid chromatographic tandem mass spectrometry (LC/MS-MS) method to quantify, in a single DNA sample, the nucleoside forms of seven DNA lesions reflecting the range of chemistries associated with inflammation: 2′-deoxyuridine, 2′-deoxyxanthosine, and 2′-deoxyinosine from nitrosative deamination; 8-oxo-2′-deoxyguanosine from oxidation; and 1,N2-etheno-2′-deoxyguanosine, 1,N6-etheno-2′-deoxyadenosine, and 3,N4-etheno-2′-deoxycytidine arising from reaction of DNA with lipid peroxidation products. Using DNA purified from cells or tissues under conditions that minimize artifacts, individual nucleosides are purified by HPLC and quantified by isotope-dilution, electrospray ionization LC/MS-MS. The method can be applied to other DNA damage products and requires 4-6 days to complete depending upon the number of samples.
Search terms: biomarker; inflammation; DNA damage; reactive oxygen species; reactive nitrogen species; oxidative stress; nitrosative stress; macrophage; neutrophil; deamination; 8-oxo-dG, 8-oxo-2′-deoxyguanosine; lipid peroxidation; etheno adducts; xathine; hypoxanthine; uracil; mass spectrometry; chromatograpy; LC/MS-MS
Introduction
The relationship between chronic inflammation and human diseases such as cancer has moved from association to essentially cause-and-effect,1,2 though efforts to predict the risk of diseases associated with inflammation have lagged due to weaknesses in our ability to quantify the chemical and molecular events that link inflammation with disease. The strongest link entails a scenario in which local infection, tissue damage or autoimmune dysfunction signals the infiltration and activation of macrophages and neutrophils to eradicate the offending stimuli.3 Activation of the phagocytes causes them to secrete a host of bioactive molecules, such as nitric oxide (NO) and superoxide (O2●-) by macrophages and hypochlorous acid (HOCl) by neutrophils (Figure 1).4 Further reactions of NO, O2 and O2●- lead to the formation of a variety of reactive nitrogen species such as nitrous anhydride (N2O3), nitrogen dioxide radical (NO2●), peroxynitrite (ONOO-) and nitrosoperoxycarbonate (ONOOCO2-).4 These reactive oxygen and nitrogen species attack virtually all types of cellular molecules to produce mutation and cell death that ultimately lead to expression of disease. Given the potential for DNA damage to play a causative role in mutation and carcinogenesis, we have developed a highly sensitive method to quantify DNA damage products as biomarkers of inflammation.
Figure 1.
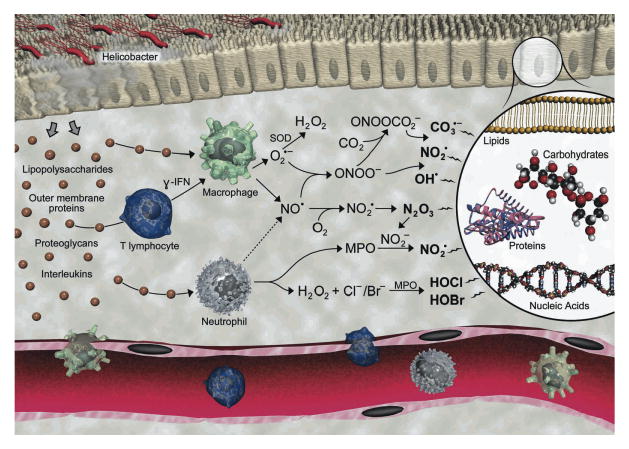
The chemical biology of inflammation. Illustration by Jeff Dixon.
Of a general three-step approach to the development of biomarkers,5 the first step involves selection of an appropriate target molecule(s) thought to be involved in the pathogenic mechanism, which poses a challenge given limited knowledge of the chemistries actually occurring at sites of inflammation. DNA is thus considered to be a major mechanistic target in the association of chronic inflammation with diseases such as cancer, such that DNA damage products may represent biomarkers of inflammation in the carcinogenic process.4,6,7 As shown in Figure 2, the major outcomes of damage to DNA by chemical mediators of inflammation are predicted to fall into three categories: nitrosation, nitration/oxidation and alkylation. The major source of nitrosative deamination of DNA bases is N2O3, which is derived from interaction of NO and O2 and causes conversion of cytosine to uracil (2′-deoxyuridine, dU), guanine to xanthine or oxanine (2′-deoxyxanthosine, dX, or 2′-deoxyoxanosine, dO, respectively) and adenine to hypoxanthine (2′-deoxyinosine, dI).8,9 DNA is also subject to oxidative and nitrative assaults mainly as a consequence of ONOOCO2- that preferentially reacts with dG to form a variety of primary and secondary oxidation and nitration products.10,11 One of the major G oxidation products, 8-oxo-2′-deoxyguanosine (8-oxodG), is approximately 1000-times more reactive than dG towards further oxidation to yield a host of more stable secondary oxidation products.12 It is thus critical to control oxidation artifacts during sample preparation and analysis to determine the correct cellular concentration of 8-oxodG. The third category of DNA lesions arises from reactions of DNA with electrophilic products derived from oxidation of other cellular molecules, such as reactions of lipid peroxidation-derived α,β-unsaturated aldehydes with DNA bases to form unsubstituted etheno adducts 1,N2-etheno-2′-deoxyguanosine (1,N2-εdG), N2,3-etheno-2′-deoxyguanosine (N2,3-εdG), 1,N6-etheno-2′-deoxyadenosine (εdA), and 3,N4-etheno-2′-deoxycytidine (εdC),13,14 as shown in Figure 2. The pyrimidopyrinone alkylation species, M1dG, can arise from reactions of DNA with both the base propenal product of 2-deoxyribose oxidation in DNA15,16 and with malondialdehyde, a product of lipid peroxidation.17 This wide spectrum of candidate chemistries and products arising from inflammation may pose a problem for selection of biomarker target molecules. To this end, we were motivated to develop a sensitive, robust and general approach to quantify DNA lesions caused by the known classes of inflammation chemistry shown in Figure 2. It should be noted that there are many different products of DNA oxidation and alkylation arising from exposure to ionizing radiation and other insults,18-24 but these species are not unlikely to form under the biological conditions of inflammation.4 Further, we have recently initiated efforts to include the cytosine and guanine halo-adducts arising from reactions of DNA with neutrophil-derived HOCl.
Figure 2.
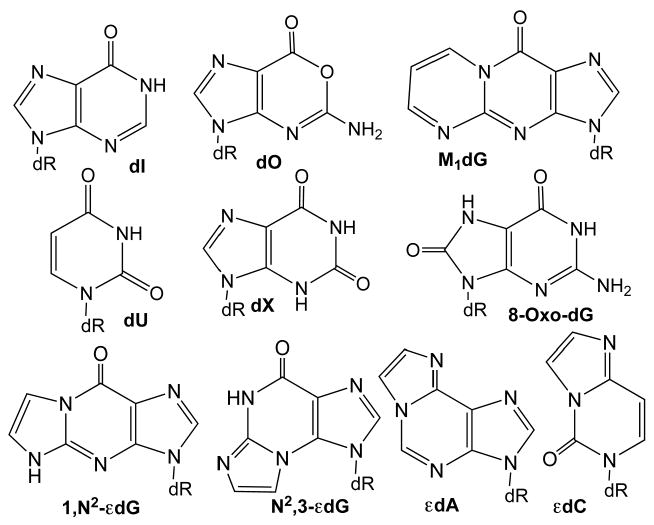
DNA damage products associated with the chemical mediators of inflammation. Abbreviations are defined in the text.
Analysis of low levels of DNA adducts in inflamed tissues is a challenging task that requires high specificity and sensitivity for unequivocal identification and quantification. The most widely used methods include immunoassays, 32P and fluorescent post-labeling methods, and methods coupling chromatography with mass spectrometry, each of which has advantages and drawbacks. 25 For example, the relative simplicity of immunoassays facilitates implementation of biomarkers in a clinical setting,26 while post-labeling methods are considered among the most sensitive for detection of very low levels of DNA adducts.27 However, both methods suffer from a lack of chemical specificity and a lack of internal standards to control for loss and artifacts in sample preparation.
