

Abstract
Chromatin immunoprecipitation (ChIP) is a powerful method for analyzing the interaction of regulatory proteins with genomic loci, but has been difficult to apply to studies on early embryos due to the limiting amount of genomic material in these samples. Here, we present a comprehensive technique for performing ChIP on blastula and gastrula stage Xenopus embryos. We also describe methods for optimizing crosslinking and chromatin shearing, verifying antibody specificity, maximizing PCR sensitivity, and quantifying PCR results, allowing for the use of as few as 50 early blastula stage embryos (approximately 5×104 cells) per experimental condition. Finally, we demonstrate the predicted binding of endogenous β-catenin to the nodal-related 6 promoter, binding of tagged Fast-1/FoxH1 to the goosecoid promoter, and binding of tagged Tcf3 to the siamois and nodal-related 6 promoters as examples of the potential application of ChIP to embryological investigations.
Keywords: chromatin, chromatin immunoprecipitation (ChIP), Xenopus, embryo, transcription factor, histone, DNA, β-catenin, Fast-1/FoxH1, Tcf3, Goosecoid, Xnr6, Siamois
Introduction
Chromatin immunoprecipitation (ChIP) has emerged as an invaluable tool for the study of the mechanisms of transcriptional control and chromatin dynamics. ChIP allows an investigator to determine whether a genomic locus is occupied by chromatin-bound factors such as transcription factors, chromatin remodeling complexes, and modified histones. The most widespread ChIP procedure uses formaldehyde-crosslinked, sheared chromatin from 106 to 107 cells as the input material for an immunoprecipitation (IP), which is followed by several rounds of washing, crosslink reversal, and DNA purification. Because the genomic DNA is sheared to an average size of 1000 base pairs or less, the IP results in the purification of discrete genomic DNA fragments that associate with the antigen of interest. Thereafter, the purified DNA from the experimental IP is queried for enrichment relative to a control IP, either by PCR—for small numbers of target genes—or by one of several genome-wide analysis methods (microarray, high-throughput sequencing, library screening). Thus, ChIP represents a powerful method for investigating in vivo protein-DNA interactions.
For molecular embryologists, however, the typical ChIP protocol poses a number of challenges. Embryos represent heterogeneous populations of cells containing limiting amounts of genomic material. In addition, fractionation of embryos under the denaturing conditions commonly used in ChIP releases a large amount of non-chromatin-associated proteins, such as yolk, that complicate sample preparation and can increase nonspecific background. Consequently, molecular embryologists have been slow to adopt ChIP as a routine assay, especially for the analysis of early embryos. However, ChIP analysis of early embryos could help forge new frontiers in developmental biology. Whether by providing for the enhanced analysis of transcription factor function in gene regulatory networks, or by investigating the function of histone modifications and how their patterns unfold during embryogenesis, numerous new avenues of investigation will require the establishment of a ChIP protocol amenable to embryonic tissues.
Our goal was to develop a ChIP procedure that would be sensitive enough to detect transcription factor occupancy at promoters in cleavage-stage Xenopus laevis embryos (stage 7.5 to 8: approximately 1×103 cells per embryo). In addition, we wanted the protocol to be amenable to typical embryological manipulations, such as microinjection, and therefore optimized the protocol to use as few as 50 embryos per sample from these early stages. Finally, we wanted to circumvent several foreseeable problems with sample preparation by minimizing non-chromatin proteins in the ChIP samples, optimizing the crosslinking and sonication steps, and optimizing DNA extraction and PCR conditions to maximize sensitivity. While this protocol was in development, a number of ChIP experiments on Xenopus laevis embryos were reported (Jallow et al., 2004; Kim et al., 2004; Morgan et al., 2004; Messenger et al., 2005; Park et al., 2005; Ng and Gurdon, 2008), representing four different protocols. While all of these protocols are similar in principle to ours, we have concentrated on maximizing the sensitivity of this procedure for the analysis of earlier stages of development with a small number of embryos. Here, we present our optimized protocol in detail, with some examples of its implementation. By demonstrating the basic method, and describing how the protocol was optimized, we aim to facilitate the adoption of ChIP as a routine assay in the Xenopus embryological laboratory.
While our method is similar to now-standard ChIP protocols in use with other model systems (Kuo and Allis, 1999), some critical differences may be particular to the Xenopus system. First, we report optimized fixation and sonication techniques that yield chromatin crosslinked and sheared enough to detect transcription factor occupancy at promoters with at least 1000bp resolution. Second, because the standard 1% SDS lysis buffer used during sonication tended in our hands to produce low-quality chromatin from early embryos, we used a low-SDS (0.1%) radio-immunoprecipitation assay (RIPA) buffer. RIPA buffer produces high quality sheared chromatin samples while reducing yolk solubilization, thus limiting background by preventing protein precipitation. Doubling the number of washes further reduces background. Finally, we have optimized DNA purification and PCR conditions to allow for the reliable detection of as little as 30 copies of a target sequence per reaction, facilitating the use of as few as 5×104 cells in the starting sample, an improvement of two orders of magnitude from the typical ChIP protocol. We also demonstrate approaches for quantitative PCR and statistical analysis of ChIP results, which can offer several advantages over endpoint PCR detection strategies for detecting differences between samples. Interestingly, several protocols for ChIP using either cultured cells, early mouse embryos, or tissue biopsies have been described that use as few as 1×102 cells (O'Neill et al., 2006; Acevedo et al., 2007; Dahl and Collas, 2008), suggesting that, with modification, the sensitivity of this procedure could be enhanced even further. The protocol we present here is well suited to the Xenopus embryologist: it facilitates the use of microinjected embryos and explanted tissues by decreasing the amount of genomic material required to obtain meaningful data. Thus, we present this work with the hope that it will help advance the use of ChIP in embryological experiments and lead to new avenues of research in developmental biology.
Results and Discussion
In this section, we discuss the critical parameters for optimization and validation as well as general guidelines for ChIP in Xenopus. A step-by-step protocol follows in “Experimental Procedures”. The most critical parameter in our experience is the method used to generate the sheared chromatin sample. Several factors need to be considered: extent of crosslinking, duration/strength of sonication, and yield. In general, the greater the extent of crosslinking, the greater the amount of sonication that will be required to generate ideal (<1000bp) fragments. However, prolonged crosslinking will render chromatin impervious to fragmentation (Orlando et al., 1997), and over-sonication will result in reduced overall yield of genomic DNA. In addition, the target antigens should be considered. For example, nucleosomes can be immunoprecipitated with sheared genomic DNA without crosslinking (O'Neill and Turner, 2003), while transcription factors and secondarily-associated protein complexes may require extended crosslinking times (or different crosslinking reagents) to achieve sufficient co-immunoprecipitation of genomic DNA (Zeng et al., 2006).
These factors should also be considered when customizing this protocol to particular applications. The following procedure was used to optimize crosslinking and shearing for blastula and gastrula stage Xenopus embryos. All sonication steps were performed using a Branson Sonifier 250 equipped with a 1/4″ microtip horn, set at 20% output. Other makes of sonicators will have different efficiencies, making these optimization steps even more critical.
