

1. Structure
Encoded by the VMD2 gene on chromosome 11q13 Best-1 is the prototypic member of the RFP family of proteins which are more commonly called “bestrophins”. The protein family was originally identified in Caenorhabditis elegans based on a conserved amino acid (aa) motif Arg-Phe-Pro (RFP). In humans there are four members of the bestrophin family numbered sequentially Best-1 through Best-4. Bestrophins all contain a conserved domain of ~310 aa which begins at their respective N-termini and contains four putative transmembrane helices (TM, Fig. 1). In Best-1, hydropathy plots predict that the extracellular loop between TM-1 and TM-2 is 20 amino acids in length. A large cytoplasmic loop separates TM-2 and TM-3, however, the precise location of TM-3 is unknown as computer predictions and experimental data are not in agreement. A highly variable cytosolic domain which follows TM-4 distinguishes the members of the family from each other. Best-1 reportedly forms homo-oligomers, however the stoichiometry of these oligomers has not been fully resolved.
Fig. 1.
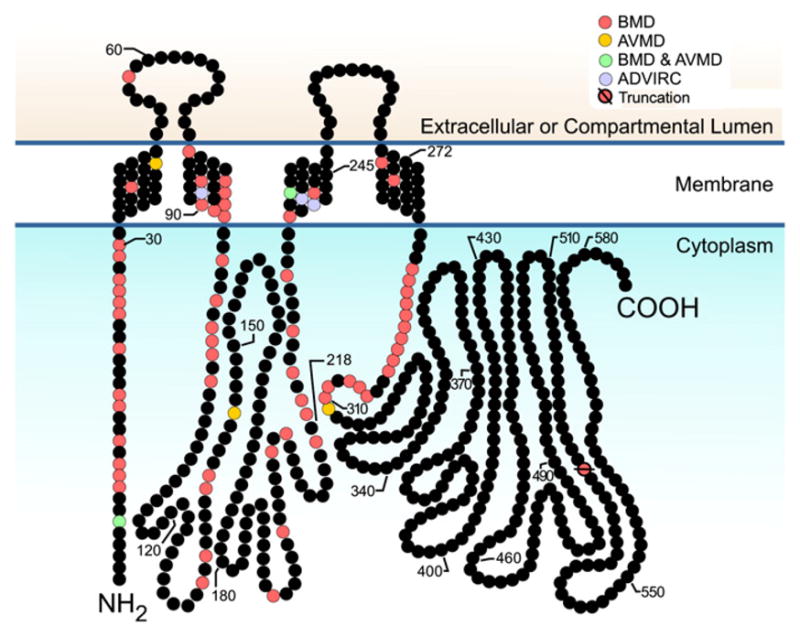
Putative structure of human Best-1. The protein is predicted to form four transmembrane helices with both the N- and C-termini within the cytoplasm. Individual mutations associated with BMD, AVMD, or ADVIRC are indicated.
2. Function
Best-1 has a very limited tissue distribution with mRNA having been identified only in the retinal pigment epithelium (RPE), testis, placenta, and brain, and protein having been detected only in the RPE where it is localized to the basolateral plasma membrane. The light peak (LP) of the electrooculogram (EOG) is generated by a Cl− conductance across the basolateral plasma membrane of the RPE. Since LP defects are a characteristic of Best vitelliform macular dystrophy (BMD), a disease caused by mutations in Best-1, it was hypothesized that Best-1 functions as a Ca++ sensitive Cl− channel (CaCC) that generates the LP. Whole cell patch clamp studies of Best-1 and other bestrophins heterologously expressed in cultured cells support this hypothesis (Sun et al., 2002). Further support comes from experiments in which replacement of key amino acids appears to alter the channel ion selectivity (reviewed in Hartzell et al., 2005). The LP, however, exhibits increased luminance sensitivity in Vmd2 knock-out mice and alterations in the Ca++ response evoked by ATP without any obvious effects on Cl− conductances (Marmorstein et al., 2006). Furthermore, the LP is desensitized when Best-1 is overexpressed in rats. Thus, Best-1 appears as an antagonist of the EOG light peak, not the generator. Recently, Rosenthal et al. (2006) found that Best-1 can modify the kinetics of voltage dependent Ca++ channels (VDCCs). Interestingly, the BMD associated mutations W93C and R218C altered VDCC kinetics different from each other and wild-type Best-1. The relationship between Best-1’s function as a CaCC and its ability to alter VDCC kinetics and Ca++ signaling requires further study.
3. Disease involvement
Mutations in the VMD2 gene resulting in changes to the primary structure of Best-1 have been identified in three diseases; BMD, (http://www3.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=153700), adult-onset vitelliform dystrophy (AVMD, http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=608161), and autosomal dominant vitreoretinalchoroidopathy (ADVIRC, http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=193220). All of the above diseases exhibit a dominant pattern of inheritance. No disease caused by VMD2 with a recessive pattern of inheritance has been identified to date, and studies of Vmd2 deficient mice indicate that the absence of Best-1 is well tolerated (Marmorstein et al., 2006). At least 95 different mutations causing BMD and/or AVMD have been described. These are summarized at the VMD2 mutation database (http://www.uni-wuerzburg.de/humangenetics/vmd2.html). Of these mutations (Fig. 1), 92 are single aa substitutions or deletions occurring at one of 68 different positions in the conserved RFP-domain of the protein. One is at a splice site and two are frame shifts. In ADVIRC, 3 mutations resulting in aa substitutions and possibly exon skipping have been described. All three amino acids are in TM domains. Mutations in these three aa have not been attributed to BMD or AVMD. With only two exceptions all of the mutations causing BMD, AVMD, and ADVIRC are found in four clusters occurring in the cytoplasmic region of the protein near each TM helix, or within the TM helix itself (see Fig. 1).