Alternatively, the MS-based methods provide unequivocal identification and the most rigorous quantification by employing isotopically-labeled internal standards.28 Gas chromatography coupled with mass spectrometry (GC/MS) has proven to be a highly sensitive approach to quantification of many types of DNA lesions, though the approach is not as readily applicable to nucleoside forms of base damage as liquid chromatography-based methods and there is some controversy regarding artifacts associated with the method.29,30 Coupling reversed phase HPLC with tandem mass spectrometry (LC/MS-MS) provides excellent separation of nucleosides, readily derived from DNA by enzymatic means, with highly sensitive quantification of 2′-deoxynucleoside species by virtue of the characteristic loss of the deoxyribose moiety during collision-induced dissociation.31 This combination of specificity and sensitivity with ease is unmatched by other bioanalytical methods. The major drawbacks to the use of tandem mass spectrometry are the high cost of the instruments, a requirement for relatively large amounts of DNA (10-100 μg) and the relatively low throughput of the analyses. A logical scenario that respects both the strengths and the drawbacks of MS-based analytical methods is to make an initial investment in developing DNA damage biomarkers of inflammation in animal models and limited human studies using LC/MS-MS technology, followed by development of monoclonal antibodies against those biomarkers found to correlate with inflammation for application in widespread clinical use. To demonstrate the flexibility of our analytical method, we describe the application of the method using two different triple quadrupole mass spectrometry systems commonly available on university campuses with chemistry and bioscience departments and in academic medical centers.
A critical facet of any bioanalytical method for a rare biomolecule involves minimizing background noise or interfering signals for more abundant undamaged molecules. For example, the deaminated nucleobase products dX, dO, dI and dU are one mass unit larger than the canonical nucleobases from which they are derived (dG, dA and dC) such that isotope natural abundance could create an M+1 signal for the highly abundant canonical nucleoside that could interfere with detection of the higher molecular weight deamination product. The addition of a pre-purification step reduces the potential for these problems and increases the sensitivity of the assay. We have chosen to pre-purify damaged nucleosides by HPLC. Pre-purification can also be achieved by immunoaffinity purification but the availability of antibodies is limiting, with HPLC offering the advantage of purifying a battery of DNA lesions simultaneously. After pre-purification, the analytes are subject to a second, different chromatographic step in LC/MS-MS that further reduces interference. For example, the molecular weight of εdC is identical to that of dA such that even slight contamination with dA in the first HPLC resolution could overwhelm the billion-fold lower εdC signal entirely. Even with an on-line immunoaffinity chromatography purification step, the problem of residual dA in the analysis of εdC caused Roberts et al. to convert dA to dI using adenosine deaminase.32 We have taken the more general approach of using a second and different chromatographic resolution step in which we are able to shift the retention time of εdC before that of dA making it possible for it to be quantified without interference from tailing of the dA peak.
A second challenge to the problem of the sensitivity and specificity of a method is the adventitious formation of DNA damage products during DNA isolation and processing steps. We have observed adventitious formation of dI and dU during DNA isolation and hydrolysis as a result of endogenous nucleobase deaminases and deaminase activities contaminating commercial enzyme preparations.9,33 This problem was completely controlled by adding the dA and dC deaminase inhibitors coformycin and tetrahydrouridine, respectively, to all buffers used during DNA isolation and processing. Oxidative artifacts, such as adventitious oxidation of dG to form 8-oxodG, is a major problem that has been shown to depend more on the operator and less on the method34 and can be minimized by addition of the metal-chelating desferroxamine and the antioxidant butylated hydroxytoluene during DNA isolation and hydrolysis steps.
With all of these considerations, we describe a rigorous LC/MS-MS method (Scheme 1) to quantify, in a single DNA sample, seven DNA damage products that reflect the spectrum of different chemistries associated with chronic inflammation.35 The method yields results in accord with reliable published analyses of individual lesions in animal tissues.36,37
Scheme 1.
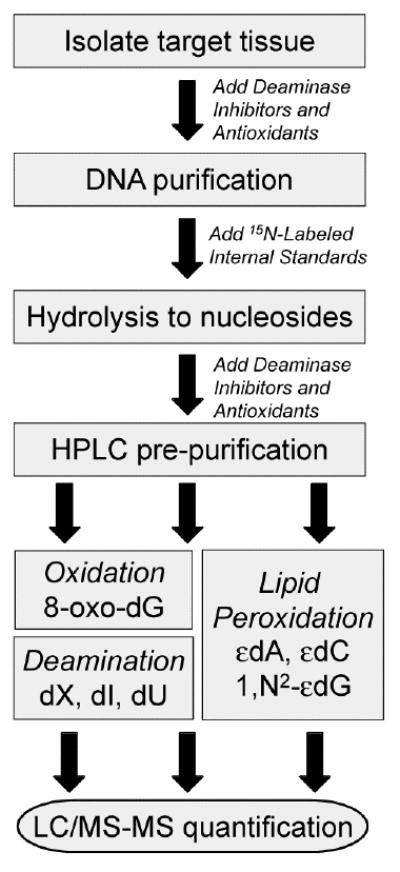
Flow chart for the LC/MS-MS quantification of DNA damage products in cells and tissues
Experimental Design
There are several critical issues to consider before embarking on the quantification of damage products in cells or animal tissues. After choosing an appropriate animal model and target tissue for analysis, The first aspect of experimental design involves the choice of animal model and target tissue for analysis, with the requirement for inflammation studies of an inflammation-inducing stimulus, such as an infection, a non-inflamed control group, and, if at all possible, some evidence for pathology in the target organ. The studies should be designed to provide three or more animals per treatment group, which accommodates statistical treatments of the resulting biomarker quantification (e.g., Student's t-test). Since the largest source of variance in the resulting biomarker levels lies in the individual tissue samples, it is recommended to design the experiment with more than three animals per treatment group. Tissues available in limiting amounts, such as the spleen in mice, may need to be pooled from several mice to provide a single analytical sample, which increases the number of mice per treatment group needed to provide a total of three independent analyses. Tissues from euthanized animals should be snap-frozen in liquid nitrogen as soon as possible after isolation and the DNA isolated immediately upon thawing the tissue. While the protocol is written for tissue samples, the method is readily adapted to studies in cultured cells by simply designing the experiment with large enough cell cultures to provide 100 μg of DNA.
Another important issue to consider in the experimental design is the availability of instruments. We demonstrate the method with two mass spectrometer systems that provide slightly different sensitivities for the various DNA damage products. While the method can be applied to any triple-quadrupole instrument with minor adjustments of the mass spectrometer parameters, the parameters must be determined in experiments first with unlabeled standards and then with samples consisting of purified DNA spiked with labeled and unlabeled internal standards to account for contaminants in the enzyme and buffer preparations and other sources unique to the local environment.
Materials
Reagents
[15N3]-2′-Deoxycytidine (Cambridge Isotope, cat. no. NLM-3897-0)
[15N5]-2′-Deoxyadenosine (Cambridge Isotope, cat. no. NLM-3895-0)
[15N5]-2′-Deoxyguanosine (Cambridge Isotope, cat. no. NLM-3899-0)
1,N2-Etheno-2′-deoxyguanosine (AREL Biosynth. Inc., Salt Lake City, UT)
▲ CRITICAL Etheno adducts are light sensitive and should be stored in tinted vials to prevent degradation of the standard
1,N6-Etheno-2′-deoxyadenosine (Sigma Chemical Co., cat. no. E4132)
▲ CRITICAL Etheno adducts are light sensitive and should be stored in tinted vials to prevent degradation of the standard
2′-Deoxyadenosine (Sigma Chemical Co., cat. no. D7400)
2′-Deoxycytidine (Sigma Chemical Co., cat. no. D3897)
2′-Deoxyguanosine (Sigma Chemical Co., cat. no. D7145)
2′-Deoxyinosine (Sigma Chemical Co., cat. no. D5287)
2′-Deoxyuridine (Sigma Chemical Co., cat. no. D5412)
3,N4-Etheno-2′-deoxycytidine (AREL Biosynth. Inc., Salt Lake City, UT)
▲ CRITICAL Etheno adducts are light sensitive and should be stored in tinted vials to prevent degradation of the standard
8-oxo-7,8-dihydro-2′-deoxyguanosine (Sigma Chemical Co., cat. no. H5653)
▲ CRITICAL Due to its low redox potential, 8-oxoguanosine is prone to secondary oxidation and standard should not be exposed to heat and stored at -80° C for long-term storage
Acetonitrile (VWR, cat. no. BJ015)
Alkaline phosphatase (17 units/μl; Sigma Chemical Co., cat. no. P5521)
Ammonium acetate (Sigma Chemical Co., cat. no. 17836)
Butylated hydroxytoluene (Sigma Chemical Co., cat. no. W218405)
Chloroacetaldehyde (∼50 wt% in H2O; Aldrich cat. no. 317276)
! CAUTION This compound is extremely toxic and carcinogenic, should be handled in a fume hood.