Crosslinking Optimization
To optimize crosslinking time, we performed a fixation time course on gastrula stage (stage 10) embryos. Using this stage ensures that enough DNA will be recovered for analysis by agarose gel electrophoresis. We collected embryos fixed in 1% formaldehyde / PBS for 15, 30, 45, and 60 minutes, as well as control, non-fixed embryos. Samples were prepared according to the ChIP Day 1 protocol up to step 8 (see Experimental Procedures). We sonicated the samples minimally using conditions (3× 20 seconds, 20% output, 20% duty cycle) that would solubilize the genomic DNA from the insoluble pellet—effectively shearing native DNA— but would minimally shear crosslinked DNA. Post-sonication supernatants were adjusted to 1% SDS, 10mM EDTA prior to crosslink reversal and DNA purification, as described in the Experimental Procedures, except that following RNase treatment, DNA was ethanol precipitated, resuspended in 50μl H2O and analyzed by agarose gel electrophoresis.
While genomic DNA from embryos fixed for 15 minutes showed no resistance to shearing as compared to control, DNA from embryos fixed for as little as 30 minutes showed evidence of crosslinking, indicated by the detection of slower-migrating, sonication-resistant DNA (Figure 1A). Subsequently, the extent of crosslinking was increased incrementally until the 60-minute time point. Although 60-minute fixation times have been reported (Orlando et al., 1997), typical ChIP protocols performed on yeast and cultured cell systems fix samples for 10 to 15 minutes (Luo et al., 1998; Kuo and Allis, 1999), with similar amounts of formaldehyde in PBS or culture medium. This result suggests that the kinetics of nucleoprotein crosslinking by formaldehyde is different in the case of the Xenopus embryo, perhaps due to a greater non-chromatin protein to DNA ratio compared to other systems. Therefore, it will also be important to optimize crosslinking times for later embryonic stages when this ratio begins to approach typical somatic levels.
Figure 1. Crosslinking and Sonication Optimization.

(A) Minimally-sonicated, gastrula stage (stage 10) DNA from a 1% formaldehyde/1×PBS time course was resolved by 2% agarose gel electrophoresis. Onset of crosslinking is observed by the recovery of sonication-resistant, high molecular weight DNA (>1kb). (B) 60-minute crosslinked chromatin from gastrula stage embryos was fully sonicated for one to four rounds of 20 seconds each and resolved by 1.2% agarose gel electrophoresis. Three rounds of sonication balances optimal yield (90%) with maximal shearing (<1kb) of the crosslinked chromatin.
General Sonication Guidelines
Perform sonication on ice to prevent overheating. Place the sample to be sonicated in a beaker filled with ice, and hold this under the sonicator horn during shearing.
Sonicate in short bursts. 20-second rounds of sonication prevent sample overheating. Let the samples rest for at least 1 minute before the next round.
Center the horn in the sample and avoid contact with the walls of the microcentrifuge tube. This will improve reproducibility.
Avoid sample foaming, which happens when the tip of the horn draws air into the sample because it was brought too close to the surface.
Shearing Optimization
To determine whether we could generate sufficiently small average DNA fragment size with a longer fixation time, we crosslinked gastrula stage (stage 10) embryos for 60 minutes and performed one to four rounds of full-strength sonication (20 seconds each, 20% output, 100% duty cycle) and repeated the crosslink reversal and DNA purification as described below (see Experimental Procedures). One round of sonication generated a majority of <1000bp fragments, but we also noted a population of high molecular weight DNA that was reduced with each successive round of sonication (Figure 1B). By four rounds of sonication, the high molecular weight DNA was virtually undetectable, but overall DNA yield was also reduced by 25%. Therefore, we concluded that three 20-second rounds of sonication balances optimal yield (90%) with average fragment length (<1000bp). The efficiency and specificity of this method of crosslinking and shearing was further validated as described below.
Chromatin Immunoprecipitation in Blastula Stage Embryos
We tested the ChIP protocol by scanning for occupancy of the transcription factor β-catenin within a 2.5kb upstream portion of the Xenopus nodal-related 6a (Xnr6) locus that contains several predicted Tcf/Lef binding sites (Figure 2A). Wnt/β-catenin pathway activity is required for Xnr6 expression (Takahashi et al., 2000; Rex et al., 2002; Xanthos et al., 2002; Yang et al., 2002), so we predicted that some of these sites would be occupied by β-catenin in blastula stage embryos. In addition, we performed ChIP for a euchromatin marker, di-acetylated [K9/K14] histone H3 (AcH3), reasoning that an active locus should be positive for AcH3 (Roh et al., 2005). A portion of the related Xnr1 locus that is not predicted to bind β-catenin was analyzed as an additional control (Figure 2A).
Figure 2. Chromatin immunoprecipitation in blastula-stage Xenopus embryos.

Schematic representations of the Xnr6a and Xnr1 genomic loci (A) demonstrate the locations of predicted Tcf/Lef consensus sequences relative to the ChIP PCR amplicons. (B) These primer sets amplify standard genomic DNA by PCR with similar efficiencies, with a limit of detection at approximately 30 haploid genomes. (C) Blastula-stage embryos (Stage 9) were processed for ChIP using either anti-acetylated histone H3, or anti-β-catenin antisera (with the corresponding negative controls). The PCR products were visualized by 3% agarose gel electrophoresis and ethidium bromide staining. Co-immunoprecipitation of associated genomic DNA is observed when the PCR signal is greater for the experimental IP (AcH3, β-catenin) than in the control IP (IgG, Serum). Note that the Xnr1 (-221) product occasionally amplifies as a doublet (seen in panel C): this represents genetic variation at this locus within our colony.
PCR conditions were optimized to ensure detection of immunoprecipitated DNA present in limiting quantities. To maximize detection, we performed nested PCR, using two rounds of 20 cycles each to amplify target sequences from genomic DNA standards. Nested PCR has the twofold advantage of replenishing the polymerase, primers, and dNTPs available for amplification while increasing priming specificity by using a second, internal primer set for the second round of amplification. By this method, we are able to detect PCR products from as little as 100pg of genomic DNA, corresponding to approximately 30 haploid genomes (Figure 2B). Radiolabeling the second, inner PCR reaction further increases sensitivity (not shown).
We performed ChIP on blastula stage embryos with anti-AcH3 and anti-β-catenin (Figure 2C). All loci tested were associated with AcH3, while only the Xnr6 (-118) and (-2349) amplicons were bound by β-catenin. Notably, an intermediate amplicon (-1280) was negative for β-catenin binding. Therefore, the fixation and sonication method in this ChIP protocol sufficiently fragments chromatin, and has at least a ∼1000bp resolution. We expect that this approach will be useful for scanning intergenic regions for transcription factor binding sites and occupancy of modified histones. Additionally, these results confirm an expected result, namely, that Xnr6 is a direct target of the Wnt/β-catenin signaling pathway.
