

Abstract
Protein kinase B (PKB or Akt) is an important component of intracellular signaling pathways regulating growth and survival. Signaling through PKB is frequently deregulated in cancer, and inhibitors of PKB therefore have potential as antitumor agents. The optimization of lipophilic substitution within a series of 4-benzyl-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-4-amines provided ATP-competitive, nanomolar inhibitors with up to 150-fold selectivity for inhibition of PKB over the closely related kinase PKA. Although active in cellular assays, compounds containing 4-amino-4-benzylpiperidines underwent metabolism in vivo, leading to rapid clearance and low oral bioavailability. Variation of the linker group between the piperidine and the lipophilic substituent identified 4-amino-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamides as potent and orally bioavailable inhibitors of PKB. Representative compounds modulated biomarkers of signaling through PKB in vivo and strongly inhibited the growth of human tumor xenografts in nude mice at well-tolerated doses.
Introduction
The serine/threonine protein kinase B (PKB,a also known as Akt) plays an important role in signaling within cells, promoting both cell proliferation and survival.(1) PKB is a key downstream component in the phosphatidylinositol-3 kinase (PI3K) signaling pathway.(2) The binding of extracellular growth factors to tyrosine receptor kinases at the cell surface leads to activation of PI3K, which in turn produces phosphatidylinositol-3,4,5 triphosphate (PI(3,4,5)P3) anchored to the inner side of the plasma membrane. Binding of PKB to PI-3,4,5-P3 through the pleckstrin homology (PH) domain of the enzyme promotes activation of the kinase by phosphorylation on Ser473 and Thr308.3,4 Activated PKB signals through phosphorylation of several enzyme or transcription factor substrates, including GSK3β, FKHRL1, BAD, and mTOR, to promote proliferation, protein translation, progression through the cell cycle, and antiapoptotic survival.1,2
Unregulated signaling in the PI3K−PKB−mTOR pathway is a common molecular pathology in many human cancers.(5) PKB itself is overexpressed or activated in several cancers, such as prostate, breast, and ovarian carcinomas, and PKB is therefore an attractive target for cancer therapy.6−10 Efforts in targeting PKB have increased in recent years, and a number of inhibitor chemotypes with well-defined interaction to the protein have been described in the literature.7−10 These cover a range of mechanisms from ATP- or substrate-competitive inhibition through to allosteric modulation of kinase activity. Several classes of ATP-competitive small molecule inhibitors of PKB have been described, including pyridines,(11) azepanes,(12) indazole-4,7-diones,(13) isoquinoline-5-sulfonamides,(14) 6-phenylpurines,(15) 4-phenylpyrazoles,(16) pyrrolo[2,3-d]pyrimidines,17,18 thiophenecarboxamides,(19) and aminofurazans.(20) However, only a limited number of chemotypes have been reported to have entered early phase clinical trials, including the aminofurazan 1 (GSK690693)(21) (Figure 1). A challenge in the development of selective ATP-competitive inhibitors of PKB has been the extensive conservation of the ATP binding sites of the AGC kinase family.(22) An alternative approach to achieve highly selective inhibition of PKB has been developed using ATP noncompetitive inhibitors that target an allosteric site between the kinase and PH-domains of the enzyme.7,8,23,24 An allosteric PKB inhibitor is in clinical development.(25)
Figure 1.

Structures of the ATP-competitive inhibitors 1 and 2.
Our laboratory has previously reported the development of a hit from fragment screening15,16 into 4-(4-chlorobenzyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-4-amine 2 (CCT128930) (Figure 1), a potent ATP-competitive inhibitor of PKBβ. Crucially, 2 also showed inhibition of relevant molecular biomarkers in the PI3K−PKB−mTOR pathway in cells.(17) This compound was 28-fold selective for PKB compared to the structurally homologous kinase PKA and showed good overall selectivity for PKB and other AGC kinases in a wider kinome profile. Although the selectivity and cellular potency of 2 were sufficient to merit investigation of its in vivo profile, the compound had high clearance in vivo and low oral bioavailability. In this article, we describe modifications to 2 leading initially to compounds with higher selectivity for PKB and ultimately to the identification of 4-amino-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamides as selective and orally bioavailable inhibitors of PKB with in vivo antitumor activity.
Results and Discussion
The design of ATP-competitive inhibitors selective for PKB against PKA is challenging because these enzymes are very closely related with high sequence homology in the ATP-binding site (∼80%).(22) X-ray crystallographic analysis of the modes of binding of 2 in PKA and a PKA-PKB chimeric protein representative of PKB(26) suggested that 2 exhibited productive binding of the chlorobenzyl group within a lipophilic pocket formed by P-loop residues in PKB.(17) However, in PKA, the presence of a single amino acid difference (Met282 PKB, Leu173 PKA) in the ribose binding site resulted in a change of conformation of the bound ligand, directing the lipophilic 4-chlorobenzyl group into a less favorable, solvent exposed region. On the basis of this explanation for the observed selectivity of 2, we attempted the synthesis of a wider range of substituted analogues to investigate if higher selectivity could be obtained (Table 1).
Table 1. Inhibition of PKBβ and PKA by Substituted 4-Benzyl-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-4-amines.

| no. | Rn | PKBβ IC50 (nM)a |
PKA IC50 (nM)b |
selectivity for PKB against PKA (fold)c |
|---|---|---|---|---|
| 2 | 4-Cl | 6.0 (±1.5)d | 168 (±36)d | 28 |
| 3 | 3-Cl | 46 | 280 | 6 |
| 4 | 2-Cl | 44 (±35)e | 2150 (1700, 2600)f | 49 |
| 5 | 4-OCF3 | 35 (19, 50)f | 660 | 19 |
| 6 | 3-OCF3 | 19 | 600 | 32 |
| 7 | 2-OCF3 | 84 | 2800 | 33 |
| 8 | 3-OMe | 15 | 720 | 48 |
| 9 | 2-OMe | >1000 | ndg | ndg |
| 10 | 4-tBu | 27 (±18)h | 3400 (3200, 3600)f | 126 |
| 11 | 2,3-Cl2 | 35 | 520 | 15 |
| 12 | 2,4-Cl2 | 8.5 (7, 10)f | 1300 (1300, 1300)f | 153 |
| 13 | 2,5-Cl2 | 16 | 770 | 48 |
| 14 | 2,6-Cl2 | 20 | 3300 | 165 |
| 15 | 3,4-Cl2 | 18 (9, 26)f | >300 | >16 |
| 16 | 2-Cl, 4-F | 90 | 1200 | 13 |
| 17 | 25 | ndg | ndg | |
| 18 | 7.0 | 490 | 70 |
Inhibition of PKBβ kinase activity in a radiometric filter binding assay,(17) single determination. Standard inhibitor N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinoline sulfonamide (H-89) gave mean IC50 (±SD) = 590 (±220) nM (n = 20).
Inhibition of PKA kinase activity in a radiometric filter binding assay,(16) single determination. Standard inhibitor N-(2-methylaminoethyl)-5-isoquinoline sulfonamide (H-8) gave mean IC50 (±SD) = 5300 (±2010) nM (n = 14).
Ratio of IC50 values (PKA/PKB).
Mean (±SD), n = 3 determinations.
Mean (±SD), n = 4 determinations.
Mean of two determinations, individual values in parentheses.
nd = not determined.
Mean (±SD), n = 5 determinations.