Coformycin (DTP/NCI Open Chemical Repository, http://dtp.nci.nih.gov/dtpstandard/chemname/index.jsp)
Deoxyribonuclease 1 (Sigma, cat. no. D4263)
Desferrioxamine (Sigma, cat. no. D9533)
DNA Isolation Kit for Cells and Tissues (Roche; cat. no. 11 814 770 001)
Ethanol (Pharmco, cat. no. 111ACS200)
Glacial acetic acid (Aldrich, cat. no. 317276)
HPLC-grade water (VWR, cat. no. BJ365)
Isopropanol (VWR, cat. no. MK303216)
Liquid nitrogen (Airgas purity: 99.998%)
Lysis buffer (Roche, cat. no. 11 814 770 001L2)
Nuclease P1 (2 units/μl, US Biological, cat. no. N7000)
Phosphodiesterase I (US Biological, cat. no. 20240Y)
Potassium carbonate (Sigma Chemical Co., cat. no. 209619)
Protein precipitation solution (Roche, cat. no. 11 814 770 001L5)
Proteinase K (Roche, cat. no. 11 814 770 001L3)
RNase (Roche, cat. no. 11 814 770 001L4)
Sodium acetate (Sigma Chemical Co., cat. no. 71183)
Tetrahydrouridine (Calbiochem, cat. no. 584222)
Zinc chloride (Sigma Chemical Co., cat. no. 96468)
Equipment
15 ml Falcon tubes (VWR, cat. no. 21008-918)
50 ml Falcon tubes (VWR, cat. no. 21008-951)
A triple quadrupole mass spectrometer system, such as the Agilent 6410 (Agilent Technologies, Santa Clara, CA) or the API 3000 (Applied Biosystems, Foster City, CA)
Autoclave
Autosampler (Agilent Technologies, Santa Clara, CA)
Avanti J-25 centrifuge (Beckman)
Blue screw caps (Agilent Technologies, cat. no. 5182-0717)
Bunsen burner
HPLC column heater (Agilent Technologies, Santa Clara, CA)
Digital heat block (VWR)
Diode array detector, G1315B (Agilent Technologies, Santa Clara, CA)
DU-640 UV-Vis spectrophotometer (Beckman)
Agilent 1100 HPLC system (Agilent Technologies, Santa Clara, CA)
Phenomenex Synergi 4μ Hydro-RP 80A 250×4.6 mm HPLC column (Phenomenex, cat no. 00G-4375-E0)
Thermo Hypersil Gold 3μ 150×2.1 mm HPLC column (Thermo, cat no.25003-152130)
Microcentrifuge (Eppendorf, centrifuge 5415D)
Microcon YM-10 10,000 Da MWCO filters and collection tubes (Millipore, cat. no. 42407)
PowerGen 1800 model Mechanical homogenizer (Fisher)
Razors
Scissors
Screw cap vial (Agilent Technologies, cat. no. 5182-0715)
Temperature controlled water bath (VWR)
Tweezers
Vial insert, 100 μl glass with polymer feet (Agilent Technologies, cat. no. 5181-1270)
▲ CRITICAL Glass inserts must be used for trace level analysis to prevent contamination coming from the less expensive polypropylene inserts.
Vortexer (e.g., VWR Vortex Genie)
Synthesis of Internal Standards
Note that syntheses should always be attempted first with unlabeled material to check the procedure and optimize the reaction conditions prior to starting syntheses with more expensive isotopically labeled reagents. Always verify the structures of synthesized standards by mass spectrometry, UV/VIS spectroscopy and NMR spectroscopy, depending on the availability of these instruments. The yield for each of the following syntheses is expected to be at least 70% overall, but will vary depending upon experience and other factors.
▲ CRITICAL The concentrations of the standards can be determined either by UV spectroscopy using known extinction coefficients or by [1H] NMR spectroscopy with a known amount of dimethyl sulfoxide as an internal standard.38
a. [15N5]-1,N2-Etheno-2′-deoxyguanosine
[15N5]-1,N2-εdG is synthesized using the procedure of Kusmierek et al.,39 which is summarized as follows. In a fume hood, add 8.5 μl of chloroacetaldehyde (57 μmol) to 166 μl N,N′-dimethylformamide containing 7 mg K2CO3 (50 μmol) and 10 mg [15N5]-2′-deoxyguanosine (34 μmol). After stirring the suspension for 3 h at ambient temperature, add an additional 4.66 mg of K2CO3 (33.3 μmol) and 5 μl of chloroacetaldehyde (33 μmol) and stir for an additional 12 h at ambient temperature. The product is purified by reversed phase HPLC (Phenomenex Synergi Hydro-RP 80A; 250 × 4.6 mm; 4μ particle size) starting with 3% acetonitrile in 8 mM ammonium acetate at a flow rate of 600 μl/min, ramping to 30% acetonitrile over 30 min, holding for 10 min at 30% acetonitrile and ramping down to 3% acetonitrile over 12 min, with 10 further minutes of column equilibration. Product elution time can be verified using commercially available 1,N2-εdG and the product characterized by UV-vis spectroscopy and mass spectrometry. Quantification of the standard is performed by UV spectroscopy using an extinction coefficient of 11,800 M-1cm-1 at 284 nm and pH 7.39
b. [15N3]-3,N4-Etheno-2′-deoxycytidine
[15N3]-εdC is synthesized by the procedure of Zhang et al.40 Briefly, 10.5 mg [15N3]-2′-deoxycytidine (46 μmol) and 2 mg monobasic potassium phosphate (15 μmol) are dissolved in 400 μl of 25% chloroacetaldehyde in water. The pH is adjusted to 3.5 by adding a 1 M potassium hydroxide and the solution is stirred at ambient temperature for 48 h, followed by adjustment of the pH to 7 with 20% potassium carbonate. Product is purified by reversed phase HPLC as described above, with elution time confirmed with commercially available εdC and the product characterized by UV-vis spectroscopy and mass spectrometry. Quantification of the standard is performed by UV spectroscopy using an extinction coefficient of 12,000 M-1cm-1 at 272 nm.40
c. [15N5]-1,N6-Etheno-2′-deoxyadenosine(εdA)
[15N5]-εdA is synthesized using the procedure described by Doerge et al.41 Dissolve 12.6 mg [15N5]-2′-deoxyadenosine (0.05 mmol) in 3 ml of 100 mM potassium phosphate buffer (pH 6) and then add 40 μl of 50% chloroacetaldehyde (0.26 mmol) and stir at ambient temperature for 24 h followed by 2h at 37 °C; both incubations should be performed in the dark (wrap the reaction vessel in aluminum foil). Product is purified by HPLC as described above with elution time verified using commercially available εdA and the product characterized by UV-vis spectroscopy and mass spectrometry. Quantification of the standard is performed by UV spectroscopy using an extinction coefficient of 10,300 M-1cm-1 at 260 nm.42,43
d. [15N5]-7,8-dihydro-8oxo-2′-deoxyguanosine
[15N5]-8-oxo-dG is synthesized by the procedure of Singh et al.44 Dissolve 2.2 mg [15N5]-dG monohydrate (7.7 μmol) in 1.5 ml water by gentle warming at 37 °C and add 50 μl of 50 M ascorbic acid (25 μmol), 30 μl of 0.1 M CuSO4 (3 μmol) and 100 μl of 30% hydrogen peroxide, followed by stirring at ambient temperature for 2 h. Product is purified by HPLC described as above and characterized by UV-vis spectroscopy and mass spectrometry. Quantification of the standard is performed by UV spectroscopy using an extinction coefficient of 10,300 M-1cm-1 at 293 nm.44
e. [15N4]-Labeled and unlabeled 2′-deoxyxanthosine (dX) and 2′-deoxyoxanosine (dO)
dX and dO standards are synthesized by a modification of the method of Suzuki et al.45 Briefly, incubate 10 mM 2′-deoxyguanosine or [15N5]-2′-deoxyguanosine with 100 mM NaNO2 in 0.3 M sodium acetate buffer (pH 3.7) at 37 °C for 6 h. dX and dO are purified by HPLC using a LUNA C18 reversed-phase column (250 × 3 mm, 5 μm particle size, 100 Å pore size; Phenomenex, Torrance, CA) with elution performed at a flow rate of 0.4 ml/min with 1% acetonitrile in 50 mM ammonium acetate (pH 7.4) for the first 5 min, followed by a linear gradient of 1-25% acetonitrile for 5 min; holding at 25% for 10 min; then a reversal of the gradient to 1% for 5 min; and finally eluting at 1% acetonitrile over the last 5 min. Fractions containing the products are dried under vacuum and redissolved in water followed by desalting on a second HPLC system consisting of a Haisil HL C18 reversed phase column (250 × 4.6 mm, 5 μm particle size, 100 Å pore size; Higgins Analytic Inc, Mountain View, CA) eluted with 5% acetonitrile in water at a flow rate of 0.4 ml/min, with elution confirmed using commercially available standards. Quantification of the standards is performed by UV spectroscopy using the following extinction coefficients: dX, 7,800 M-1cm-1 at 260 nm; dO, 5,100 M-1cm-1 at 260 nm.45
f. 2′-Deoxyinosine (dI) and 2′-deoxyuridine (dU)
There are two approaches to synthesis of 15N-labeled and unlabeled dI and dU. One exploits the presence of dA and dC deaminase activities in commercial preparations of alkaline and acid phosphatases, respectively.9 dA and dC are incubated with 0.5 U alkaline phosphatase (Sigma Chemical) and acid phosphatase (Roche), respectively, per μg of nucleoside in supplied buffers at 37 °C for 3 h followed by removal of enzymes by filtration on a Microcon YM-30 column (Millipore Corporation). Alternatively and more reliably, dI and dU are prepared by reacting 10 mM dA or dC with 100 mM NaNO2 in 0.3 M sodium acetate buffer (pH 3.7) at 37 °C for 6 h. Using either approach, the products are purified by HPLC using a HAISIL HL C18 reversed-phase semi-preparative column (250 × 10 mm, 5 μm particle size, 100 Å pore size) eluted with 5% acetonitrile in water at a flow rate of 4 ml/min, with elution confirmed using commercially available standards. Products are characterized by UV-vis spectroscopy and MS. Quantification of the standards is performed by UV spectroscopy using the following extinction coefficients: dI, 12,800 M-1cm-1 at 249 nm; dU, 10,100 M-1cm-1 at 262 nm.46
▲ CRITICAL To avoid contamination of the labeled and unlabeled internal standards (and ensuing loss of isotopic purity), either purify the two species on separate HPLC systems (separate columns will not suffice since most carryover contamination involves the injector port and sample loop) or wash the HPLC column and the injection port/sample loop (requires dismantling the port) with a series of solvents of different polarity (e.g., acetonitrile, 50% methanol in water, pure water). The isotopic purity is best assured by purchasing labeled materials with high isotope content (>98%) and will be apparent during mass spectrometric analysis of the product.