Controls for Antibody Specificity
Several control experiments confirmed the specificity of our β-catenin antibody under the conditions used for ChIP. To demonstrate antibody specificity, we both confirmed that the antibody could be competed by the immunizing peptide and tested that depletion of β-catenin would reduce the amount of co-immunoprecipitated chromatin. When possible, this latter approach is a powerful method for antibody validation, as it will reveal off-target antibody recognition that could be overlooked by peptide competition alone. First, we optimized conditions for peptide competition of antibody-antigen binding using western blotting of protein recovered from immunoprecipitated chromatin. The β-catenin antibody was raised against the 145 N-terminal amino acids of the Xenopus laevis β-catenin protein. We therefore used a 6×His-tagged peptide corresponding to the immunizing peptide to compete for β-catenin binding in the ChIP assays, either by pre-incubation of the peptide with the antibody prior to the addition of sheared chromatin (Figure 3A, lane 2) or by addition of the peptide to the sheared chromatin before the antibody (Figure 3A, lane 3). For this experiment, the ChIP protocol was followed through Day 2, step 9, whereupon samples were analyzed by 8% SDS-PAGE followed by western blotting (see Experimental Procedures). Both methods of peptide competition reduced immunoprecipitated β-catenin in the ChIP samples, confirming the specificity of the antiserum. Notably, a 1/5th embryo equivalent was loaded in the input lane, while 2 embryo equivalents were loaded in the IP lanes, but similar band intensities for β-catenin are observed by western. This indicates that, following ChIP for β-catenin, as little as 10% of the available antigen is recovered, although these conditions effectively deplete lysates of antigen under native conditions (not shown).
Figure 3. Controls for antibody specificity.

(A) The specificity of the β-catenin antiserum was confirmed by performing ChIP on blastula-stage (stage 9) embryos and competing with an excess of immunizing peptide (lane 2: pre-incubation of antibody with peptide; lane 3, addition of peptide directly to sheared chromatin sample). Following IP, samples were processed as described in the text and separated by 8% SDS-PAGE. Immunoprecipitated β-catenin was analyzed by a standard western blot using the β-catenin antiserum. (B) Peptide competition of the β-catenin ChIP and (C) knockdown of β-catenin by morpholino injection (20ng/cell at the 2-cell stage) confirms the specificity of the interaction between β-catenin and the Xnr6 promoter. The competitions in (B) and (C) were performed by pre-incubation of the peptide with the antiserum for 1 hour before addition to the sheared chromatin sample.
Additionally, we tested whether the immunizing peptide would compete for co-immunoprecipitation of the Xnr6 genomic locus. Indeed, when the β-catenin antibody is competed with the immunizing peptide, only background levels of Xnr6 (-118) are co-immunoprecipitated, thus confirming the specificity of this interaction (Figure 3B). Likewise, only background levels of signal were detected with a negative control locus, Myosin Light Chain 2 (Xmlc2) (Park et al., 2005). Finally, in Figure 3C, we demonstrate that knockdown of β-catenin by microinjection of a morpholino oligonucleotide (Heasman et al., 2000) also results in a loss of β-catenin binding to the Xnr6 locus, comparable to the reduction seen by peptide competition. This latter result is notable, insofar as the experiment was performed with a single set of microinjected embryos (100 total, plus 100 non-injected controls), demonstrating that this approach is amenable to typical embryological manipulations in common use within the Xenopus community. These observations validate the specificity of the ChIP protocol and the observation that β-catenin binds to the Xnr6 genomic locus in blastula stage embryos.
Quantitative PCR Analysis
To determine whether ChIP samples produced by this protocol would be amenable to quantitative PCR analysis, we designed an experiment to evaluate the binding of Fast-1 (FoxH1) to the endogenous Goosecoid promoter. Goosecoid is an organizer gene with a well-defined promoter region responsive to both Wnt and Nodal signals (Watabe et al., 1995). During early embryogenesis, the Nodal pathway signals through the DNA-bound effector Fast-1 (Shen, 2007).
Embryos were injected at the one-cell stage with mRNA encoding myc-tagged Fast-1 (250pg) alone or in combination with Xnr1 mRNA (50pg), a Xenopus Nodal-related gene. Embryos were collected at the mid-gastrula stage (stage 10.5) and processed according to the ChIP protocol, using a polyclonal anti-myc antibody to immunoprecipitate myc-Fast-1 containing complexes, followed by QPCR. As a control for non-specific binding of the antibody, uninjected embryo samples were analyzed in parallel. As a negative control, binding of myc-Fast1 to the Ef1α coding region, which is not expected to bind Fast-1, was also examined.
As shown in Figure 4, myc-Fast1 binds to the endogenous Goosecoid promoter, and not to Ef1α. While the signal from the Goosecoid promoter is high, the background from Ef1α is low, indicating that the binding of myc-Fast1 to the Goosecoid promoter is quite robust, as predicted. Similar results were obtained with injection of as little as 25pg of myc-Fast-1 mRNA (data not shown). From these results we conclude that myc-Fast-1 indeed binds to the endogenous Goosecoid promoter and that QPCR provides a sensitive and quantitative method for analyzing ChIP samples obtained using this protocol.
Figure 4. Quantitative PCR analysis of ChIP.
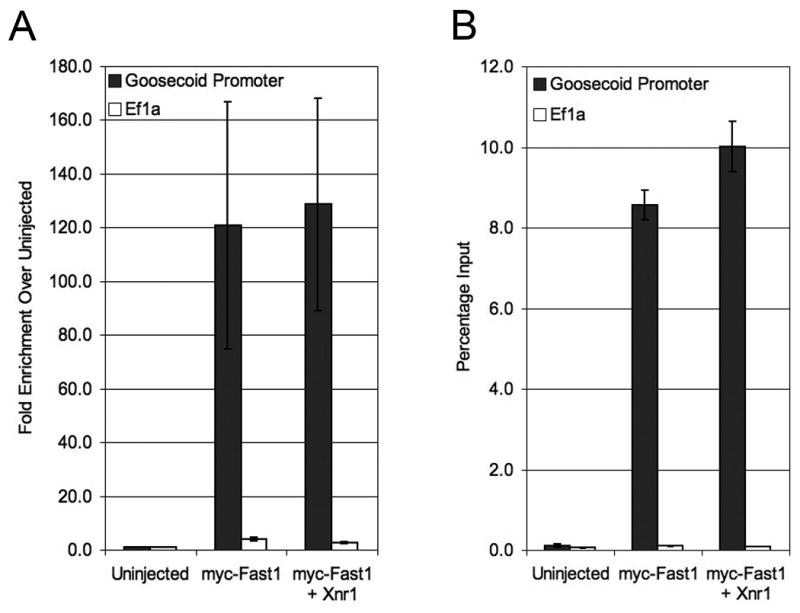
Quantitative PCR for the Goosecoid promoter (gray bars) and Ef1α (white bars) normalized to uninjected embryo control (A) or quantified as a percentage of input DNA (B). Graphs represent average relative quantification for four independent experiments. An average of 45 one-cell embryos were injected with myc-Fast1 (250pg) alone or in combination with Xnr1 (50pg) and harvested at gastrula stage (stage 10.5) for ChIP analysis according to this protocol. QPCR was performed using SYBR green and relative quantification was performed using the ΔΔC(t) method. Error bars shown represent standard error.
Figure 4 presents two approaches to data normalization. Figure 4A represents the fold enrichment when experimental samples are normalized to uninjected control samples and Figure 4B shows the quantity of each immunoprecipitated target sequence as a percentage of total input DNA. In this experiment, PCR amplification was linear over a range of 5% (the highest amount tested) to 0.01% input material. By comparison to genomic DNA standards, this corresponds to a range between 65 and 0.1ng genomic DNA (data not shown). Thus, in these experiments, single round QPCR is as sensitive as the nested PCR shown in Figure 2B. As such, we have not investigated whether QPCR is amenable to a nested PCR approach, but it is conceivable that a limited, initial (conventional) PCR amplification could enhance the sensitivity of a subsequent QPCR analysis.