Variation of the substituents on the benzyl group of 2 in general lead to somewhat reduced affinity for PKB. Exceptions were the 2,4-dichlorobenzyl and 2-napthyl analogues 12 and 18, respectively, which inhibited PKB with similar potencies to 2. An interesting influence of the substituents on the selectivity of the compounds for PKB versus PKA was seen. While translocation of the 4-chloro group of 2 to the 3-position (3) reduced both affinity and selectivity, approximately 40-fold selectivity was recovered in the 2-chlorobenzyl analogue 4. Replacement with more electron-rich 2-, 3-, or 4-substituents (5−8) gave compounds with selectivities in a similar range (ca. 20−48-fold), although the 2-methoxy analogue 9 was surprisingly less potent at PKB. Gratifyingly, combination of the 2- and 4-chloro substituents in the analogue 12 increased the selectivity to ca. 150-fold while retaining nanomolar potency at PKB. The 2,6-dichloro substitution pattern 14 gave similarly high selectivity for PKB, although this was not seen with other dihalobenzyl analogues 13, 15, and 16. Introduction of a larger, lipophilic 4-tert-butyl substituent 10 also gave a high selectivity for PKB (ca. 126-fold). An intermediate level of selectivity (ca. 70-fold) was seen for the 2-napthyl derivative 18.
Where the selectivity of PKB over PKA was increased for the compounds in Table 1, this was due to reduced inhibitory activity against PKA rather than an increase in affinity for PKB and was associated with increased lipophilicity of the benzyl group. This structure−activity relationship was broadly consistent with the rationale proposed from the comparison of 2 bound to PKA and PKA-PKB chimera, in which the benzyl substituent interacts poorly with PKA relative to PKB, and is directed toward solvent. The ability to bind to PKB was minimally compromised for the analogues with larger substituents. The X-ray crystal structure of the PKB-selective analogue 10 bound to PKBβ was determined and showed a very similar binding mode to that of 2(17) (Figure 2). The tert-butyl substituent occupied the lipophilic pocket formed by the P-loop of PKB, with the 4-amino substituent interacting with Glu236 and the backbone carbonyl of Glu279 in the ribose pocket.
Figure 2.

Crystal structures of (A) 10 bound to PKBβ and (B) 21 bound to PKBβ. (C) Overlay of the bound conformations of 2 (gold), 10 (green), and 21 (blue).
As an alternative to substituent variation in the 4-amino-4-benzylpiperidine series, we also investigated compounds with varied chain length between the 4-aminopiperidine and 4-chlorophenyl groups (Table 2). The ether 19 was as potent as 2 against PKB but had no selectivity against PKA, which we speculated was due to the more flexible linker group. While the amide 20 had reduced affinity for PKB, the isomeric amide 21 retained activity for PKB and showed some selectivity (ca. 14-fold) over PKA.
Table 2. Development of 4-Amino-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide PKB Inhibitors.

| no. | R | PKBβ IC50 (nM)a |
PKA IC50 (nM)a |
selectivity for PKB against PKA (fold)a |
|---|---|---|---|---|
| 19 | 8.0 | 13 | 1.6 | |
| 20 | 69 | ndb | ndb | |
| 21 | 4-Cl | 2.2 (±1.2)c | 30 | 14 |
| 22 | 3-Cl | 7.0 | 82 | 12 |
| 23 | 2-Cl | 36 | 64 | 1.8 |
| 24 | 4-F | 14 | 94 | 6.7 |
| 25 | 4-CF3 | 11 | 110 | 10 |
| 26 | 4-CF3O | 29 | ndb | ndb |
| 27 | 4-tBu | 210 | ndb | ndb |
| 28 | 2,4-Cl2 | 4.9 | 120 | 24 |
| 29 | 3,4-Cl2 | 5.7 | 45 | 7.9 |
| 30 | 2,4-F2 | 11 | 79 | 7.2 |
| 31 | 2-Cl, 4-F | 8.0 | 44% @ 30 nM | ndb |
| 32 | 2-F, 4-Cl | 4.0 | 60% @ 10 nM | ndb |
| 33 | 25 | 84 | 3.4 | |
| 34 | 250 | ndb | ndb | |
| 35 | 680 | ndb | ndb |
Inhibition of PKB β kinase activity in a radiometric filter binding assay,(17) single determination. Standard inhibitor N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide (H-89) gave mean IC50 (±SD) = 590 (±220)nM (n = 20).
nd = not determined.
Mean (±SD), n = 3 determinations.
A set of analogues of the amide 21 were investigated using substituent patterns corresponding to those studied for the 4-amino-4-benzylpiperidines (Table 2). Most compounds were potent against PKB, but selectivity was generally decreased against PKA when compared with the 4-benzylpiperidines shown in Table 1. Variation of the position of the chlorine atom in the aromatic ring showed that 4-substitution as in 21 was optimal. Other 4-substituents (24−27) showed a decrease in PKB inhibitory activity with increasing size, and the 4-tert-butyl analogue 27 in particular was less active than the rest of the analogues in this set. This contrasted with the structure−activity relationship seen for the 4-benzylpiperidines, and we ascribed these differences to the presence of the longer and relatively inflexible amide spacer which could result in larger 4-substituents being unable to interact as favorably with PKB. As with the 4-benzylpiperidines, the 2,4-dichlorobenzyl amide 28 gave improved selectivity (ca. 24-fold) for PKB over PKA. Other less lipophilic 2,4-dihalobenzyl amides (30−32) retained activity at PKB but with reduced selectivity. In some cases, increases in PKA activity for the benzyl amides were seen relative to nonamide comparators. Although constrained by the amide, the longer linker will allow the lipophilic substituent to attain a different range of conformations compared to the simple 4-benzylpiperidines (Figure 2), resulting in the recovery of productive contacts to the P-loop of PKA.
Methylation of the amide NH of 21 to give compound 33, and the conformationally constrained tertiary amides 34 and 35, led to loss of potency against PKB. The crystal structure of 21 bound to PKBβ showed the inhibitor bound in very similar fashion to 2 and 10, with the 4-amino group forming interactions with Glu236 and the backbone carbonyl of Glu279, while the 4-chlorophenyl ring was located in the P-loop lipophilic pocket (Figure 2B). As observed for 2 and 10, the inhibitor’s basic amino group formed a favorable close contact with the sulfur of Met282 (ca. 3.5 Å), an interaction which is lost in PKA. It is possible that the proximity of the electron-rich sulfur residue compensates for loss of hydration of the protonated amine on binding.(17) A possible additional interaction was also observed to the amide spacer of 21 with close approach (ca. 3.3 Å) of the amide NH in the inhibitor and the side chain of Asp293. The 10-fold drop in activity for the N-methyl amide 33 relative to 21 may reflect the disruption of this conformation in that complex.