Clinically, BMD and AVMD are characterized by vitelliform lesions in the ocular fundus. At early stages, the yellow lesion has an appearance similar to that of an egg-yolk, which, as the disease advances becomes “scrambled”. In BMD this lesion may occur as early as the first decade while in AVMD it is undetected until the fourth or fifth decade. BMD and AVMD are distinguished clinically by electrophysiological testing. The electroretinogram (ERG) of patients with both BMD and AVMD is typically normal, however, the ratio of the LP to dark trough of the EOG is markedly diminished in BMD. The histopathologic consequences of BMD and AVMD are similar and include accumulation of lipofuscin, RPE hypertrophy, sub-retinal and occasional sub-RPE deposits.
The fundus appearance of ADVIRC includes an abnormal zone of hyper- and hypo-pigmentation between the equator. Cystoid macular edema is often observed. While EOG abnormalities have been reported in ADVIRC they are typically accompanied by ERG abnormalities as well. The histopathology of ADVIRC includes RPE cells of markedly irregular thickness and pigmentation, accumulation of lipofuscin in RPE cells, loss of photoreceptor outer segments, RPE atrophy, and proliferation of glial cells resulting in preretinal membranes.
4. Future studies
Understanding the function of Best-1 and how mutations in the protein cause disease is essential to developing treatment strategies for BMD, AVMD, and ADVIRC. While data supporting the hypothesis that Best-1 is a CaCC are compelling, two mutations have been shown to cause both AVMD and BMD. Furthermore from our studies of Vmd2 knock-out mice, we conclude that Best-1 is not the LP generator, but rather functions as a modifier of the light peak luminance response possibly via its ability to alter RPE Ca++ responses. Thus, further studies are necessary to determine whether Best-1 forms a channel pore, and/or as data regarding VDCCs and the Vmd2 deficient mouse would suggest, is a regulatory component of Ca++ signaling. Along this line, it is important to understand the mechanism by which Best-1 alters the kinetics of VDCCs and intracellular Ca++ responses. Reconciling any relationship between CaCC activity and Ca++ modulatory activity should have a high priority. While every member of the bestrophin family tested to date has been associated with de novo CaCC activity, and all exhibit unique I/V relationships, the sensitivity of bestrophins to CaCC specific inhibitors (i.e. niflumic acid) and the single channel characteristics have not yet been reported. A comprehensive description of the pharmacology of putative bestrophin channels, single channel recordings, and experiments using planar lipid bilayers would seem essential. Perhaps the greatest challenge will be to understand the relationship between Best-1 dysfunction and the histopathological consequences of diseases associated with the VMD2 gene. Based on the available data, the relationship between Best-1 dysfunction and the accumulation of lipofuscin and/or pigment defects in the RPE, as well as the interaction of Best-1 with the LP are still open questions that must be addressed.
Footnotes
The gene VMD2 has recently been redesignated Best-1 by the HUGO nomenclature committee. Similarly, the Best-1 homologues VMD2L1, VMD2L2, and VMD2L3 have been re-designated Best-2, Best-4, and Best-3 respectively. We apologize to the authors of the many important studies that are referred to but which we could not cite due to space limitations imposed by the journal.
References
- Hartzell C, Putzier I, Arreola J. Calcium-activated chloride channels. Annu Rev Physiol. 2005;67:719–758. doi: 10.1146/annurev.physiol.67.032003.154341. [DOI] [PubMed] [Google Scholar]
- Marmorstein LY, Wu J, McLaughlin PJ, Yocom J, Karl MO, Neussert R, Wimmers S, Stanton JB, Gregg RG, Strauss O, Peachey NP, Marmorstein AD. The light peak of the electroretinogram is dependent on voltage gated calcium channels and antagonized by bestrophin (best-1) J Gen Physiol. 2006;127(5):577–589. doi: 10.1085/jgp.200509473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal R, Bakall B, Kinnick T, Peachey N, Wimmers S, Wadelius C, Marmorstein A, Strauss O. Expression of bestrophin-1, the product of the VMD2 gene, modulates voltage-dependent Ca2+ channels in retinal pigment epithelial cells. FASEB J. 2006;20:178–180. doi: 10.1096/fj.05-4495fje. [DOI] [PubMed] [Google Scholar]
- Sun H, Tsunenari T, Yau KW, Nathans J. The vitelliform macular dystrophy protein defines a new family of chloride channels. Proc Natl Acad Sci USA. 2002;99:4008–4013. doi: 10.1073/pnas.052692999. [DOI] [PMC free article] [PubMed] [Google Scholar]