Finally, β-catenin binds to chromatin indirectly through interaction with Tcf/Lef family members, raising the possibility that prolonged crosslinking (60 min) is only required for indirect binding proteins, whereas proteins that bind directly to DNA may be crosslinked more efficiently. We therefore evaluated the effect of crosslinking time on the recovery of Tcf3 at target gene promoters by ChIP. Tcf3 is a member of the Tcf/Lef family of transcription factors that is predicted to bind directly the promoters of β-catenin target genes during early embryogenesis (Molenaar et al., 1996). Embryos were injected with 25pg Myc-tagged Tcf3 per blastomere at the two-cell stage and fixed at stage 10 for 15, 30, 45, or 60 minutes. Following ChIP, samples were analyzed by QPCR for recovery of promoter sequences for the β-catenin target genes Siamois and Xnr6. Tcf3 has been shown previously to bind the Siamois promoter in gastrula stage embryos, and thus serves as a positive control in this experiment (Park et al., 2005). Recovery of Xmlc2 was also measured as a negative control for Tcf3 binding. Figure 5 demonstrates that recovery of Tcf3-bound promoter sequences is maximal with 60 minutes of crosslinking time, and that crosslinking for 30 minutes or less leads to little or no recovery of target gene promoter sequences. Notably, crosslinking for up to 60 minutes does not result in a large increase of non-specific enrichment for the negative control locus. In addition, a 45-minute crosslinking time is also sufficient to recover bound promoters (albeit less efficiently), with the advantage of slightly less signal from the negative control ChIP. These results underscore the observation in Figure 1A that extended crosslinking times are required to perform ChIP for DNA-bound transcription factors in early Xenopus embryos.
Figure 5. Effect of crosslinking times on ChIP.

Anti-Myc-Tag ChIP was performed on sets of 37 gastrula stage (stage 10), non-injected or Myc-Tcf3 injected (50pg) embryos, which were crosslinked for 15, 30, 45, or 60 minutes, as indicated. Samples were analyzed by QPCR for enrichment of Xmlc2 (-118), Xnr6 (-118), and Siamois (-303), represented as % Input recovery. Crosslinking times between 45 and 60 minutes are required for the specific enrichment of the promoters for both Siamois (black bars) and Xnr6 (hatched bars) by Myc-Tcf3 ChIP compared to the Xmlc2 promoter (gray bars). All non-injected (negative control) ChIPs yielded a comparatively low signal and are represented as white bars. Recovery of input material was similar for each time point.
Further considerations
In addition to the factors described above, several additional considerations should be made when designing a Xenopus ChIP experiment. Particularly, since embryos represent a heterogeneous population of cells, certain protein:DNA interactions may occur in only a small fraction of the experimental sample. This will result in an overall reduction in the number of available binding events for analysis. In cases where a minimal amount of starting material is used, this heterogeneity could lead to “false negative” results. Several approaches are available to circumvent this problem: increasing the amount of starting material, increasing the amount of material in the PCR analysis, and enriching for the subpopulation of cell types with the predicted protein:DNA interaction. Xenopus embryos are particularly well suited for this latter approach. For example, using explanted tissues containing the lineage of interest can enrich for particular cell types. Alternatively, molecular techniques for expanding cell lineages can be used to troubleshoot negative results. Finally, in certain cases (and also when a suitable antibody is unavailable), it may be possible to overexpress the DNA binding factor of interest throughout the embryo to increase the number of protein:DNA binding events available for analysis. However, care must be taken in the interpretation of overexpression experiments, particularly due to the difficulty in controlling the spatio-temporal expression of injected mRNAs. In this case, it may be possible to refine further overexpression approaches by driving tissue-specific expression of the DNA-binding factor of interest, either by plasmid DNA injection or transgenesis.
Concluding Remarks
We have reported a ChIP assay protocol amenable to early Xenopus embryo research, have demonstrated examples of its implementation, and have outlined methods to optimize several aspects of the protocol. We strongly encourage repeating the optimization experiments shown in Figure 1 when different conditions or embryo stages are used, as we find this is the most critical aspect of the protocol in our hands. We expect that ChIP will become a useful tool within the Xenopus community, allowing for the identification of direct protein-DNA interactions, which, in the past, have been indirectly surmised, usually by plasmid-based reporter gene assays or by the combination of overexpressed, hormone inducible chimeras and cycloheximide treatment. In addition, this technique will allow for many unanswered questions in developmental biology to be addressed, including the systematic analysis of gene regulatory networks and the investigation of the function of chromatin modifications during early vertebrate embryogenesis.
Experimental Procedures
General Approach and Design
ChIP experiments are designed similarly to common immunoprecipitations (IPs), which include both experimental and control IPs and a sample of the pre-IP (input) material, generally 1%. This protocol has been used successfully with as few as 50 stage 8, 1000-cell embryos (5×104 cells total) per IP (or an equivalent amount of explanted tissues from later-stage embryos). Therefore, 100 embryos are required for a basic ChIP if early blastula stage is used. If preliminary experiments demonstrate weak signal strength, the number of embryos can be increased at the investigator's discretion.
ChIP requires high-quality antibodies that will specifically immunoprecipitate the antigen in 0.1% SDS and maintain the interaction throughout several washes of increasing stringency. Because the IP material is also highly crosslinked, certain antigens may be masked to a greater extent than under native conditions. Since this protocol is amenable to the use of microinjected samples, N- or C- terminal tagged proteins of interest can be used if an antibody doesn't perform as expected, or if one is unavailable. We have successfully used both Myc- and HA-tagged proteins in this way, and recommend the use of polyclonal rather than monoclonal antisera, when possible.
ChIP Protocol
This protocol was developed to detect occupancy of transcription factors at target gene promoters during blastula and early gastrula stages. It is divided into five sections, each of which can be completed easily in one day: “Sample Preparation,” “Days 1-3,” and “PCR.” Points that can be shortened to accelerate sample preparation will be noted.
Sample Preparation
Embryos are cultured to the desired stage and fixed in formaldehyde. This will crosslink proteins to one another and to DNA (Orlando et al., 1997). Fixation is quenched by washing with glycine, yielding a sample ready for ChIP.
Materials
Standard Xenopus embryo culture and microinjection materials (Sive et al., 2000)
Phosphate Buffered Saline (PBS): (Per liter) 8g NaCl, 0.2g KCl, 1.44g Na2HPO4, 0.24g KH2PO4, pH 7.4 (Sambrook and Russell, 2001).
Formaldehyde (37% stock, Molecular Biology Grade, Thermo Fisher BP531); 1% Formaldehyde in PBS working solution: 676μl formaldehyde per 25ml PBS
0.125M Glycine in PBS: Mix 235mg Glycine (Sigma G7125) per 25ml PBS
1.5ml microcentrifuge tubes (Eppendorf 02236411)
Time
Following culture to the desired stage, up to 1 hour to crosslink, and 20 minutes to quench, wash, and distribute samples to tubes.
Procedure
At the desired stage, embryos are fixed/crosslinked in 1% formaldehyde/PBS for up to 1 hour (see Results and Figure 1a). Agitation is not necessary. Crosslinking is stopped by a 10-minute wash in 0.125M Glycine/PBS, followed by three washes in PBS. Fixed embryos are transferred to microcentrifuge tubes (50 per tube), excess PBS is removed, and they may be either frozen at -80°C for at least 3 months or processed for ChIP immediately (Day 1 protocol).