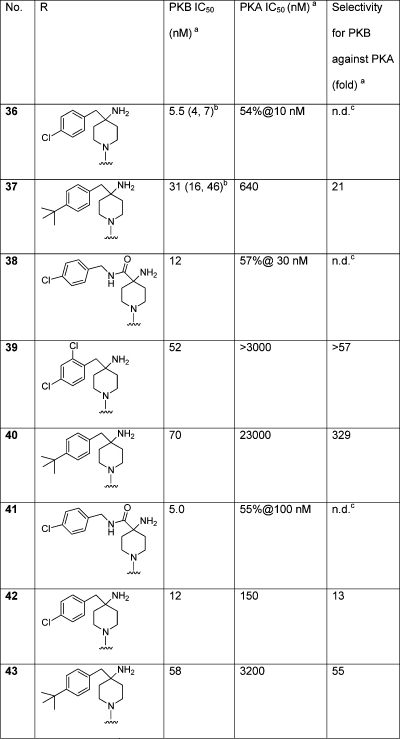
The effect of substituting the pyrrolo[2,3-d]pyrimidine bicycle by 7-azaindole, oxopurine, and pyrazolo[3,4-b]pyridine was investigated for the most potent and selective piperidine moieties (Table 3). The bicyclic heteroaromatic groups form hydrogen bonds to a part of the kinase domain, known as the hinge region, that links the distinct N- and C-terminal lobes. 7-Azaindole was the original hinge-binding fragment from which this compound series was derived.15,17 The carbonyl functionality of 8-oxopurine was expected to make additional interactions with PKB, particularly the residue Thr213 at the entrance to the hydrophobic pocket of the kinase which differs between PKB and PKA. For a similar reason, the pyrazolo[3,4-b]pyridine bicycle was selected to provide an additional polar atom in the ligand in this region.
Table 3. 4-Azaindole, 8-Oxopurine, and Pyrazolo[3,4-b]pyridine Hinge Binders Substituted with Selected 4-Aminopiperidines.

 |
Inhibition of PKBβ kinase activity in a radiometric filter binding assay,(17) single determination. Standard inhibitor N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinoline sulfonamide (H-89) gave mean IC50 (±SD) = 590 (±220) nM (n = 20).
Mean of two determinations, individual values in parentheses.
nd = not determined.
The azaindole 36, the direct analogue of 2, showed similar potency but no selectivity for PKB over PKA. The 4-amidopiperidine containing azaindole 38 was also unselective. Introduction of the 4-tert-butyl substituent to give 37 increased the selectivity, mirroring the structure−selectivity relationship seen with the pyrrolo[2,3-d]pyrimidines 2 and 27, but only to ca. 20-fold. The 7-azaindoles were therefore associated with generally lower selectivity for PKB over PKA than the pyrrolo[2,3-d]pyrimidines. We believe this reduction in selectivity arises from the replacement of a nitrogen (N-3) in the pyrrolo[2,3-d]pyrimidines by a carbon in the azaindoles. This changes the preferred conformation and orientation of the piperidine ring relative to the bicycle and thus the vectors of the basic amine and lipophilic substituents. Because selectivity in this series arises from efficiently exploiting a single amino acid difference (Leu/Met) between PKA and PKB, selectivity is particularly sensitive to the positioning of the amine group relative to this residue. The 8-oxopurines 39−41 offered similar or improved selectivity compared to their pyrrolo[2,3-d]pyrimidine congeners but with somewhat lower potency at PKB. On the basis of the binding modes of purine inhibitors in this series,(17) the 8-oxopurine carbonyl group is positioned to accept a hydrogen bond from the side-chain of Thr213. Because the equivalent residue is valine in PKA, this would be expected to contribute to selectivity for PKB. Targeting this difference in the ATP binding sites of PKB and PKA has been noted to increase the selectivity of other inhibitor chemotypes.(11g) The pyrazolo[3,4-b]pyridine hinge-binding group provided ligands 42 and 43 with intermediate potency and selectivity between the azaindole and pyrrolo[2,3-d]pyrimidine analogues.
The inhibitory profiles of the PKB-selective compounds 2, 10, and 21 were investigated in a wider panel of 22 kinases(27) (Figure 3). As previously reported,(17)2 mainly showed activity (>80% at 1uM) against PKB, PKA, and two other enzymes (ROCK2 and p70S6K), all from the AGC kinase subfamily. Some activity (40−80% @ 1 μM) was seen against four other kinases in the panel. The tert-butyl analogue 10 was remarkably selective, showing significant activity against only two kinases, namely PKB and p70S6K, in this panel. The amide 21 had a similar profile to 2, showing some activity (>80% at 1uM) against the same enzymes (PKB, PKA, ROCK2, and p70S6K). Lower levels of activity were seen for some other AGC kinases but not for kinases outside this subfamily. The pattern of selectivity was reflected in the Gini coefficients for the compounds,(28) which takes into account the total levels of inhibitory activity across the panel, with compound 10 showing a higher overall selectivity (Figure 3).
Figure 3.

Selectivity profiles and Gini coefficients(28) (G) of 2 (A), 10 (B), and 21 (C) against a panel of 22 human kinases, measured at 1 μM concentration of the test compound. Kinase dendrogram(29) reproduced courtesy of Cell SignalingTechnology, Inc. (www.cellsignal.com).
The antiproliferative activity of selected inhibitors was assessed in the PC3 M human prostate cancer and U87MG glioblastoma human cancer cell lines, which are known to have PTEN deletion and an activated PI3K-PKB pathway30,31 (Table 4). A specific readout of targeted PKB inhibition in cells was also obtained by quantifying inhibition of phosphorylation of the downstream substrate GSK3β by cell ELISA.(32) The majority of the compounds were active in the antiproliferative assays, although this assay may sometimes include contributions from off-target activities. The more selective pyrrolo[2,3-d]pyrimidines 10 and 12 showed similar potencies in the cellular assays to the lead compound 2. The potent but unselective ether-linked analogue 19 was also active, but a fall in antiproliferative activity was observed for the amides 21 and 28 compared to their nonamide counterparts. N-Methylation of the amide in 33 to reduce polarity and increase cell permeability did not substantially improve the cellular activity relative to 21, but the activity of 33 may be compromised by the 3-fold drop in affinity for PKB (Table 2). The 7-azaindole analogues 36 and 37 were similar to their pyrrolo[2,3-d]pyrimidine counterparts 2 and 10, while the 7-azaindole 38 was less active in cells than its comparator 21. The 8-oxopurine 40 retained similar cellular activity to the analogous pyrrolo[2,3-d]pyrimidine 10. The cellular activity of these compounds, which are highly selective for PKB over PKA, and over other kinases in the case of 10, argues for the antiproliferative effects of the inhibitors being primarily driven by inhibition of PKB. The 8-oxopurine amide analogue 41 had no activity in the cellular assays despite good PKB affinity. It is likely that low cell penetration is encountered for this scaffold, the most polar of those prepared, an effect encountered earlier in the evolution of this series.(17) Better cellular activity was seen for the pyrazolo[3,4-b]pyridines 42 and 43, but these compounds offered no advantage over the activity of 2, 10, and 21.
Table 4. Cellular Activities of Selected Compounds in PC3M (Prostate) and U87MG (Glioblastoma) Human Tumour Cell Linesa.
| PC3M |
U87MG |
|||
|---|---|---|---|---|
| no. | SRB GI50 (μM)b | ELISA IC50 (μM)c | SRB GI50 (μM)d | ELISA IC50 (μM)e |
| 2 | 12 | 3.0 | 5.0 | 0.66 |
| 10 | 8.9 (7.7, 10)f | 4.0 | 5.7 | ndg |
| 12 | 8.5 (8.0, 8.9)f | 1.9 | 5.9 | ndg |
| 14 | 20 | 6.7 | 14 | ndg |
| 19 | ndg | ndg | 5.1 | 0.59 |
| 21 | >50h | 2.3 (2.3, 2.3)f | 17 | 0.93 |
| 28 | 26 | 3.8 | 17 | ndg |
| 32 | ndg | ndg | 20 | 2.8 |
| 33 | 29 | 4.2 | 34 | ndg |
| 36 | 9.3 | 0.37 (0.36, 0.37)f | 4.6 | ndg |
| 37 | ndg | ndg | 3.7 | 3.2 |
| 38 | ndg | ndg | 19 | 7.2 |
| 40 | ndg | ndg | 13 | 8.8 |
| 41 | ndg | ndg | >50 | >50 |
| 42 | ndg | ndg | 16 | 2.7 |
| 43 | ndg | ndg | 7.0 | 6.5 |
Inhibition of PKBβ kinase activity in a radiometric filter binding assay,(17) single determination. Standard inhibitor N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinoline sulfonamide (H-89) gave mean IC50 (±SD) = 590 (±220) nM (n = 20).