Procedure
Note that the overall method is outlined in Scheme 1.
Tissue harvesting
▲ CRITICAL All experiments employing vertebrate animals must be approved by an appropriate institutional committee on animal care and employ the most humane procedures possible.
Following euthanasia by CO2 asphyxiation, necropsy is performed using sterile instruments.
-
The organs of interest are promptly chopped into 100-200 mg pieces, with each piece placed in a polypropylene microcentrifuge tube and immediately flash frozen in liquid nitrogen. Note that the amount of tissue needed from a specific organ will vary since the yield of DNA differs for each organs. Table 1 lists the suggested amounts of various mouse tissues that will yield at least 100 μg of DNA, the amount needed for the analysis of the DNA damage products.
DNA Extraction (DNA Isolation Kit for Cells and tissues; Roche; Cat number 11 814 770 001)
-
Transfer the previously snap-frozen tissue into a 50 ml conical polypropylene tube (Falcon) filled with 5 ml of lysis buffer (included with kit) containing all appropriate inhibitors and antioxidants to minimize adventitious damage (e.g., 5 μg/ml coformycin, 50 μg/ml tetrahydrouridine, 100 μM desferrioxamine, 100 μM butylated hydroxytoluene).
▲ CRITICAL STEP The addition of appropriate deaminase inhibitors and antioxidants minimizes artifacts arising during sample workup, which could lead to an over-estimation of adduct levels.
Process the tissue sample using a mechanical homogenizer (Fisher PowerGen 1800 model) at half-maximal speed for at least 10 s.
Transfer the homogenized sample to a 15 ml conical polypropylene tube (Falcon) for easier sample manipulation.
Add 3 μl of proteinase K solution (included in kit), vortex for 5 s to mix and incubate at 65 °C for 1 h in a water bath or temperature-controlled heating block.
Remove from heat source, loosen the cap to vent and cool the tube to 37 °C
-
Add 400 μl of RNase solution (included with kit), vortex for 5 s to mix and incubate at 37 °C for 1 h.
▲ CRITICAL STEP The amount of RNase used and the incubation time are longer than specified by the kit manufacturer. This ensures complete digestion of the RNA to avoid interference with the UV quantification of total DNA prior to analysis.
Add 2.1 ml of protein precipitation solution (included with kit) containing 5 μg/ml coformycin, 50 μg/ml tetrahydrouridine, 100 μM desferrioxamine, and 100 μM butylated hydroxytoluene, and vortex the sample thoroughly for 10 s.
Cool the sample on ice for 5 min.
-
Centrifuge the samples at ≥ 27000 × g for 20 min.
▲ CRITICAL STEP The force of centrifugation is very important to ensure formation of a hard protein pellet (i.e., resistant to flaking or oozing when decanting or removing the supernatant) and to ensure complete removal proteins. Be careful to transfer the samples to tubes capable of withstanding the high centrifugal forces used in this step; standard conical polypropylene tubes (Falcon) will collapse at these forces.
Following centrifugation, carefully transfer the supernatant into a new 15 ml conical polypropylene tube (Falcon) and add 0.7 volumes of isopropanol to each sample.
Invert the tubes gently to mix and precipitate the DNA.
-
There are two options for recovering the DNA depending upon the DNA yield:
-
Low DNA yields
If there are no visible DNA fibers, then centrifuge the sample at 1400 × g for 10 min to pellet the precipitated DNA.
Remove the supernatant carefully by aspiration making sure not to disturb the DNA pellet.
Add 10 ml of ice-cold 70% ethanol to wash the pellet; tap the tube to dislodge the pellet to make sure it is thoroughly washed.
Centrifuge the sample at 1400 × g for 10 min and carefully remove the ethanol supernatant by aspiration.
-
High DNA yields
If precipitated DNA fibers are visible, then spool the DNA using a polypropylene pipette tip and place in a 1.5 ml polypropylene microcentrifuge tube.
Add 1 ml of ice-cold 70% ethanol to wash the pellet and centrifuge at 1400 × g for 10 min
Carefully remove the ethanol supernatant by aspiration.
-
Dry the pellet in a fume hood for > 20 min to evaporate all of the ethanol.
-
Dissolve the DNA in 1 ml of sterile (autoclaved) deionized water.
▲ CRITICAL STEP The DNA must be completely dissolved in the water before proceeding and complete solvation of quantities of DNA greater than 100 μg may require several hours of incubation at ambient temperature (or 37 °C) with shaking.
-
Measure the DNA concentration by absorbance at 260 and 320 nm. The absorbance at 260 nm should be corrected by subtracting absorbance at 320 nm. Using a 1 cm path length, 1 absorbance unit at 260 nm (A260 unit) = 50 μg/ml double-stranded DNA. The measured DNA sample should be diluted as needed to maintain absorbance values at 260 nm within the linear range of most UV-vis spectrophotometers, which is generally between 0.1 and 1 absorbance units.
■ PAUSE POINT The sample in the capped vial can be stored at -20 °C for at least 2 weeks and longer at the recommended temperature of -80 °C.
Table 1.
Quantities of mouse tissues needed to DNA adduct analysis
| Organ | Wet weight yielding 100 μg DNA (mg) | Notes |
|---|---|---|
| Spleen | 100 | Pool spleens from control mice when spleen weighs <100 mg |
| Liver | 50 | Prone to RNA contamination; minimized by doubling amount of RNAse during DNA isolation |
| Kidney | 100 | Longer homogenization may be required to obtain high yields |
| Colon | 50 | |
| Lung | 100 |
Hydrolysis of DNA to nucleotides and addition of isotopically labeled internal standards
-
18
Prepare samples containing 100 μg of DNA in 1.5 ml polypropylene microcentrifuge tubes. Note that this quantity of DNA is more than actually needed for quantification of many of the DNA lesions, but it ensures that all damage products will be present at levels exceeding the limits of quantification.
-
19
Dry the DNA completely under vacuum taking care to minimize drying time to avoid artifacts.
▲ CRITICAL STEP Subjecting the DNA to prolonged drying time, heating, or contamination during the drying step can create artifacts due to oxidation and other reactions. There are two approaches to avoid these problems. One is to empirically determine the minimal time needed to dry the sample in a particular solvent evaporation system. Alternatively, solvent removal can be stopped when a minimal (∼2-3 μl) volume of water remains in the tube. In both cases, the solvent evaporation should be performed at ambient or lower temperature in a very clean system. The effects of different drying methods can be assessed by comparing the levels of DNA damage products in samples subjected to different drying conditions.
-
20
Using the volumes specified in Table 2, prepare a “cocktail” containing all enzymes and cofactors needed for hydrolysis of the DNA into nucleosides as well as all inhibitors and antioxidants to minimize artifacts and appropriate internal standards for adduct measurement. Multiply the quantities of each reagent noted in Table 2 by the number of samples plus one additional sample to be used as a parallel control. Combine the reagent volumes and mix by vortexing. Note that the final pH of this solution is higher than the pH optimum for nuclease P1, which is intended to minimize depurination of 2′-deoxyxanthosine (dX) under more acidic conditions, yet still preserve nuclease P1 activity. Also, note that the amount of isotopically labeled internal standard added to each sample has been determined empirically to produce a reasonable signal intensity that correlates with both background and elevated levels of damage products expected for the amount of DNA used in each sample (see Table 9).