Day 1
Samples are homogenized and crosslinked chromatin is sheared to <1000bp fragments by sonication. Samples are then pre-cleared and the primary antibody incubation is performed overnight.
Materials
RIPA buffer (4°C, 1.25ml per set of 50 embryos): 50mM Tris-HCl, pH 7.4, 1% Igepal CA-630 (NP-40) (Sigma I3021), 0.25% Na-Deoxycholate, 150mM NaCl, 1mM EDTA, 0.1% SDS, 0.5mM DTT, 5mM Na-Butyrate, Protease Inhibitor Cocktail (Sigma P8340), Phosphatase Inhibitor Cocktail I (Sigma P2850), Phosphatase Inhibitor Cocktail II (Sigma P5726).
20ml 5% Bovine Serum Albumin (BSA fraction V, Sigma A9647) in PBS (see above)
15ml conical tubes with caps (Thermo Fisher 14-959-70C)
Recombinant Protein G-Agarose (100μl per set of 50 embryos) (Invitrogen 15920-010)
Kontes Pellet Pestles (Thermo Fisher K749521-1590)
Cold centrifuge capable of 14,000g
Kimwipes (Thermo Fisher 06-666)
Nutator or end-over-end rotator (at 4°C) to mix samples
Sonicator (Branson Sonifier 250 or equivalent, with a microtip horn)
Safe-Lock 1.5ml microcentrifuge tubes (Eppendorf 2260002-8)
TES Buffer: 50mM Tris-HCl pH 8.0, 10mM EDTA, 1% SDS (store at room temperature)
Control and Experimental Antibodies
Time
1.5 hours for homogenization and shearing, 1 hour preclear, overnight primary incubation
Procedure
Thaw crosslinked embryos for 15 minutes on ice.
Prepare blocked protein-G agarose while the embryos are thawing. Dispense enough protein-G agarose for each IP (plus an extra 50μl) into each of two 15ml conical tubes. Mark the volume's height on the side of the tube with a marker. Add 10ml 5%BSA/PBS to each tube and incubate at 4°C with mixing for at least 1 hour prior to use.
Add 600ul 4°C RIPA buffer to the fixed embryos.
Homogenize the fixed embryos with a pellet pestle: gently disrupt the embryo pellet, and then vigorously homogenize each sample for 30 seconds. Proceed through all the samples, using different pestles for each set of embryos, and then repeat the homogenization once again. The sample will become a uniform gray color with a fine particulate consistency. No large embryo fragments should remain.
Incubate embryos on ice for at least 10 minutes beginning from the time of initial homogenization.
Centrifuge at 14,000g for 10 minutes at 4°C.
Discard the supernatant and wipe the wall of the tube above the pellet with a kimwipe to remove lipid residue.
Add 650μl 4°C RIPA to the pellet and re-homogenize vigorously, ensuring that no coarse fragments of pellet remain.
Sonicate sample (see Results and Discussion).
Centrifuge for 10 minutes at 4°C at 14,000g.
Transfer the supernatant (600μl recovery is typical) into pre-chilled, clean 1.5ml microcentrifuge tubes. The supernatant contains the sheared chromatin and should appear medium-yellow without any debris. The pellet should be compact, and will have a top layer of dark pigment, with a bottom yellowish, yolky layer.
To prepare Input Samples: In a safe-lock tube, combine 195μl TES buffer and 5μl sheared chromatin. These samples will be processed once the IPs are complete. Freeze at -80°C.
Centrifuge one of the 15ml conicals containing the blocked protein-G agarose at 1000g for 5 minutes. Remove excess 5%BSA/PBS to the level of the original bead volume (marked on the side of the tube).
Gently resuspend the blocked beads by low speed vortexing.
Pre-clearing: Dispense 50μl blocked beads to each sample of sheared chromatin, mixing the beads by pipetting before removing them from the 15ml conical to ensure equal distribution between samples. Mixing by vortexing each time is not recommended.
Incubate the samples at 4°C with mixing for 1 hour.
Centrifuge at 1000g for 1 minute at 4°C.
Transfer 580μl pre-cleared, sheared chromatin to a pre-chilled 1.5ml tube. Avoid transferring any beads.
Begin the immunoprecipitation: Add the appropriate amount of antibody or control serum to each sample. Amounts will need to be determined empirically. In the examples here, 1μg affinity purified IgG, or 5μl whole serum, per sample was optimal.
Incubate samples overnight at 4°C with mixing. (To shorten sample preparation time, the antibody incubation could be reduced to a minimum of 1 hour, followed by continuation with the Day 2 Protocol. Appropriate minimum conditions will have to be determined empirically.)
Day 2
Immunocomplexes are precipitated, washed extensively, and eluted. Crosslinks are reversed and proteins are digested.
Materials
Blocked protein-G agarose (see above)
Wash Buffer I: 20mM Tris-HCl pH 8.0, 0.1% SDS, 1% Triton X-100, 2mM EDTA, 150mM NaCl
Wash Buffer II: (Wash Buffer I, with 500mM NaCl)
Wash Buffer III: 10mM Tris pH 8.0, 0.25M LiCl, 1% Igepal CA-630 (NP-40), 1% Na-Deoxycholate, 1mM EDTA
Wash Buffer IV: (TE buffer) 10mM Tris-HCl pH 8.0, 1mM EDTA
1ml syringe fitted to a vacuum aspirator
20-gauge syringe needles
26-gauge syringe needles
TES Buffer (see above)
Safe-lock tubes (see above)
Hybridization oven or water bath set at 65°C
Proteinase K/Glycogen Solution: 15mg/ml Proteinase K, 6mg/ml GlycoBlue (Ambion AM9515), PBS, 30% Glycerol
Time
1 hour for immune complex precipitation, 2 hours for the washes and elution, followed by an overnight crosslink reversal/proteinase K digestion.
Procedure
Centrifuge samples at 14,000g for 5 minutes at 4°C and transfer supernatants (560μl) to pre-chilled 1.5ml microcentrifuge tubes. This will remove insoluble precipitates that may have formed during the overnight incubation.
Repeat steps 13-17 of the Day 1 procedure with the second 15ml conical tube of pre-blocked protein-G agarose.
-
To Wash: Each wash consists of a 1-minute spin at 1000g in a 4°C centrifuge to pellet the immunocomplexes, removal of supernatant via aspiration with a 20-gauge needle, addition of 1ml wash buffer (see below), and incubation at 4°C with mixing for 5 minutes.
Wash for a total of 8 times, using 2 washes each with buffers I through IV.
If high background is observed, it may help to do 3 washes with each of the buffers, or to increase the wash time to 10 minutes.
Following the washes, aspirate the supernatant with a 26-gauge needle, inserting it into the beads at the end to completely remove any residual wash buffer. If necessary, perform a quick additional spin and repeat aspiration to ensure all of the supernatant has been removed.
Add 220μl TES buffer to the beads and vortex vigorously for 5 seconds.
Elution: Incubate the samples at 65°C for 10 minutes, vortexing each sample vigorously for 5 seconds every 2 minutes.
During this time, the frozen input samples should be thawed and vortexed to resuspend any precipitated SDS. IP and input samples are processed in the same manner for the rest of the procedure.
Centrifuge the samples at 14,000g at room temperature for 1 minute.
Transfer 200ul of the eluted IP supernatant to a safe-lock tube. Do not transfer any beads.