Cell growth inhibition by sulforhodamine B colorimetric assay,(33) single determination in PC3 M human prostate cancer cells. Standard inhibitor H-89 gave mean (±SD) IC50 = 18 (±6.0) μM in this assay.
Cellular ELISA for inhibition of GSK3β phosphorylation in PC3 M cells,(32) Standard inhibitor H-89 gave mean (±SD) IC50 = 15 (±2.0) μM.
Cell growth inhibition by sulforhodamine B colorimetric assay,(32) single determination in U87MG human glioblastoma cancer cells. Standard inhibitor H-89 gave mean (±SD) IC50 = 15 (±2.3) μM in this assay.
Cellular ELISA for inhibition of GSK3β phosphorylation in U87MG cells.(32) Standard inhibitor 2-(4-morpholino)-8-phenyl-4H-1-benzopyran-4-one (LY294002) gave IC50 = 8.1 (±3.0) μM.
Mean of n = 2 determinations, individual values in parentheses.
nd not determined.
n = 2 determinations.
The inhibitory effect of compounds 2, 10, and 21 toward five human cytochrome P450 isoforms (1A2, 2D6, 3A4, 2C9, and 2C19) was assessed in microsomal preparations.(34) In general, no significant inhibition was observed (IC50 > 10 μM) for most of the isoforms tested. Compound 2 showed inhibition of the 2D6 isoform (IC50 = 0.66 μM), but this was not observed for compounds 10 and 21. Rather, these examples showed moderate inhibition (IC50 ca. 1 μM) for the 2C9 isoform only.
The pharmacokinetic properties of compounds from this series were investigated in mice, including the selective pyrrolo[2,3-d]pyrimidine inhibitors 2, 10, and 21 (Table 5). The 4-(4-tert-butylbenzyl)-4-aminopiperidine 10 showed a similar profile to that for 2.(17) Following iv dosing at 10 mg/kg, compound 10 was widely distributed but was very quickly cleared from the general circulation with a plasma clearance higher than that of mouse liver blood flow. Although compound 2 had shown low oral bioavailability (Foral = 8.5%), no measurable drug levels were detectable for 10 following oral dosing. Oxidative metabolites of 2 and 10, corresponding to masses of M + 16 and M + 32, were detected by mass spectrometry in plasma samples from these experiments. The fragmentation patterns in the mass spectra of the M + 16 metabolite of 2, when compared to those for the parent drug, suggested that oxidation was occurring at C-2 in the piperidine ring.
Table 5. Pharmacokinetic Parameters of Compounds 2, 10, and 21 in Mice.
| plasma PK parametersa |
2 | 10 | 21 |
|---|---|---|---|
| IV dose (mg kg−1) | 25 | 10 | 5 |
| T1/2 (h) | 0.95 | 0.79 | 0.90 |
| Vss (L) | 0.25 | 0.27 | 0.06 |
| Clp (L h−1) | 0.33 | 0.31 | 0.08 |
| Foral (%)b | 8.5 | <1c | 58 |
Pharmacokinetic parameters were calculated using the program WinNonLin, fitted to model 201 with default settings.
Following oral administration of same dose.
Plasma levels below limit of detection following oral dosing.
A striking difference was observed in the in vivo pharmacokinetic properties of the inhibitors containing the 4-amino-4-amidopiperidine moiety, such as 21, compared to the 4-benzyl-4-aminopiperidines 2 and 10. The plasma clearance of 21 was approximately 3-fold lower than that of 2 and 10, while the volume of distribution was also reduced for the more polar amide scaffold. Importantly, compound 21 showed very good oral bioavailability in mice (Foral = 58%). While lower first pass metabolism and subsequent reduced clearance may contribute to the improved oral bioavailabilty of 21, the difference in basicity between 2 and 21 may also play a part. Calculated pKa values(35) for the protonation of the 4-amino group varied between 8.8 and 9.3 for 2, depending on the methodology, compared to a range of 6.5−7.4 for 21. Thus the 4-amino-4-amidopiperidines would be expected to be significantly less protonated than 2 or 10 in the gut, leading to enhanced passive absorption. The solubilities of 2 and 21 were determined in aqueous buffer at pH 7 and 6.5. Interestingly, the solubility of 2 showed a strong pH dependence, with S = 0.26 mg/mL at pH 6.5 but negligible solubility at pH 7, suggesting a much greater aqueous solubility for the protonated than the unprotonated form. In contrast, the solubilty of 21 was less affected by pH (S = 0.1 mg/mL at pH 7, S = 0.04 mg/mL at pH 6.5). Thus better solubility for the unprotonated form may also contribute to the improved bioavailability of 21.
Earlier reported studies on the efficacy of some indazole-derived PKB inhibitors in human tumor xenograft models had suggested that mechanism-related effects of PKB inhibition could underlie the toxicity observed with these compounds.(12a) We were therefore keen to test selective inhibitors from the novel pyrrolo[2,3-d]pyrimidine series in vivo. The efficacy and pharmacodynamic effects of the orally bioavailable inhibitor 21 and the close analogue 32 were studied in mice bearing established subcutaneous U87MG human glioblastoma xenografts (Figure 4). Doses of 21 up to 200 mg kg−1 (5 days dosing in 7) were well tolerated with no effects on mouse body weight (not shown). Efficacy was measured by comparison of the estimated volume of tumors in treated and control groups during the study (Figure 4) and by comparison of the final tumor weights in the treated and control groups (T/C). Very strong inhibition of tumor growth was seen with T/C = 23%. Additionally, 44% of treated tumors had regressed in volume at the completion of the experiment. In a parallel pharmacokinetic and pharmacodynamic study, high levels of 21 were found in plasma and tumor samples (20 and 43 μM, respectively) at 4 h after a single dose. Clear inhibition of PKB signaling in the tumors was observed using an electrochemiluminescence immunoassay to measure levels of phospho-GSK3β in tumor lysates(32) (Figure 4). Thus despite the somewhat reduced cellular antiproliferative activity for the more polar scaffold of 21 compared to 2, the good tolerability and reduced clearance of 21 enabled oral dosing to achieve drug levels above the concentrations at which mechanism-based and antiproliferative effects were seen in vitro in cells, resulting in inhibition of the target in vivo and reduction of tumor growth. Measurement of tumor pharmacodynamic changes in other kinase-mediated pathways would be required to establish if inhibition of other targets can contribute to the efficacy of the compounds, however the selectivity profile of the compounds argues for a major contribution from PKB inhibition. Similar effects on in vivo biomarkers and reduction in growth (T/C = 32%) of U87MG tumor xenografts were seen following treatment with the closely related compound 32, also dosed orally at 200 mg/kg (Figure 4). Details of the efficacy, pharmacodynamic effects, and tumor pharmacokinetics of 21 (CCT129524) in a broader range of tumor xenograft models will be reported separately.