-
21
Add 55 μl of the “cocktail” to each dried DNA sample and re-dissolve the DNA by vortexing; add 55 μl of the “cocktail” to an empty microcentrifuge tube as a control to be processed in parallel.
▲ CRITICAL STEP Given the inaccuracy associated with pipetting small volumes (≤1 μl), the use of a stock solution divided among all of the samples minimizes pipetting errors that could ultimately affect quantification of the damage products.
-
22
Incubate the samples for 3 h at 37 °C.
Table 2. Volumes of stock solutions added to DNA hydrolysis cocktail.
| Stock Solution of Reagent | Volume Added (μl) |
|---|---|
| 30 mM Sodium acetate (pH 6.8) | 30 |
| 10 mM ZnCl2 (cofactor for nuclease P1) | 10 |
| 2 unit/μl Nuclease P1 in sodium acetate (pH 6.8) | 2 |
| 4000 units/ml DNase I in deionized water | 2 |
| 20 μM [15N4]-dX | 0.5 |
| 20 μM [15N3]-dU | 0.5 |
| 1 μM [15N4]-dI | 1 |
| 1 μM [15N5]-8-oxo-dG | 1 |
| 100 nM [15N5]-1,N6-εdA | 1 |
| 100 nM [15N3]-εdC | 1 |
| 100 nM [15N5]-1,N2-εdG | 1 |
| 10 mg/ml Tetrahydrouridine in deionized water | 0.5 |
| 1 mg/ml Coformycin in deionized water | 1 |
| 50 mM Desferrioxamine in deionized water | 3 |
| 100 mM Butylated hydroxytoluene in deionized water | 0.5 |
| Total volume | 55 |
Table 9.
Expected background levels
| Damage product | Background level (per 10X nucleotides) |
|---|---|
| 2′-Deoxyxanthosine | 1-10 per 106 |
| 2′-Deoxyinosine | 1-10 per 106 |
| 2′-Deoxyoxanosine | <1 per 107 1 |
| 8-Oxo-7,8-dihydro-2′-deoxyguanosine | 1-10 per 107 |
| 1,N2-Etheno-2′deoxyguanosine | 1-5 per 108 |
| 1,N6-Etheno-2′-deoxyadenosine | 5-15 per 108 |
| 3,N4-Etheno-2′-deoxycytidine | 5-10 per 108 |
2-Deoxyoxanosine has not been detected in double-stranded DNA at the LOD of 1 per 107 nt.
Further DNA hydrolysis and dephosphorylation of nucleotides
-
24
Prepare a second “cocktail” for another round of DNA hydrolysis and dephosphorylation to form nucleosides. Multiply the quantities of each stock reagent shown in Table 3 by the number of samples plus one additional sample to be used as a parallel control. Combine the reagent volumes and mix by vortexing.
▲ CRITICAL STEP Addition of phosphodiesterase I at this step, along with the alkaline phosphatase, ensures complete hydrolysis of the DNA sample to nucleosides.
-
25
Add 45 μl of the cocktail to each DNA sample and the control (empty tube) and incubate at 37 °C overnight. Note that the “cocktails” prepared in steps 20 and 24 have been optimized to keep sample volumes at the 100 μl maximal injection volume for an Agilent 1100 series HPLC autosampler.
! CAUTION The addition of deaminase inhibitors is critical here to inhibit adventitious formation of dI and dU.
Table 3. Volumes of stock solutions added to dephosphorylation cocktail.
| Stock Solution of Reagent | Volume Added (μl) |
|---|---|
| 30 mM Sodium acetate (pH 7.8) | 40 |
| 1 mg/ml Coformycin in deionized water | 1 |
| 17 units/μl Alkaline phosphatase in deionized water | 2 |
| 100 units/ml Phosphodiesterase I in deionized water | 2 |
| Total volume | 45 |
Removal of enzymes
-
26
Rinse a 10,000 MW cut-off spin filter (Amicon) by adding 300 μl of deionized water and centrifuging for 45 min at 16,400 × g.
-
27
Replace the collection tube containing rinse water with a fresh collection tube.
-
28
Transfer the hydrolyzed DNA sample to the washed spin filter and centrifuge for 45 min at 16,400 × g.
HPLC pre-purification of nucleosides
-
29
Transfer the filtrate from step 28 into a 100 μl insert tube (Agilent, cat. no. 5181-1270), place the insert in a 2 ml screw top vial (Agilent, cat. no. 5182-0715) and cap the vial.
■ PAUSE POINT The sample in the capped vial can be stored at -20 °C for at least 2 weeks and longer at the recommended temperature of -80 °C.
-
30
Prepare the HPLC system by equilibrating the column temperature to 32 °C and equilibrating the Phenomenex Synergi 4μ Hydro-RP C18 column 80A 250 mm × 4.6 mm (cat no. 00G-4375-E0) with 99% 8 mM ammonium acetate and 1% acetonitrile for at least 20 min at 0.500 ml/min.
-
31
Test the column by injecting a 1 μL volume of standards containing the damaged nucleosides at a concentration of 10 μM and the normal nucleobases at a concentration of 100 μM and running the system through the elution cycle as described in Table 4.
! CAUTION Injecting excessive amounts of standards will result in carryover of unlabeled material into the unknown samples, which will result in an over-estimation of adduct levels during LC/MS-MS quantification. Inject only enough of each standard to produce a minimally detectable UV absorbance. Check for carryover by injecting water and observing the LC/MS-MS signals, as described in the section starting at step 37.
▲ CRITICAL STEP Use this step to evaluate column performance prior to injecting DNA samples. For new columns, this step should be performed before preparing DNA samples. If the column does not provide adequate resolution (e.g., 8-oxo-dG is not well resolved from dT), then try reversing the column orientation (i.e., the outlet becomes the inlet). If that does not solve the problem, then return the column to the manufacturer and get a replacement column from a different lot.
-
32
Inject 100 μl of water and run through the elution cycle to eliminate carryover of the standards
-
33
Inject the hydrolyzed DNA samples (∼100 μl) onto the HPLC system using the autosampler and elute the nucleosides by applying the gradient described in Table 4.
-
34
Fractions of eluate containing the various nucleosides are collected on the basis of predetermined retention times, with the following example based on our experience: dX: start collecting at 13.0 min and stop when dC begins to elute; dU: 23.2-25.0 min; dI: 34.0-36.5 min; 8-oxo-dG: 48.2-50.7 min; 1,N2-εdG: 63.7-65.8 min; εdC and εdA: 68.0-72.0 min.
As shown in Figure 3, εdC and εdA do not always separate well, so it may be necessary to collect a single fraction containing both species. εdC and εdA will separate from each other on the Thermo-Hypersil Gold column during LC/MS-MS analysis.
▲ CRITICAL STEP Due to the abundance of dA, the fraction containing εdC and εdA contain a large amount of dA even though dA elutes 6-7 min earlier on the Phenomenex column. The presence of dA poses a problem for quantification of εdC since both εdC and dA produce the same molecular ion (M+H) at m/z 252 and the same fragment ion [(M+1) -116(sugar)] at m/z 136 at the unit resolution at which the triple quadrupole mass spectrometer operates. A complete base-line HPLC separation of εdC and dA is thus crucial. This is achieved in the LC/MS-MS step using a Thermo-Hypersil Gold column operated at a column temperature of 10 °C in order to provide complete baseline separation of εdC, dA and εdA.
-
35
Wash the collected fractions twice by drying under vacuum, redissolving in 300 μl of deionized water and drying under vacuum again.
▲ CRITICAL STEP In order to minimize organic solvent reaching the vacuum pump, a cold trap set at -100 °C must be installed between the SpeedVac and pump
-
36
Dissolve each dried sample in 45 μl of deionized water.
▲ CRITICAL STEP It is important to wash the sample several times and dry completely after each wash to evaporate as much of the volatile ammonium acetate as possible. Like any salt, ammonium acetate has the potential to suppress ionization, and thus detection of the analyte during MS analysis, and to foul the ionization source. This is particularly important for systems that do not have a diverter valve to route the void volume of salts to waste, since the salts can accumulate in the ionization source over time.
■ PAUSE POINT The sample can be stored at -20 °C for at least 2 weeks or longer at the recommended temperature of -80 °C.
Table 4.
Acetonitrile gradient for HPLC pre-purification of nucleosides, step 31
| Time (min) | Gradient1 |
|---|---|
| 0 – 45 | 1% to 3.5% |
| 45-50 | 3.5% to 6% |
| 50-55 | 6% to 8% |
| 55-75 | 8% |
| 75-82 | 100% |
| 82-93 | 1% |
Percent acetonitrile in 8 mM ammonium acetate (v/v); flow rate 0.5 ml/min; column temperature 32 °C
Figure 3.