Add 7μl Proteinase K/Glycogen solution to each sample.
Incubate the samples at 65°C overnight to reverse crosslinks and digest proteins. (Other protocols suggest that a 4-hour incubation is sufficient to reverse crosslinks and digest. One could try this to shorten this step.)
Day 3
DNA is purified from the samples, RNase-treated, and finally re-purified through a spin-column.
Materials
Phenol/Chloroform/Isoamyl Alcohol (Thermo Fisher BP1752)
Sterile MilliQ (or equivalent) H2O
TE-Saturated Chloroform (Mix equal parts TE buffer (10mM Tris-HCl pH8.0, 1mM EDTA) and Chloroform (Thermo Fisher BP1145) and allow the phases to separate. Use the lower, organic phase)
5M NaCl
100%, 70% Ethanol
RNase A/TE buffer (Dilute RNase A (100mg/ml stock solution, Roche 10109169001) to 2mg/ml in TE buffer)
Qiagen PCR Purification Kit (Qiagen 28104)
Time
1.5 hours for phenol/chloroform extraction and ethanol precipitation, 1 hour for RNase treatment, and 15 minutes for spin-column purification.
Procedure
Add 200μl phenol/chloroform/isoamyl alcohol to each sample, vortex vigorously, and centrifuge at 14,000g for 1 minute at room temperature.
Transfer 200μl of the upper, aqueous phase to a clean 1.5ml microcentrifuge tube.
Add 200μl sterile milliQ water to the lower, organic phase. Vortex the sample vigorously and centrifuge at 14,000g for 1 minute at room temperature.
Add 200μl of the second aqueous phase to the first aqueous phase.
Add 350μl TE-saturated chloroform to each sample, vortex vigorously, shake, and centrifuge at 14,000g for 1 minute at room temperature.
Transfer 350μl of the aqueous phase to a clean 1.5ml tube.
Add 14μl 5M NaCl to each sample. Vortex to mix and do a quick spin.
Add 920μl room temperature, 100% ethanol to each sample.
Mix well by inversion and incubate the samples at −80°C for 30 minutes.
Centrifuge at 14,000g for 15 minutes at 4°C. A blue pellet should form at the bottom of the tube. If not, vortex the tube and repeat centrifugation.
Discard the supernatant.
Wash samples by adding 1ml 70% ethanol, mixing by inversion, and centrifuge at 14,000g for 5 minutes at 4°C.
Discard the supernatant.
Resuspend pellets in 100μl RNase A / TE.
Incubate samples at 37°C for 1 hour.
Add 500μl Qiagen Buffer PB to each sample.
Mix well and briefly spin down.
Transfer each 600μl sample to a Qiagen PCR Purification Kit column.
Follow the kit protocol for PCR Purification, eluting in 50μl buffer EB.
The eluate from the Qiagen PCR Purification kit is now ready for analysis by PCR.
PCR
PCR Primers
For nested PCR, primer sequences were designed with MacVector 9.0, with default settings except that the primer melting temperature was reduced from 55°C to 50°C. Amplicon length is ideally between 100-180bp. Primers are defined as “nested” if the inner primer pairs have 5 or more unique 3′ nucleotides. The numbering of the primer sets represents the midpoint of the amplicon, relative to the predicted translational start site (ATG). The inner Xmlc2 primer was previously reported (Park et al., 2005). Xnr6 (-118): Outer/Forward 5′- TCT GAG GTG TGA GGT ATA TGA AAG G -3′; Outer/Reverse 5′- TGG GGC TCT TGA AAA CTG AAA TG -3′; Inner/Forward 5′- GGT AGA TGA AAG GCT GAC AGG TGT G -3′; Inner/Reverse 5′- GGC TGT TGA AAA CTG AAA TGA AGC -3′. Xnr6 (-1280): Outer/Forward 5′- AAA AGG AGT CTA TGA GAA GTG GC -3′; Outer/Reverse 5′- TGA GAA TAC AGT AAG GAG GGG C -3′; Inner/Forward 5′- GGA TAA TGG GTT TCT GGA TAA CTG -3′; Inner/Reverse 5′- GGT GAT GCT AAA GGT GAG ATG G -3′. Xnr6 (-2349): Outer/Forward 5′- ACA CCC CCT GCT CCC CCG -3′; Outer/Reverse 5′- GCA AAA CAA TCC CAC CCA G -3′; Inner/Forward 5′- GGT ACT TCC GCC ACT GAA AG -3′; Inner/Reverse 5′- AGA CCC CTA TCC AGA AAA TCT C -3′. Xnr1 (-221): Outer/Forward 5′- TCT GAG GTG TGA GGT ATA TGA AAG G -3′; Outer/Reverse 5′- TGG GGC TCT TGA AAA CTG AAA TG -3′; Inner/Forward 5′- GGT AGA TGA AAG GCT GAC AGG TGT G -3′; Inner/Reverse 5′- GGC TGT TGA AAA CTG AAA TGA AGC -3′. Xmlc2 (-118): Outer/Forward 5′- TGG GAT ATT TTA CTG AAC ACA ATG -3′; Outer/Reverse 5′- CGT CCT GTG CCA CCT AAT G -3′; Inner/Forward 5′- GAA TGT TAG CCC TTG TGC TCT T -3′; Inner/Reverse 5′- GGA AAG TTC TCT TGA TCA TTT TA -3′.
For QPCR, primers were 18 to 30 nucleotides long with an approximate melting temperature of 60°C. Amplicon length was between 80-120bp. The following QPCR primer sets were used. Goosecoid (-224): Forward 5′- AAT GAC AGC CAA CAG CTC AGA GGA CA -3′; Reverse 5′- TCG CAG ACT CTC CCT GTA GTT ATT CAC A -3′. EF1α (-335): Forward 5′- GTC TCG GCC CCT AAA TAT GA -3′; Reverse 5′- CAG CTC CCA GCT CTT TTG TC -3′. Siamois (-303): Forward 5′- GGG ACT TTG AAG TCT TGC CA -3′; Reverse 5′- TCT GAT GAC ACG TGT TTC CC -3′. For QPCR of Xnr6 (-118) and Xmlc2 (-118), the “outer” primer sets were used (see above).
Nested PCR
ChIP DNA is subjected to two sequential PCR reactions of 20 cycles each using Promega GoTaq Flexi DNA Polymerase (M8295). For the first, “outer,” PCR reaction, the final conditions are: 25μl total volume, containing 2μl ChIP DNA sample, 1× Clear Reaction Buffer, 1.5mM MgCl2, 0.1mM dNTP (each), 0.63U GoTaq Polymerase, and 4ng/μl each “outer” primer.
PCR was performed in a MJ Research Tetrad thermal cycler. Cycling parameters are as follows: 95°C for 3 minutes, followed by 20 cycles of [95°C for 30 seconds, 50°C for 30 seconds, and 72°C for 60 seconds], ending with 72°C for 10 minutes. The low annealing temperature is to accommodate the low C/G content of Xenopus laevis intergenic DNA.
The second, “inner” PCR reactions are identical to the “outer” PCR reactions except the template is 2μl of the complete “outer” PCR reaction, and the “inner” primer sets are used instead of the “outer” ones. In addition, the green PCR reaction buffer that comes with the polymerase can be used instead of the clear one, since it doubles as a loading buffer for electrophoresis. PCR products (15-20μl) are visualized by 3% agarose electrophoresis in 1× TAE with ethidium bromide. To facilitate handling of high-percentage agarose, a 1 to 3, low-melt to normal agarose mix is used.