Figure 4.

Effect of 21 (A) and 32 (C) (both dosed at 200 mg/kg po 5 days per week) on the growth of U87MG human glioblastoma xenografts in CrTac:Ncr-Fox1(nu) athymic mice. Effect of 21 (B) and 32 (D) on PKB phosphorylation of GSK3β in U87MG human tumor xenografts at 4 h (B) or 6 h (D) following a single dose of 200 mg/kg po.
Conclusions
A series of 4-benzyl-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-4-amines provided potent inhibitors of PKBβ. The selectivity for inhibition of PKBβ over the closely related kinase PKA was increased by introducing larger lipophilic substituents to the benzyl group. This strategy exploited the subtly different binding modes for the ligands between the two targets, arising from a single amino acid residue difference within the ATP-binding site of the enzymes. The 4-amino-4-benzylpiperidine scaffold underwent metabolism in vivo, leading to rapid clearance and poor oral bioavailability. This was overcome by modification of the piperidine scaffold to give orally bioavailable 4-amino-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamides, exemplified by the potent and selective PKB inhibitor 21. Compound 21 showed good selectivity for inhibition of PKB over a range of other human kinases, with some activity observed for related AGC kinases. The observation of strong tumor growth inhibition and biomarker modulation in vivo with well tolerated doses of 21 supports the further evaluation of compounds from this series as potential anticancer therapeutics.
Experimental Section
Synthetic Chemistry
Substituted 4-amino-4-benzylpiperidine intermediates were prepared from 4-cyano-4-benzylpiperidines as previously described for 2 using a Curtius rearrangement sequence to install the 4-amino substituent.(17) A more convenient reagent combination for this transformation was found by treating 4-benzyl-4-carbamoylpiperidines with bis(trifluoroacetoxy)iodobenzene,(36) as exemplified for the preparation of 10 (Scheme 1). Alternatively, the reactive tert-butyl sulfinimine formed from N-Boc-piperidin-4-one and tert-butylsulfonamide was reacted in situ with benzylic Grignard reagents to give the 4-amino-4-benzylpiperidine scaffolds directly.(37) Hinge-binding groups were introduced to the piperidines through SNAr reaction of 4-chloro-7H-pyrrolo[2,3-d]pyrimidine, 6-chloro-7H-purin-8(9H)-one, or 4-fluoro-1-(triisopropylsilyl)-1H-pyrrolo[2,3-b]pyridine,(38) which occurred selectively at the more reactive and less hindered secondary nitrogen atom. Additionally, the piperidines were reacted with ethyl 4-chloro-1H-pyrazolo[3,4-b]pyridine-5-carboxylate(39) followed by decarboxylation to give the pyrazolo[3,4-b]pyridine hinge-binder. Through these means the 4-benzyl-4-aminopiperidine analogues 2−18, 36, 37, 39, 40, 42, and 43 were prepared.
Scheme 1.

Reagents and conditions (yields for R = 4-tBu): (i) LDA, 4-tert-butylbenzyl bromide, THF, −78 °C, 79%; (ii) AcOH, H2SO4, reflux, then Boc2O, NaOH, dioxane-H2O, 71%; (iii) PhI(TFA)2, MeCN-H2O, 46%; (iv) tert-butylsulfinamide, Ti(OEt)4 (2 equiv), THF, reflux, then 4-tert-butylbenzylmagnesium bromide (5 equiv), THF, rt, 26%; (v) 4M HCl, dioxane, MeOH, rt, 100%; (vi) 4-chloro-7H-pyrrolo[2,3-d]pyrimidine, Et3N, n-BuOH, 100 °C, 86%; (vii) 4-fluoro-1-(triisopropylsilyl)-1H-pyrrolo[2,3-b]pyridine, Et3N, NMP, 160 °C, 37%; (viii) 6-chloro-7H-purin-8(9H)-one, Et3N, n-BuOH 100 °C, 50%; (ix) ethyl 4-chloro-1H-pyrazolo[3,4-b]pyridine-5-carboxylate, Et3N, n-BuOH, 100 °C, 66%; (x) KOH, H2O, 120 °C, 47%.
To prepare the ether-linked analogue 19, 1-(tert-butoxycarbonyl)-4-(tert-butoxycarbonylamino)piperidine-4-carboxylic acid 47 was reduced to the alcohol 48 with lithium aluminum hydride and O-benzylated to give 49 after double N-deprotection (Scheme 2). The piperidine 49 was reacted with 4-chloro-7H-pyrrolo[2,3-d]pyrimidine to give the test compound 19. Alternatively, formation of the primary amide from 47 and reduction with borane in THF gave the 4-aminomethylpiperidine 50. Acylation with 4-chlorobenzoyl chloride and deprotection produced the amide 51, which was coupled to the pyrrolopyrimidine hinge-binder to give 20. The isomeric amide 21 was prepared from an initial coupling of 4-chlorobenzylamine and 47 to give the amide 52. Deprotection to 53 and introduction of the pyrrolopyrimidine gave 21. Analogues of 21 with different substitution of the amide were prepared by varying the amine in the first step of this sequence. The 4-carbamido-4-aminopiperidine 53 was reacted with 4-fluoro-1-(triisopropylsilyl)-1H-pyrrolo[2,3-b]pyridine(38) and 6-chloro-7H-purin-8(9H)-one to give the analogues 38 and 41, respectively.
Scheme 2.

Reagents and conditions: (i) LiAlH4, THF, 0 °C then rt, 47%; (ii) NaH, 4-chlorobenzyl bromide, DMF, 0 °C then rt, 22%; (iii) 4M HCl, dioxane, MeOH, rt; 100%; (iv) 4-chloro-7H-pyrrolo[2,3-d]pyrimidine, Et3N, n-BuOH, 100 °C, 69−82%; (v) 1-hydroxybenzotriazole, 1-ethyl-3-(3-dimethylamino-propyl)carbodiimide, DMF, NH3 aq, rt, 97%; (vi) BH3, THF, 0 °C then 60 °C, 6%; (vii) 4-chlorobenzoyl chloride, Et3N, CH2Cl2, rt, 35%; (viii) R1R2NH, HATU, iPr2NEt, DMF, rt, (R1R2NH = 4-chlorobenzylamine, 86%).
General Synthetic Chemistry
Reactions were carried out under N2. Organic solutions were dried over MgSO4 or Na2SO4. Starting materials and solvents were purchased from commercial suppliers and were used without further purification. Microwave reactions were carried out using Biotage Initiator 60 or CEM microwave reactors. Flash silica chromatography was performed using Merck silica gel 60 (0.025−0.04 mm). Ion exchange chromatography was performed using Isolute Flash SCX-II (acidic) or Flash NH2 (basic) resin cartridges. 1H NMR spectra were recorded on a Bruker AMX500 instrument at 500 MHz using internal deuterium locks. 13C NMR spectra were recorded on a Bruker AMX500 instrument at 125 MHz. Chemical shifts (δ) are reported relative to TMS (δ = 0) and/or referenced to the solvent in which they were measured. Combined HPLC-MS analyses were recorded using a Waters Alliance 2795 separations module and Waters/Micromass LCT mass detector with electrospray ionization (+ve or −ve ion mode as indicated) and with HPLC performed using Supelco DISCOVERY C18, 50 mm × 4.6 mm or 30 mm × 4.6 mm i.d. columns, at a temperature of 22 °C with gradient elution of 10−90% MeOH/0.1% aqueous formic acid at a flow rate of 1 mL/min and a run time of 3.5 or 10 min as indicated. Compounds were detected at 254 nm using a Waters 2487 dual λ absorbance detector. All tested compounds gave >95% purity as determined by this method. All purified synthetic intermediates gave >95% purity as determined by this method except where indicated in the text. High-resolution mass spectra were measured on an Agilent 6210 ToF HPLC-MS with a Phenomenex Gemini 3 μm C18 (3 cm × 4.6 mm i.d.) column.