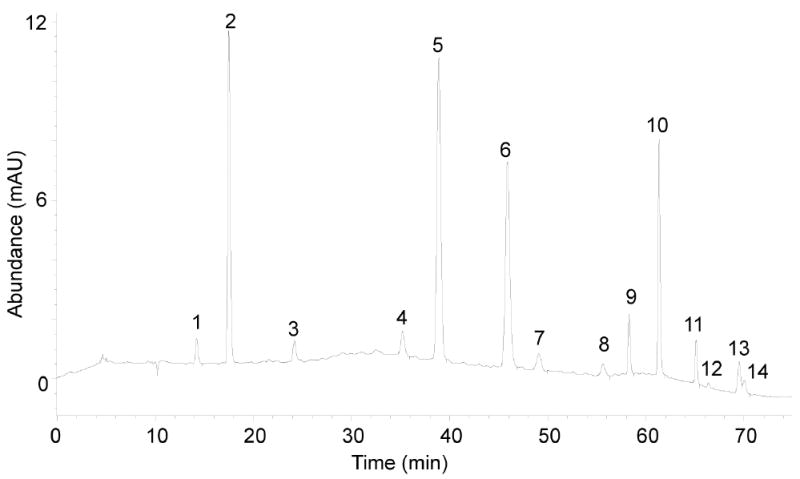
Reversed-phase HPLC separation of normal and modified nucleoside standards. (1) dX, (2) dC, (3) dU,(4) dI, (5) dG, (6) dT, (7) 8-oxo-dG, (8) dO, (9) N2,3-εdG, (10) dA, (11) 1,N2-εdG, (12) M1dG, (13) εdC, (14) εdA.
Preparation of samples for calibration curves
-
37
Prepare samples to be used for calibration curves to convert mass spectrometer signals to meaningful concentrations of damaged nucleosides in the DNA samples.
In individual microcentrifuge tubes in triplicate, add aliquots of stocks of individual unlabeled standards to yield concentrations noted in Table 5 in a final volume of 45 μl.
Add aliquots of individual labeled internal standards to yield concentrations noted in Table 2 in a final volume of 45 μl.
Dry the samples under vacuum and redissolve in 45 μl of deionized water.
Table 5.
Limits of detection, DNA requirements and parameters for preparation of calibration curve samples
| Lesion | LOD (fmol) | Minimum DNA (μg) | Unlabeled standard (nM) | Internal standard (nM) |
|---|---|---|---|---|
| dX | 10 | 50 | 62.5-10001 | 250 |
| dU | 50 | 50 | 62.5-1000 | 250 |
| dI | 5 | 10-20 | 6.25-100 | 25 |
| 8-oxo-dG | 5 | 50 | 6.25-100 | 25 |
| 1,N2-εdG | 10 | 100 | 0.625-10 | 2.5 |
| εdC | 0.5 | 50 | 0.625-10 | 2.5 |
| εdA | 1 | 10-20 | 0.625-10 | 2.5 |
Prepare 5 concentrations differing by 2-fold (e.g., 62.5, 125, 250, 500, 1000)
The concentrations of internal standards in the calibration curve samples should be identical to those used for the other DNA samples. The range of unlabeled nucleoside concentrations should encompass the expected values in background and treatment conditions.
Quantification of nucleosides by LC/MS-MS
Two issues must be noted here. First, analyses of the various DNA damage products must be performed separately to accommodate the different mass spectrometer parameters for each nucleoside (e.g., dI collected from all samples will be analyzed together). Individual calibration curve samples can be positioned at the end of each nucleoside analysis to avoid carryover contamination of the unknown samples. Second, the protocol describes analyses performed with two different triple quadrupole mass spectrometers to demonstrate the flexibility of the method. It can be readily adapted to any triple quadrupole system with minor adjustments due to machine-specific variations in sensitivity.
-
38
For each sample, inject 20 μl of the 45 μl sample volume onto the LC/MS-MS system, using conditions appropriate for each modified nucleoside (Tables 6 and 7).
The 20 μl portion of each sample will ensure a signal over the limit of quantification and for a duplicate analysis.
-
39
Each nucleoside is analyzed by LC/MS-MS with slightly different HPLC and MS conditions, as noted in Tables 6 and 7, using a reversed phase Thermo Hypersil Gold C18 column (150 × 2.1 mm; 3 μm particle size) eluted at a flow rate of 200 μl/min using 0.1% acetic acid in deionized water (solvent A) and 0.1% acetic acid in HPLC grade acetonitrile (solvent B) as mobile phases. To demonstrate the flexibility of the method, we describe the analysis of different adducts on two different triple quadrupole mass spectrometers. The two mass spectrometers provide different sensitivities for each adduct, so we have optimized parameters for two MS systems depending on which system provided the lowest limit of quantification for a nucleoside:
-
Deamination (dX, dI, dU) damage products
The HPLC column is coupled to an API 3000 tandem quadrupole MS with turbo ion spray.
MS is operated in positive ion mode with the first and third quadrupoles (Q1 and Q3, respectively) fixed to unit resolution.
The voltages and source gas are optimized for maximal sensitivity, and the parameters are as described in Table 6.
-
Oxidative (8-oxo-dG) and etheno-adducts of dA, dC and dG
The HPLC is coupled to an Agilent 6410 triple quadrupole mass spectrometer with an ion spray ion source.
MS is operated in positive ion mode with the first quadrupole (Q1) fixed to wide resolution and the third quadrupole (Q3) fixed to unit resolution.
The voltages and source gas are optimized for maximum sensitivity, and the parameters are as described in Table 7.
-
-
40
Using the software available with each system, obtain the peak area for each nucleoside and for its corresponding internal standard.
-
41
Calculate the peak area ratio for target nucleosides (unlabeled) and internal standards
-
42
Generate calibration curves by plotting the peak area ratios for the calibration curve samples as the independent variable against the concentration ratio of the unlabeled to labeled standards in the calibration curve samples.
The calibration curves should be linear, typically with correlation coefficients, r2, > 0.995.
-
43
Determine the concentration ratio for the unknown samples by interpolation from the linear calibration curves.
-
44
Calculate the amount of damaged nucleoside present in each sample by multiplying the concentration ratio by the concentration of labeled internal standard added during digestion. The final quantity of damaged nucleoside is best expressed as the number of damaged nucleosides per total nucleoside content (e.g., 106, 107, 108, etc.) calculated from the amount of DNA used to prepare the sample.
TROUBLESHOOTING Table 8
Table 6.
LC/MS-MS parameters for the API 3000 triple quadrupole mass spectrometer
| Mass Spectrometer Parameters | dX | dI | dU | 8-oxo-dG |
|---|---|---|---|---|
| 14N Q1→Q3 m/z1 | 269→153 | 253→137 | 229→113 | 284→168 |
| 15N Q1→Q3 m/z | 273→157 | 257→141 | 231→115 | 289→173 |
| Nebulizer gas (psi)) | 8 | 8 | 8 | 8 |
| Curtain gas (L/min) | 8 | 8 | 8 | 8 |
| Collision gas (L/min) | 6 | 4 | 4 | 8 |
| Ion Spray Voltage (V) | 4000 | 4500 | 4500 | 4000 |
| Temperature (°C) | 400 | 350 | 350 | 400 |
| Turbo ion spray gas (psi) | 800 | 800 | 400 | 800 |
| Declustering potential (V) | 20 | 20 | 20 | 20 |
| Focusing potential (V) | 100 | 100 | 100 | 100 |
| Entrance potential (V) | 5 | 5 | 5 | 5 |
| Collision Energy (V) | 10 | 10 | 10 | 20 |
| Collision cell exit potential (V) | 10 | 10 | 10 | 10 |
| HPLC Parameters2 | dX | dI | dU | 8-oxo-dG |
| %Solvent B | 1.3 | 0.7 | 1 | 1.5 |
| Retention time (min)3 | 8-10 | 10-12 | 3-5 | 11-13 |
m/z values monitored in MRM mode for the transition of the parent ion to the fragment ion; 14N refers to the analyte under study; 15N refers to the isotopically labeled internal standard.
Thermo Hypersil Gold C18 column used in step 39
Retention time is expected to range over the values indicated
Table 7.
LC/MS-MS parameters for Agilent 6410 triple quadrupole mass spectrometer
| Mass Spec. Parameters | εdA | εdC | 1,N2-εdG | 8-oxo-dG |
|---|---|---|---|---|
| 14N Q1→Q3 m/z1 | 276→160 | 252→136 | 292→176 | 284→168 |
| 15N Q1→Q3 m/z | 281→165 | 255→139 | 297→181 | 289→173 |
| Gas temperature (°C) | 350 | 350 | 350 | 350 |
| Gas flow (L/min) | 10 | 10 | 10 | 10 |
| Nebulizer gas (psi) | 20 | 20 | 20 | 30 |
| Capillary voltage (V) | 4000 | 4000 | 3500 | 4000 |
| Fragmentor potential (V) | 70 | 70 | 85 | 80 |
| Collision energy (V) | 12 | 12 | 14 | 10 |
| Delta EMV (V) | 700 | 700 | 1000 | 700 |
| Column temperature (°C) | 10 | 10 | 40 | 40 |
| HPLC Parameters2 | εdA | εdC | 1,1,N2-εdG | 8-oxo-dG |
| % Solvent B | 0.8 | 0.8 | 2 | 3 |
| Retention time (min)3 | 19.5-21.5 | 15-17 | 14.5-16.5 | 5.5-7.5 |
m/z values monitored in MRM mode for the transition of the parent ion to the fragment ion; 14N refers to the analyte under study; 15N refers to the isotopically labeled internal standard
Thermo Hypersil Gold C18 column used in step 39
Retention time is expected to range over the values indicated
Table 8.