Radiolabeling the “inner” PCR reaction can increase the limit of detection. To radiolabel, add 0.1μl α32P-dCTP (3pmol at 3000Ci/mmol) to each “inner” PCR reaction. PCR products (10μl) are then resolved by 6% polyacrylamide electrophoresis in 0.5× TBE and visualized with a Phosphorimager.
It may be useful to include genomic DNA standards to assess the efficiency of PCR amplification. For the experiment in Figure 2B, genomic DNA was isolated from adult male liver (∼0.5mg fragment) similarly to the ChIP DNA purification, except samples were ethanol precipitated following RNase treatment and resuspended in 200μl TE buffer. The DNA concentration was determined by absorbance at 260nm. The estimated mass of a karyotypically “haploid” Xenopus laevis genome (3.175pg) (Wickbom, 1945) (Green and Sessions, 1991) was used to calculate approximate copy number. The possible amplification by PCR of duplicated genomic loci was not considered in this estimate.
Quantitative PCR and Analysis
The QPCR reaction conditions are: 20μl total volume with 1× SYBR green master mix (Applied Biosystems, 4367659), 1μl ChIP DNA sample, and 5ng/μl each primer. If the QPCR yield is low, the amount of ChIP DNA per reaction can be increased. Amplification was performed with an Applied Biosystems Step One Plus machine, using the standard SYBR green program with an initial melt stage at 95°C for 10 minutes, followed by at least 40 cycles of 95°C for 15 seconds and 60°C for one minute. The run is finished by a melt curve from 95°C to 60°C to ensure PCR product purity.
Data are analyzed by the ΔΔC(t) (or Livak) method (reviewed in (Taneyhill and Adams, 2008)). Once a threshold cycle (C(t)) number is calculated for each sample, ΔC(t) is calculated by normalizing the IP values to the input values for each condition by subtracting input values from each corresponding ChIP value [ΔC(t) = ChIP C(t) − Input C(t)] (Table 1). ΔΔC(t) is next calculated by subtracting the ΔC(t) for uninjected samples from the ΔC(t) for experimental samples [ΔΔC(t) = ΔC(t)experimental − ΔC(t) uninjected]. Once ΔΔC(t) is determined, the fold change between samples can be determined by using the formula [Fold Change (FC) = 2[-ΔΔC(t)]] (Taneyhill and Adams, 2008). To calculate the quantity of IP-ed DNA as a percentage of the original input material, the following formula is used: [% Input = 2 [-ΔC(t)] x initial percentage input] (Table 1) (Frank et al., 2001; Taneyhill and Adams, 2008). Once several experiments have been analyzed, data can be combined to calculate standard error and be subjected to further statistical analysis
Table 1. Example of ChIP QPCR data analysis for one experiment.
| Goosecoid Promoter | average C(t) | standard deviation | ΔC(t)1 | ΔΔC(t) uninjected2 | fold change uninjected3 | % of input 4 |
|---|---|---|---|---|---|---|
| Input Uninjected | 29.7189 | 0.1963 | 4.4050 | 0.0000 | 1.0000 | 0.0472 |
| ChIP Uninjected | 34.1239 | 0.1043 | ||||
| Input myc-Fast1 | 30.9308 | 0.2736 | -3.1914 | -7.5964 | 193.5257 | 9.1351 |
| ChIP myc-Fast1 | 27.7393 | 0.1264 | ||||
| Input myc-Fast1 + Xnr1 | 31.0805 | 0.4056 | -3.1726 | -7.5766 | 191.0190 | 9.0168 |
| ChIP myc-Fast1 + Xnr1 | 27.9079 | 0.1602 | ||||
| Ef1α Control | average C(t) | standard deviation | ΔC(t)1 | ΔΔC(t) uninjected2 | fold change uninjected3 | % of input4 |
| Input Uninjected | 29.7538 | 0.2540 | 4.5108 | 0.0000 | 1.0000 | 0.0439 |
| ChIP Uninjected | 34.2646 | 0.4304 | ||||
| Input myc-Fast1 | 31.0208 | 0.1089 | 2.9121 | -1.5987 | 3.0287 | 0.1329 |
| ChIP myc-Fast1 | 33.9330 | 0.3681 | ||||
| Input myc-Fast1 + Xnr1 | 30.6189 | 0.2831 | 3.4157 | -1.0951 | 2.1363 | 0.0937 |
| ChIP myc-Fast1 + Xnr1 | 34.0346 | 0.1988 |
ΔC(t) = ChIP C(t) − Input C(t) (Taneyhill and Adams, 2008)
ΔΔC(t) = ΔC(t) − ΔC(t) uninjected (Taneyhill and Adams, 2008)
Fold Change = 2 [-ΔΔC(t)] (Taneyhill and Adams, 2008)
% of Input = 2 [-ΔC(t)] × (% I) (Frank et al., 2001)
Cloning of additional Xnr6a genomic sequence
Additional genomic sequence for the Xnr6a locus was obtained by screening a Xenopus laevis genomic library (kind gift of Dr. Steven Klein) with a probe corresponding to the 5′ end of the available Xnr6 sequence (Genbank AY050648, bases 292-712). A 15kb clone containing the Xnr6a genomic locus was isolated, sequenced, and deposited in Genbank (accession number: FJ468558).
Western Blotting
Following elution of the immunoprecipitates (Day 2, step 9) a portion of the eluted material can be analyzed by western blot to determine IP efficiency. Western blotting is performed according to standard protocols, except that samples are mixed 1:1 with standard 2× Laemmli Sample Buffer (4% SDS, 20% Glycerol, 0.125M Tris-HCl pH 6.8, 0.004% Bromophenol Blue, 0.2M Dithiothreitol) and heated to 95°C for 30 minutes to reverse the crosslinks. A portion of the input material can also be included as a control.
Antibodies
The rabbit anti-β-catenin antiserum was raised against the 145 N-terminal amino acids of Xenopus β-catenin (Cocalico Laboratories; Reamstown, PA). Anti acetylated H3 (di-acetylated H3K9/K14, (06-599)) and polyclonal anti-myc (06-549) were purchased from Millipore. Purified normal rabbit IgG was purchased from Pierce (31235).
Acknowledgments
We would like to acknowledge Robert Orford and Richard Meehan for helpful discussions while we developed this protocol and the numerous investigators with whom we shared this protocol prior to publication for encouragement, helpful comments and criticism. We also thank Dr. S. Mahmoud A. Najafi for his help in the initial generation of the anti-β-catenin antiserum. This work was supported by National Institutes of Health grants T32-GM007229 (SAB), T32-HD007516 (SAB and CDR), R01-GM64768 (DSK), and R01-GM76621 (PSK).