General Methods for Preparation of 4-Amino-4-benzylpiperidines
4-(4-tert-Butylbenzyl)piperidin-4-amine (45)
Method A. n-BuLi (2.32 M in hexanes, 11.8 mL, 27.4 mmol) was added to a solution of iPr2NH (3.8 mL, 27.4 mmol) in THF (75 mL) at −78 °C under N2. After 10 min, a solution of tert-butyl 4-cyanopiperidine-1-carboxylate (5.00 g, 23.8 mmol) in THF (30 mL) was added. The cloudy solution was stirred for 1 h at −78 °C. 1-(Bromomethyl)-4-tert-butylbenzene (5.2 mL, 28.5 mmol) was added and the clear yellow−brown solution was warmed to rt and stirred for 15 h. Water (500 mL) was added, and the mixture was extracted with Et2O (2 × 250 mL). The organic layers were combined, washed with brine (200 mL), dried, and concentrated. Recrystallization from Et2O−hexane gave tert-butyl 4-(4-tert-butylbenzyl)-4-cyanopiperidine-1-carboxylate (6.69 g, 79%). LC-MS (3.5 min) m/z 379 [M + Na+], Rt = 2.96 min. 1H NMR (CDCl3) δ 1.33 (s, 9H), 1.47 (s, 9H), 1.48−1.52 (m, 2H), 2.85 (s, 2H), 2.95−3.04 (m, 2H), 4.08−4.16 (m, 2H), 7.20−7.22 (m, 2H), 7.36−7.38 (m, 2H). 13C NMR (CDCl3) δ 28.4, 31.3, 34.5, 34.7, 39.2, 41.0, 45.4, 80.0, 122.0, 125.4, 130.0, 131.2, 150.5, 154.5 ppm. A solution of tert-butyl 4-(4-tert-butylbenzyl)-4-cyanopiperidine-1-carboxylate (5.40 g, 15.2 mmol) in AcOH (14 mL) and conc H2SO4 (7 mL) was heated at 50 °C for 3 h and then at 90 °C for 2 h. The mixture was cooled and cautiously basified to pH 14 by the addition of 2 M NaOH aq (350 mL). Boc2O (4.96 g, 22.7 mmol) in dioxane (100 mL) was added, and the mixture was stirred for 24 h. The mixture was extracted with EtOAc (3 × 100 mL). The extracts were dried and concentrated. Flash column chromatography, eluting with EtOAc, gave tert-butyl 4-(4-tert-butylbenzyl)-4-carbamoylpiperidine-1-carboxylate 44 (4.02 g, 71%). LC-MS (3.5 min) m/z 397 [M + Na+], Rt = 2.86 min. 1H NMR (CDCl3) δ 1.31 (s, 9H), 1.46 (s, 9H), 1.49−1.56 (m, 2H), 2.81 (s, 2H), 3.00−3.07 (m, 2H), 3.82−3.90 (m, 2H), 7.07−7.08 (m, 2H), 7.29−7.30 (m, 2H). 13C NMR (CDCl3) δ 28.4, 31.3, 33.5, 34.4, 41.0 (br), 45.6, 46.7, 79.6, 125.1, 129.9, 133.1, 149.7, 154.9, 176.9 ppm.
Bis(trifluoroacetoxy)iodobenzene (5.05 g, 11.8 mmol) was added to a suspension of 44 (4.00 g, 10.7 mmol) in MeCN (17 mL) and water (17 mL). After 17 h, water (70 mL) and conc HCl (15 mL) were added. After a further 1 h, the mixture was washed with Et2O (100 mL) and the aqueous layer was concentrated to give 4-(4-tert-butylbenzyl)piperidin-4-amine bis hydrochloride 45 (1.57 g, 46%). LC-MS (10 min) m/z 247 [M + H+], Rt = 1.25 min. 1H NMR (CD3OD) δ 1.33 (s, 9H), 2.14−2.17 (m, 4H), 3.16 (s, 2H), 3.47−3.49 (m, 4H), 7.24−7.26 (m, 2H), 7.47−7.49 (m, 2H). 13C NMR (CD3OD) δ 30.9, 31.7, 35.4, 40.7, 41.7, 54.5, 127.2, 131.2, 131.7, 152.2 ppm.
Method B. Ti(OEt)4 (0.42 mL, 2.00 mmol) was added to a solution of tert-butyl 4-oxopiperidine-1-carboxylate (0.205 g, 1.03 mmol) and 2-methylpropane-2-sulfinamide (0.130 g, 1.07 mmol) in THF (5 mL) at rt. The resulting pale-yellow solution was refluxed for 5 h and then cooled to 0 °C. A solution of (4-tert-butylbenzyl)magnesium bromide in Et2O, prepared from Mg (0.121 g, 5.00 mmol), 4-tert-butylbenzyl bromide (0.62 mL, 5.00 mmol), and Et2O (5 mL), was added and the mixture was stirred at rt for 15 h. The reaction was quenched with methanol (5 mL) and absorbed onto silica gel. Flash column chromatography, eluting with EtOAc−hexanes 1:1, gave tert-butyl 4-(4-tert-butylbenzyl)-4-(1,1-dimethylethylsulfinamido)piperidine-1-carboxylate 46 (0.121 g, 26%). LC-MS (10 min) m/z 473 [M + Na+], Rt = 8.84 min. 1H NMR (CDCl3) δ 7.32−7.28 (m, 4H), 3.90−3.71 (br m, 2H), 3.35 (br s, 1H), 3.17 (d, J = 13.5 Hz, 1H), 3.15−2.98 (br m, 2H), 2.68 17 (d, J = 13.5 Hz, 1H), 2.36−2.33 (m, 1H), 1.79−1.72 (m, 1H), 1.59−1.54 (m, 1H), 1.46 (s, 9H), 1.42−1.35 (m, 1H), 1.32 (s, 9H), 1.25 (s, 9H). 4 M HCl in dioxane (4 mL) was added to 46 (0.111 g, 0.246 mmol) in MeOH (4 mL) at rt. After 16 h, the mixture was concentrated and purified by ion exchange on acidic resin, eluting with MeOH and then 2 M NH3 in MeOH to give 45 (free amine) (0.061 g, 100%). LC-MS (10 min) m/z 247 [M + H+], Rt = 1.25 min. 1H NMR (CD3OD) δ 1.33 (s, 9H), 1.39−1.41 (m, 2H), 1.60−1.65 (m, 2H), 2.71 (s, 2H), 2.85−2.96 (m, 4H), 7.14−7.15 (m, 2H), 7.34−7.36 (m, 2H). 13C NMR (CD3OD) δ 31.8, 35.2, 39.1, 43.0, 47.8, 50.9, 126.0, 131.5, 131.7, 135.4, 150.8 ppm.