Troubleshooting table
| Problem | Possible reason(s) | Solution(s) |
|---|---|---|
| Low DNA yields | → Incomplete lysis of tissue or cells | → Optimize cell or tissue disruption conditions (e.g., increase time or intensity of mechanical homogenization) |
| → DNA yield is sometimes tissue dependent, some tissues yield higher DNA amount than others | → Increase sample size | |
| RNA contamination | → Inadequate RNA degradation during DNA isolation | → Increase RNase during extraction and prolong the incubation time |
| Inconsistent retention times during HPLC pre-purification or LC/MS-MS | → Column not equilibrated prior to purification | → Increase column equilibration time |
| → Inconsistent column pressures due to inadequate degassing of mobile phases | → Use a degassing apparatus on the HPLC or Sonicate buffers for 20 min | |
| → Problems with the stationary phase in the column (e.g., use of high aqueous isocratic conditions during LC/MS/MS analysis leads to column collapse) | → Regenerate column by running 100% acetonitrile overnight at 0.100 ml/min; replace column, if necessary | |
| dI levels are higher than expected | → Deaminase contamination | → Include or increase coformycin during all stages of DNA isolation and processing |
| → Contamination of dI with dA during HPLC pre-purification or LC/MS-MS; m/z values differ by 1, so dA isotopomer signals (M+1) contribute to dI signal | → Decrease the % organic solvent to resolve the dA and dI peaks | |
| dX and dO levels are higher than expected | → Contamination of dX or dO with dG during HPLC pre-purification or LC/MS-MS; m/z values differ by 1, so dG isotopomer signals (M+1) contribute to dX/dO signal | → Decrease the % organic solvent to resolve the dG and dX or dO peaks |
| 8-Oxo-dG levels are higher than expected | → Artifactual formation during sample work-up or in vacuo drying | → Include desferrioxamine and butylated hydroxytoluene during cell processing, DNA extraction and hydrolysis steps; process samples on ice; minimize sample evaporation time |
| → As 8-oxo-dG can be formed from 2′-deoxyguanosine (dG) in the ion source of the mass spectrometer it is important that dG contamination separates well from 8-oxo-dG prior to entering the MS | → Decrease the % organic solvent to resolve dG from 8-oxo-dG | |
| εdC levels are higher than expected | → Contamination of εdC with dA due to similar HPLC retention times and identical m/z values | → Optimize the separation of εdC and dA in the pre-purification step. Also, optimize the HPLC step of the LC/MS-MS analysis to such that εdC elutes 2-3 min before dA. |
Anticipated Results
The baseline adduct levels for all of the damaged nucleosides are listed in Table 9. Examples of the quantities of the DNA damage products observed in spleen, liver and kidney from a mouse model of nitric oxide overproduction and inflammation can be found in Tables 1, 2 and 3, respectively, in the publication by Pang et al.35
● Timing
Steps 1-2: tissue harvesting, 1-3 h (depending on sample number)
Steps 3-17: DNA extraction and quantitation, 1 day
Steps 18-25: DNA hydrolysis to nucleoside form, 1 day
Steps 26-28: removal of enzymes, 2 h
Steps 29-36: HPLC pre-purification of nucleosides, 2 h for equilibration and standard run and 1.5 h per sample to be purified
Steps 37-40: LC/MS-MS quantification of DNA damage products, depends on number of samples and adducts. A good estimate is 20 min per sample (or standard) per adduct plus 30 min of equilibration time prior to analysis
Contributor Information
Koli Taghizadeh, Email: kolit@mit.edu.
Jose L. McFaline, Email: jose_mc@mit.edu.
Bo Pang, Email: bpang@alnylam.com.
Matthew Sullivan, Email: matsul30@mit.edu.
Min Dong, Email: min.dong@novartis.com.
Elaine Plummer, Email: elaineturano22@gmail.com.
Peter C. Dedon, Email: pcdedon@mit.edu.
References
- 1.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–7. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ohshima H, Bartsch H. Chronic infections and inflammatory processes as cancer risk factors: possible role of nitric oxide in carcinogenesis. Mutat Res. 1994;305:253–64. doi: 10.1016/0027-5107(94)90245-3. [DOI] [PubMed] [Google Scholar]
- 3.Suerbaum S, Michetti P. Helicobacter pylori infection. N Engl J Med. 2002;347:1175–86. doi: 10.1056/NEJMra020542. [DOI] [PubMed] [Google Scholar]
- 4.Dedon PC, Tannenbaum SR. Reactive nitrogen species in the chemical biology of inflammation. Arch Biochem Biophys. 2004;423:12–22. doi: 10.1016/j.abb.2003.12.017. [DOI] [PubMed] [Google Scholar]
- 5.Halliwell B. Effect of diet on cancer development: is oxidative DNA damage a biomarker? Free Radic Biol Med. 2002;32:968–74. doi: 10.1016/s0891-5849(02)00808-0. [DOI] [PubMed] [Google Scholar]
- 6.Shacter E, Weitzman SA. Chronic inflammation and cancer. Oncology (Huntingt) 2002;16:217–26. 229. [PubMed] [Google Scholar]
- 7.Ohshima H, Tatemichi M, Sawa T. Chemical basis of inflammation-induced carcinogenesis. Arch Biochem Biophys. 2003;417:3–11. doi: 10.1016/s0003-9861(03)00283-2. [DOI] [PubMed] [Google Scholar]
- 8.Lewis RS, Tamir S, Tannenbaum SR, Deen WM. Kinetic analysis of the fate of nitric oxide synthesized by macrophages in vitro. J Biol Chem. 1995;270:29350–29355. doi: 10.1074/jbc.270.49.29350. [DOI] [PubMed] [Google Scholar]
- 9.Dong M, Wang C, Deen WM, Dedon PC. Absence of 2′-deoxyoxanosine and presence of abasic sites in DNA exposed to nitric oxide at controlled physiological concentrations. Chem Res Toxicol. 2003;16:1044–55. doi: 10.1021/tx034046s. [DOI] [PubMed] [Google Scholar]
- 10.Cadet J, Douki T, Ravanat JL. One-electron oxidation of DNA and inflammation processes. Nat Chem Biol. 2006;2:348–9. doi: 10.1038/nchembio0706-348. [DOI] [PubMed] [Google Scholar]
- 11.Niles JC, Wishnok JS, Tannenbaum SR. Peroxynitrite-induced oxidation and nitration products of guanine and 8-oxoguanine: structures and mechanisms of product formation. Nitric Oxide. 2006;14:109–21. doi: 10.1016/j.niox.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 12.Yu H, Venkatarangan L, Wishnok JS, Tannenbaum SR. Quantitation of four guanine oxidation products from reaction of DNA with varying doses of peroxynitrite. Chem Res Toxicol. 2005;18:1849–1857. doi: 10.1021/tx050146h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee SH, Arora JA, Oe T, Blair IA. 4-Hydroperoxy-2-nonenal-induced formation of 1,N2-etheno-2′-deoxyguanosine adducts. Chem Res Toxicol. 2005;18:780–6. doi: 10.1021/tx0497088. [DOI] [PubMed] [Google Scholar]
- 14.Nair J, Barbin A, Velic I, Bartsch H. Etheno DNA-base adducts from endogenous reactive species. Mutat Res. 1999;424:59–69. doi: 10.1016/s0027-5107(99)00008-1. [DOI] [PubMed] [Google Scholar]
- 15.Dedon PC, Plastaras JP, Rouzer CA, Marnett LJ. Indirect mutagenesis by oxidative DNA damage: formation of the pyrimidopurinone adduct of deoxyguanosine by base propenal. Proc Natl Acad Sci U S A. 1998;95:11113–6. doi: 10.1073/pnas.95.19.11113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou X, Taghizadeh K, Dedon PC. Chemical and biological evidence for base propenals as the major source of the endogenous M1dG adduct in cellular DNA. J Biol Chem. 2005;280:25377–25382. doi: 10.1074/jbc.M503079200. [DOI] [PubMed] [Google Scholar]