Grant information: National Institutes of Health; R01-GM76621 (PSK) and R01-GM64768 (DSK)
References
- Acevedo LG, Iniguez AL, Holster HL, Zhang X, Green R, Farnham PJ. Genome-scale ChIP-chip analysis using 10,000 human cells. BioTechniques. 2007:791–797. doi: 10.2144/000112625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl JA, Collas P. A rapid micro chromatin immunoprecipitation assay (microChIP) Nat Protoc. 2008:1032–1045. doi: 10.1038/nprot.2008.68. [DOI] [PubMed] [Google Scholar]
- Frank SR, Schroeder M, Fernandez P, Taubert S, Amati B. Binding of c-Myc to chromatin mediates mitogen-induced acetylation of histone H4 and gene activation. Genes & Development. 2001:2069–2082. doi: 10.1101/gad.906601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DM, Sessions SK. Amphibian cytogenetics and evolution. xv. San Diego: Academic Press; 1991. p. 456. [Google Scholar]
- Heasman J, Kofron M, Wylie C. Beta-catenin signaling activity dissected in the early Xenopus embryo: a novel antisense approach. Dev Biol. 2000:124–134. doi: 10.1006/dbio.2000.9720. [DOI] [PubMed] [Google Scholar]
- Jallow Z, Jacobi UG, Weeks DL, Dawid IB, Veenstra GJ. Specialized and redundant roles of TBP and a vertebrate-specific TBP paralog in embryonic gene regulation in Xenopus. Proc Natl Acad Sci USA. 2004:13525–13530. doi: 10.1073/pnas.0405536101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SW, Park JI, Spring CM, Sater AK, Ji H, Otchere AA, Daniel JM, McCrea PD. Non-canonical Wnt signals are modulated by the Kaiso transcriptional repressor and p120-catenin. Nat Cell Biol. 2004:1212–1220. doi: 10.1038/ncb1191. [DOI] [PubMed] [Google Scholar]
- Kuo MH, Allis CD. In vivo cross-linking and immunoprecipitation for studying dynamic Protein:DNA associations in a chromatin environment. Methods. 1999:425–433. doi: 10.1006/meth.1999.0879. [DOI] [PubMed] [Google Scholar]
- Luo RX, Postigo AA, Dean DC. Rb interacts with histone deacetylase to repress transcription. Cell. 1998:463–473. doi: 10.1016/s0092-8674(00)80940-x. [DOI] [PubMed] [Google Scholar]
- Messenger NJ, Kabitschke C, Andrews R, Grimmer D, Núnez Miguel R, Blundell TL, Smith JC, Wardle FC. Functional specificity of the Xenopus T-domain protein Brachyury is conferred by its ability to interact with Smad1. Developmental Cell. 2005:599–610. doi: 10.1016/j.devcel.2005.03.001. [DOI] [PubMed] [Google Scholar]
- Molenaar M, van de Wetering M, Oosterwegel M, Peterson-Maduro J, Godsave S, Korinek V, Roose J, Destrée O, Clevers H. XTcf-3 transcription factor mediates beta-catenin-induced axis formation in Xenopus embryos. Cell. 1996;86:391–399. doi: 10.1016/s0092-8674(00)80112-9. [DOI] [PubMed] [Google Scholar]
- Morgan MJ, Woltering JM, In der Rieden PM, Durston AJ, Thiery JP. YY1 regulates the neural crest-associated slug gene in Xenopus laevis. J Biol Chem. 2004;279:46826–46834. doi: 10.1074/jbc.M406140200. [DOI] [PubMed] [Google Scholar]
- Ng R, Gurdon J. Epigenetic memory of an active gene state depends on histone H3.3 incorporation into chromatin in the absence of transcription. Nat Cell Biol. 2008:102–109. doi: 10.1038/ncb1674. [DOI] [PubMed] [Google Scholar]
- O'Neill LP, Turner BM. Immunoprecipitation of native chromatin: NChIP. Methods. 2003:76–82. doi: 10.1016/s1046-2023(03)00090-2. [DOI] [PubMed] [Google Scholar]
- O'Neill LP, VerMilyea MD, Turner BM. Epigenetic characterization of the early embryo with a chromatin immunoprecipitation protocol applicable to small cell populations. Nat Genet. 2006:835–841. doi: 10.1038/ng1820. [DOI] [PubMed] [Google Scholar]
- Orlando V, Strutt H, Paro R. Analysis of chromatin structure by in vivo formaldehyde cross-linking. Methods. 1997:205–214. doi: 10.1006/meth.1996.0407. [DOI] [PubMed] [Google Scholar]
- Park JI, Kim SW, Lyons JP, Ji H, Nguyen TT, Cho K, Barton MC, Deroo T, Vleminckx K, Moon RT, McCrea PD. Kaiso/p120-catenin and TCF/beta-catenin complexes coordinately regulate canonical Wnt gene targets. Developmental Cell. 2005:843–854. doi: 10.1016/j.devcel.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Rex M, Hilton E, Old R. Multiple interactions between maternally-activated signalling pathways control Xenopus nodal-related genes. Int J Dev Biol. 2002:217–226. [PubMed] [Google Scholar]
- Roh TY, Cuddapah S, Zhao K. Active chromatin domains are defined by acetylation islands revealed by genome-wide mapping. Genes & Development. 2005:542–552. doi: 10.1101/gad.1272505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Russell DW. Molecular cloning : a laboratory manual. Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- Shen MM. Nodal signaling: developmental roles and regulation. Development. 2007:1023–1034. doi: 10.1242/dev.000166. [DOI] [PubMed] [Google Scholar]
- Sive HL, Grainger RM, Harland RM. Early development of Xenopus laevis : a laboratory manual. ix. Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press; 2000. p. 338. [Google Scholar]
- Takahashi S, Yokota C, Takano K, Tanegashima K, Onuma Y, Goto J, Asashima M. Two novel nodal-related genes initiate early inductive events in Xenopus Nieuwkoop center. Development. 2000:5319–5329. doi: 10.1242/dev.127.24.5319. [DOI] [PubMed] [Google Scholar]
- Taneyhill LA, Adams MS. Investigating regulatory factors and their DNA binding affinities through real time quantitative PCR (RT-QPCR) and chromatin immunoprecipitation (ChIP) assays. Methods Cell Biol. 2008;87:367–389. doi: 10.1016/S0091-679X(08)00219-7. [DOI] [PubMed] [Google Scholar]
- Watabe T, Kim S, Candia A, Rothbächer U, Hashimoto C, Inoue K, Cho KW. Molecular mechanisms of Spemann's organizer formation: conserved growth factor synergy between Xenopus and mouse. Genes & Development. 1995:3038–3050. doi: 10.1101/gad.9.24.3038. [DOI] [PubMed] [Google Scholar]
- Wickbom T. Cytological Studies on Dipnoi, Urodela, Anura, and Emys. Hereditas. 1945;31:241–345. doi: 10.1111/j.1601-5223.1945.tb02756.x. [DOI] [PubMed] [Google Scholar]
- Xanthos JB, Kofron M, Tao Q, Schaible K, Wylie C, Heasman J. The roles of three signaling pathways in the formation and function of the Spemann Organizer. Development. 2002:4027–4043. doi: 10.1242/dev.129.17.4027. [DOI] [PubMed] [Google Scholar]
- Yang J, Tan C, Darken RS, Wilson PA, Klein PS. Beta-catenin/Tcf-regulated transcription prior to the midblastula transition. Development. 2002:5743–5752. doi: 10.1242/dev.00150. [DOI] [PubMed] [Google Scholar]
- Zeng PY, Vakoc CR, Chen ZC, Blobel GA, Berger SL. In vivo dual cross-linking for identification of indirect DNA-associated proteins by chromatin immunoprecipitation. BioTechniques. 2006:694, 696, 698. doi: 10.2144/000112297. [DOI] [PubMed] [Google Scholar]