General Method for Preparation of Pyrrolo[2,3-d]pyrimidines
4-(4-tert-Butylbenzyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-4-amine (10)
A mixture of 45 bis hydrochloride salt (0.500 g, 1.57 mmol), 4-chloro-7H-pyrrolo[2,3-d]pyrimidine (0.229 g, 1.49 mmol) and Et3N (1.04 mL, 7.46 mmol) in n-BuOH (15 mL) was heated at 100 °C for 20 h. The mixture was cooled, concentrated, and the resulting solid washed with CH2Cl2 (15 mL). The residual solid was collected. Ion exchange chromatography on acidic resin, eluting with MeOHand then 2 M NH3 in MeOH, gave 10 (0.465 g, 86%). LC-MS (10 min) m/z 364 [M + H+], Rt = 4.25 min. Hi-Res LC-MS calcd for C22H30N5 364.2501, found 364.2503. 1H NMR (CD3OD) δ 1.32 (s, 9H), 1.55−1.58 (m, 2H), 1.75−1.81 (m, 2H), 2.78 (s, 2H), 3.79−3.85 (m, 2H), 4.24−4.29 (m, 2H), 6.63 (d, J = 4.0 Hz, 1H), 7.13 (d, J = 4.0 Hz, 1H), 7.37 (d, J = 8.0 Hz, 2H), 7.37 (d, J = 8.0 Hz, 2H), 8.12 (s, 1H). 13C NMR (CD3OD) 31.9, 35.3, 38.2, 43.3, 51.4, 102.7, 104.2, 122.2, 126.2, 131.6, 135.0, 150.6, 151.7, 152.4, 158.3 ppm. One 13C peak was obscured by solvent.
4-Amino-N-(4-chlorobenzyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide (21)
Dry DMF (1 mL) was added to a mixture of 47 (151 mg, 0.44 mmol) and HATU (220 mg, 0.58 mmol). iPr2NEt (0.38 mL, 2.1 mmol) was added, and the mixture was stirred for 15 min at rt. 4-Chloro-benzylamine (70 μL, 0.57 mmol) was added, and the solution was stirred for 23 h. The reaction mixture was partitioned between water (10 mL) and CH2Cl2 (3 × 10 mL). The organic extracts were dried, filtered, and concentrated. Flash column chromatography, eluting with CH2Cl2−MeOH 96:4, gave 4-tert-butoxycarbonylamino-4-(4-chloro-benzylcarbamoyl)-piperidine-1-carboxylic acid tert-butyl ester 52 (177 mg, 0.38 mmol, 86%). LC-MS (10 min) m/z 490 [M + Na+], Rt = 8.07 min. 1H NMR (CDCl3) δ 1.41 (s, 9H), 1.47 (s, 9H), 1.98 (br d, J = 12 Hz, 2H), 2.10−2.16 (m, 2H), 3.10−3.15 (m, 2H), 3.84 (b rd, J = 11 Hz, 2H), 4.42 (d, J = 5 Hz, 2H), 4.69 (s, 1H), 7.22 (d, J = 9 Hz, 2H), 7.28 (d, J = 9 Hz, 2H). 13C NMR (CDCl3) δ 28.2, 28.4, 32.2, 39.9, 42.9, 57.7, 79.8, 80.9, 128.7, 128.9, 133.2, 136.9, 154.6, 154.9, 173.4 ppm.
HCl (4 M) in dioxane (4.8 mL, 19.2 mmol) was added dropwise to a solution of 52 (96 mg, 0.20 mmol) in MeOH (4.8 mL). The solution was stirred at rt for 17 h. The mixture was concentrated to give 4-amino-piperidine-4-carboxylic acid 4-chloro-benzylamide bis hydrochloride 53 (70 mg, 0.20 mmol, 100%) that was used in the next step without further purification. 1H NMR (CD3OD) δ 2.15−2.22 (m, 2H), 2.60−2.69 (m, 2H), 3.34−3.36 (m, 2H), 3.44−3.46 (m, 2H), 4.47 (s, 2H), 7.36 (s, 4H). 13C NMR (CD3OD) δ 29.6, 41.0, 44.2, 57.5, 129.7, 130.6, 134.4, 138.3, 169.4 ppm. A degassed mixture of 53 bis hydrochloride (48 mg, 0.14 mmol), 4-chloro-7H-pyrrolo[2,3-d]pyrimidine (21 mg, 0.12 mmol), Et3N (126 μL, 0.9 mmol), and n-BuOH (1.2 mL) was stirred at 100 °C for 18 h. The mixture was concentrated and purified by ion exchange chromatography on acidic resin, eluting with MeOH then 2 M NH3 in MeOH. Preparative TLC, eluting with CH2Cl2−MeOH 9:1, gave 21 (37 mg, 0.096 mmol, 69%). LC-MS (10 min) m/z 385 [M + H+], Rt = 2.88 min. Hi Res LC-MS calcd for C19H22N6OCl 385.1544, found 385.1557. 1H NMR (CD3OD) δ 1.61 (br d, J = 14 Hz, 2H), 2.19−2.25 (m, 2H), 3.65−3.71 (m, 2H), 4.38 (s, 2H), 4.47−4.50 (m, 2H), 6.64 (d, J = 4 Hz, 1H), 7.14 (d, J = 4 Hz, 1H), 7.28 (d, J = 8 Hz, 2H), 7.32 (d, J = 8 Hz, 2H), 8.14 (s, 1H). 13C NMR (CD3OD) δ 35.6, 43.0, 43.4, 57.2, 102.5, 104.2, 122.4, 129.6, 130.0, 133.9, 139.1, 151.6, 152.4, 158.2, 179.4 ppm.
Acknowledgments
We thank Dr A. Mirza, Dr M. Liu, and M. Richards for assistance in the spectroscopic characterisation of test compounds and V. Martins for assistance in analysing PK parameters. This work was supported by funding from Cancer Research UK [CUK] grant numbers C309/A2187, C309/A8274, and C309/A8365, and The Institute of Cancer Research. We acknowledge NHS funding to the NIHR Biomedical Research Centre. This work was carried out as part of a funded research collaboration with Astex Therapeutics and intellectual property arising from the programme has been licensed to AstraZeneca plc.
Supporting Information Available
Representative experimental procedures and characterization data for compounds 3−9, 11−20, 22−43; kinase panel selectivity data for compounds 10 and 21; experimental procedures for PKBβ and PKA biochemical assays; experimental procedures for SRB and ELISA cellular assays; details of the X-ray crystallography data collection and refinement for 10-PKB, 21-PKB. This material is available free of charge via the Internet at http://pubs.acs.org.
PDB ID Codes: 2x37 (10-PKB), 2x39 (21-PKB).
Footnotes
Abbreviations: AGC, cAMP-dependent, cGMP-dependent and protein kinase C; ELISA, enzyme-linked immunosorbent assay; mTOR, mammalian target of rapamycin; PH, pleckstrin homology; PKA, protein kinase A; PKB, protein kinase B; PI3K, phosphatidylinositol-3 kinase; PI(3,4,5)P3, phosphatidylinositol-3,4,5 triphosphate; PTEN, phosphatase and tensin homologue.