- 17.Marnett LJ. Oxy radicals, lipid peroxidation and DNA damage. Toxicology. 2002;181-182:219–22. doi: 10.1016/s0300-483x(02)00448-1. [DOI] [PubMed] [Google Scholar]
- 18.Karakaya A, Jaruga P, Bohr VA, Grollman AP, Dizdaroglu M. Kinetics of excision of purine lesions from DNA by Escherichia coli Fpg protein. Nucleic Acids Res. 1997;25:474–9. doi: 10.1093/nar/25.3.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Senturker S, Dizdaroglu M. The effect of experimental conditions on the levels of oxidatively modified bases in DNA as measured by gas chromatography-mass spectrometry: how many modified bases are involved? Prepurification or not? Free Radic Biol Med. 1999;27:370–80. doi: 10.1016/s0891-5849(99)00069-6. [DOI] [PubMed] [Google Scholar]
- 20.Cooke MS, Olinski R, Evans MD. Does measurement of oxidative damage to DNA have clinical significance? Clin Chim Acta. 2006;365:30–49. doi: 10.1016/j.cca.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 21.Dizdaroglu M, Jaruga P, Rodriguez H. Identification and quantification of 8,5′-cyclo-2′-deoxy-adenosine in DNA by liquid chromatography/mass spectrometry. Free Radic Biol Med. 2001;30:774–84. doi: 10.1016/s0891-5849(01)00464-6. [DOI] [PubMed] [Google Scholar]
- 22.Rodriguez H, et al. Lymphoblasts of women with BRCA1 mutations are deficient in cellular repair of 8,5′-Cyclopurine-2′-deoxynucleosides and 8-hydroxy-2′-deoxyguanosine. Biochemistry. 2007;46:2488–96. doi: 10.1021/bi062022p. [DOI] [PubMed] [Google Scholar]
- 23.Cadet J, et al. Radiation-induced DNA damage: formation, measurement, and biochemical features. J Environ Pathol Toxicol Oncol. 2004;23:33–43. doi: 10.1615/jenvpathtoxoncol.v23.i1.30. [DOI] [PubMed] [Google Scholar]
- 24.Pouget JP, et al. Formation of modified DNA bases in cells exposed either to gamma radiation or to high-LET particles. Radiat Res. 2002;157:589–95. doi: 10.1667/0033-7587(2002)157[0589:fomdbi]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 25.Dedon P, et al. Challenges in developing DNA and RNA biomarkers of inflammation. Biomarkers Med. 2007;1:293–312. doi: 10.2217/17520363.1.2.293. [DOI] [PubMed] [Google Scholar]
- 26.Hu CW, et al. Comparison of analyses of urinary 8-hydroxy-2′-deoxyguanosine by isotope-dilution liquid chromatography with electrospray tandem mass spectrometry and by enzyme-linked immunosorbent assay. Rapid Commun Mass Spectrom. 2004;18:505–10. doi: 10.1002/rcm.1367. [DOI] [PubMed] [Google Scholar]
- 27.Randerath K, Randerath E. 32P-postlabeling methods for DNA adduct detection: overview and critical evaluation. Drug Metab Rev. 1994;26:67–85. doi: 10.3109/03602539409029785. [DOI] [PubMed] [Google Scholar]
- 28.Dizdaroglu M, Jaruga P, Birincioglu M, Rodriguez H. Free radical-induced damage to DNA: mechanisms and measurement. Free Radic Biol Med. 2002;32:1102–15. doi: 10.1016/s0891-5849(02)00826-2. [DOI] [PubMed] [Google Scholar]
- 29.Cadet J, et al. Facts and artifacts in the measurement of oxidative base damage to DNA. Free Radic Res. 1998;29:541–50. doi: 10.1080/10715769800300581. [DOI] [PubMed] [Google Scholar]
- 30.Dizdaroglu M. Facts about the artifacts in the measurement of oxidative DNA base damage by gas chromatography-mass spectrometry. Free Radic Res. 1998;29:551–63. doi: 10.1080/10715769800300591. [DOI] [PubMed] [Google Scholar]
- 31.Cadet J. Assesment of oxidative base damage to isolated and cellular DNA by HPLC-MS/MS measurement. Free Radic Biol Med. 2002;33:441–449. doi: 10.1016/s0891-5849(02)00820-1. [DOI] [PubMed] [Google Scholar]
- 32.Roberts DW, Churchwell MI, Beland FA, Fang JL, Doerge DR. Quantitative analysis of etheno-2′-deoxycytidine DNA adducts using on-line immunoaffinity chromatography coupled with LC/ES-MS/MS detection. Anal Chem. 2001;73:303–9. doi: 10.1021/ac000866n. [DOI] [PubMed] [Google Scholar]
- 33.Dong M, Dedon PC. Relatively small increases in the steady-state levels of nucleobase deamination products in DNA from human TK6 cells exposed to toxic levels of nitric oxide. Chem Res Toxicol. 2006;19:50–57. doi: 10.1021/tx050252j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.ESCODD. Comparative analysis of baseline 8-oxo-7,8-dihydroguanine in mammalian cell DNA, by different methods in different laboratories: an approach to consensus. Carcinogenesis. 2002;23:2129–33. doi: 10.1093/carcin/23.12.2129. [DOI] [PubMed] [Google Scholar]
- 35.Pang B, et al. Lipid peroxidation dominates the chemistry of DNA adduct formation in a mouse model of inflammation. Carcinogenesis. 2007;28:1807–1813. doi: 10.1093/carcin/bgm037. [DOI] [PubMed] [Google Scholar]
- 36.Nair J, et al. Etheno adducts in spleen DNA of SJL mice stimulated to overproduce nitric oxide. Carcinogenesis. 1998;19:2081–4. doi: 10.1093/carcin/19.12.2081. [DOI] [PubMed] [Google Scholar]
- 37.Cadet J, Douki T, Gasparutto D, Ravanat JL. Oxidative damage to DNA: formation, measurement and biochemical features. Mutat Res. 2003;531:5–23. doi: 10.1016/j.mrfmmm.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 38.Chen B, Bohnert T, Zhou X, Dedon PC. 5′-(2-Phosphoryl-1,4-dioxobutane) as a product of 5′-oxidation of deoxyribose in DNA: elimination as trans-1,4-dioxo-2-butene and approaches to analysis. Chem Res Toxicol. 2004;17:1406–1413. doi: 10.1021/tx049818e. [DOI] [PubMed] [Google Scholar]
- 39.Kusmierek JT, Singer B. 1,N2-ethenodeoxyguanosine: properties and formation in chloroacetaldehyde-treated polynucleotides and DNA. Chem Res Toxicol. 1992;5:634–8. doi: 10.1021/tx00029a007. [DOI] [PubMed] [Google Scholar]
- 40.Zhang W, Rieger R, Iden C, Johnson F. Synthesis of 3,N4-etheno, 3,N4-ethano, and 3-(2-hydroxyethyl) derivatives of 2′-deoxycytidine and their incorporation into oligomeric DNA. Chem Res Toxicol. 1995;8:148–56. doi: 10.1021/tx00043a020. [DOI] [PubMed] [Google Scholar]
- 41.Doerge DR, Churchwell MI, Fang JL, Beland FA. Quantification of etheno-DNA adducts using liquid chromatography, on-line sample processing, and electrospray tandem mass spectrometry. Chem Res Toxicol. 2000;13:1259–64. doi: 10.1021/tx0001575. [DOI] [PubMed] [Google Scholar]
- 42.Hillestrom PR, Weimann A, Poulsen HE. Quantification of urinary etheno-DNA adducts by column-switching LC/APCI-MS/MS. J Am Soc Mass Spectrom. 2006;17:605–10. doi: 10.1016/j.jasms.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 43.Green T, Hathway DE. Interactions of vinyl chloride with rat-liver DNA in vivo. Chem Biol Interact. 1978;22:211–24. doi: 10.1016/0009-2797(78)90126-6. [DOI] [PubMed] [Google Scholar]
- 44.Singh R, McEwan M, Lamb JH, Santella RM, Farmer PB. An improved liquid chromatography/tandem mass spectrometry method for the determination of 8-oxo-7,8-dihydro-2′-deoxyguanosine in DNA samples using immunoaffinity column purification. Rapid Commum Mass Spectrom. 2003;17:126–34. doi: 10.1002/rcm.883. [DOI] [PubMed] [Google Scholar]
- 45.Suzuki T, Yamaoka R, Nishi M, Ide H, Makino K. Isolation and characterization of a novel product, 2′-deoxyoxanosine, from 2′-deoxyguanosine, oligodeoxynucleotide, and calf thymus DNA treated with nitrous acid and nitric oxide. J Am Chem Soc. 1996;118:2515–2516. [Google Scholar]
- 46.Stimson MM, Reuter MA. Ultraviolet absorption spectra of nitrogenous heterocycles. VII. The effect of hydroxy substitution on the ultraviolet absorption of the series: hypoxanthine, xanthine and uric acid. J Am Chem Soc. 1943;65:153–155. [Google Scholar]