Supplementary Material
References
- Sale E. M.; Sale G. J. Protein kinase B: signalling roles and therapeutic target. Cell. Mol. Life Sci. 2008, 65, 113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnero A.; Blanco-Aparicio C.; Renner O.; Link W.; Leal J. F. M. The PTEN/PI3K/AKT signalling pathway in cancer, therapeutic application. Curr. Cancer Drug Targets 2008, 8, 187–198. [DOI] [PubMed] [Google Scholar]
- Milburn C. C.; Deak M.; Kelly S. M.; Price N. C.; Alessi D. R.; Van Aalten D. M. F. Binding of phoisphatidylinositol 3,4,5-trisphosphate to the pleckstrin homology domain of protein kinase B induces a conformational change. Biochem. J. 2003, 375, 531–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov D. D.; Guertin D. A.; Ali S. M.; Sabatini D. M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [DOI] [PubMed] [Google Scholar]
- Yap T. A.; Garrett M. D.; Walton M. I.; Raynaud F.; De Bono J. S.; Workman P. Targeting the PI3K−AKT−mTOR pathway: progress, pitfalls, and promises. Curr. Opin. Pharmacol. 2008, 8, 393–412. [DOI] [PubMed] [Google Scholar]
- Bellacosa A.; Kumar C. C.; Di Cristofano A.; Testa J. R. Activation of AKT kinases in cancer: implications for cancer therapeutics. Adv. Cancer Res. 2005, 94, 29–86. [DOI] [PubMed] [Google Scholar]
- Lindsley C. W.; Barnett S. F.; Yaroschak M.; Bilodeau M. T. Recent progress in the development of ATP-competitive and allosteric Akt kinase inhibitors. Curr. Top. Med. Chem. 2007, 7, 1349–1363. [DOI] [PubMed] [Google Scholar]
- Lindsley C. W.; Barnett S. F.; Layton M. E.; Bilodeau M. T. The PI3/Akt Pathway: Recent progress in the development of ATP-competitive and allosteric Akt kinase inhibitors. Curr. Cancer Drug Targets 2008, 8, 7–18. [DOI] [PubMed] [Google Scholar]
- Li Q. Recent progress in the discovery of Akt inhibitors as anticancer agents. Expert Opin. Ther. Patents 2007, 17, 1077–1130. [Google Scholar]
- Collins I. Targeted small molecule inhibitors of protein kinase B as anticancer agents. Anti-Cancer Agents Med. Chem. 2009, 9, 32–50. [DOI] [PubMed] [Google Scholar]
- a Luo Y.; Shoemaker A. R.; Liu X.; Woods K. W.; Thomas S. A.; de Jong R.; Han E. K.; Li T.; Stoll V. S.; Powlas J. A.; Oleksijew A.; Mitten M. J.; Shi Y.; Guan R.; McGonigal T. P.; Klinghofer V.; Johnson E. F.; Leverson J. D.; Bouska J. J.; Mamo M.; Smith R. A.; Gramling-Evans E. E.; Zinker B. A.; Mika A. K.; Nguyen P. T.; Oltersdorf T.; Rosenberg S. H.; Li Q.; Giranda V. L. Potent and selective inhibitors of Akt kinases slow the progress of tumors in vivo. Mol. Cancer Ther. 2005, 4, 977–986. [DOI] [PubMed] [Google Scholar]; b Woods K. W.; Fischer J. P.; Claiborne A.; Li T.; Thomas S. A.; Zhu G.; Diebold R. B.; Liu X.; Shi Y.; Klinghofer V.; Han E. K.; Guan R.; Magnone S. R.; Johnson E. F.; Bouska J. J.; Olson A. M.; de Jong R.; Oltersdorf T.; Luo Y.; Rosenberg S. H.; Giranda V. L.; Li Q. Synthesis and SAR of indazole-pyridine based protein kinase B/Akt inhibitors. Bioorg. Med. Chem. 2006, 14, 6832–6846. [DOI] [PubMed] [Google Scholar]; c Li Q; Woods K. W.; Thomas S.; Zhu G. D.; Packard G.; Fisher J.; Li T.; Gong J.; Dinges J.; Song X.; Abrams J.; Luo Y.; Johnson E. F.; Shi Y.; Liu X.; Klinghofer V.; Des Jong R.; Oltersdorf T.; Stoll V. S.; Jakob C. G.; Rosenberg S. H.; Giranda V. L. Synthesis and structure−activity relationship of 3,4′-bispyridinylethylenes: discovery of a potent 3-isoquinolinylpyridine inhibitor of protein kinase B (PKB/Akt) for the treatment of cancer. Bioorg. Med. Chem. Lett. 2006, 16, 2000–2007. [DOI] [PubMed] [Google Scholar]; d Zhu G. D.; Gong J.; Claiborne A.; Woods K. W.; Gandhi V. B.; Thomas S.; Luo Y.; Liu X.; Shi Y.; Guan R.; Magnone S. R.; Klinghofer V.; Johnson E. F.; Bouska J.; Shoemaker A.; Oleksijew A.; Stoll V. S.; De Jong R.; Oltersdorf T.; Li Q.; Rosenberg S. H.; Giranda V. L. Isoquinoline-pyridine-based protein kinase B/Akt antagonists: SAR and in vivo antitumor activity. Bioorg. Med. Chem. Lett. 2006, 16, 3150–3155. [DOI] [PubMed] [Google Scholar]; e Zhu G. D.; Gandhi V. B.; Gong J.; Luo Y.; Liu X.; Shi Y.; Guan R.; Magnone S. R.; Klinghofer V.; Johnson E. F.; Bouska J.; Shoemaker A.; Oleksijew A.; Jarvis K.; Park C.; Jong R. D.; Oltersdorf T.; Li Q.; Rosenberg S. H.; Giranda V. L. Discovery and SAR of oxindole-pyridine-based protein kinase B/Akt inhibitors for treating cancers. Bioorg. Med. Chem. Lett. 2006, 16, 3424–3429. [DOI] [PubMed] [Google Scholar]; f Thomas S. A.; Li T.; Woods K. W.; Song X.; Packard G.; Fischer J. P.; Diebold R. B.; Liu X.; Shi Y.; Klinghofer V.; Johnson E. F.; Bouska J. J.; Olson A.; Guan R.; Magnone S. R.; Marsh K.; Luo Y.; Rosenberg S. H.; Giranda V. L.; Li Q. Identification of a novel 3,5-disubstituted pyridine as a potent, selective, and orally active inhibitor of Akt1 kinase. Bioorg. Med. Chem. Lett. 2006, 16, 3740–3704. [DOI] [PubMed] [Google Scholar]; g Zhu G. D.; Gong J.; Gandhi V. B.; Woods K.; Luo Y.; Liu X.; Guan R.; Klinghofer V.; Johnson E. F.; Stoll V. S.; Mamo M.; Li Q.; Rosenberg S. H.; Giranda V. L. Design and synthesis of pyridine-pyrazolopyridine-based inhibitors of protein kinase B/Akt. Bioorg. Med. Chem. 2007, 15, 2441–2452. [DOI] [PubMed] [Google Scholar]; h Zhu G. D.; Gandhi V. B.; Gong J.; Thomas S.; Woods K. W.; Song X.; Li T.; Diebold R. B.; Luo Y.; Liu X.; Guan R.; Klinghofer V.; Johnson E. F.; Bouska J.; Olson A.; Marsh K. C.; Stoll V. S.; Mamo M.; Polakowski J.; Campbell T. J.; Martin R. L.; Gintant G. A.; Penning T. D.; Li Q.; Rosenberg S. H.; Giranda V. L. Syntheses of potent, selective, and orally bioavailable indazole-pyridine series of protein kinase B/Akt inhibitors with reduced hypotension. J. Med. Chem. 2007, 50, 2990–3003. [DOI] [PubMed] [Google Scholar]
- a Breitenlechner C. B.; Wegge T.; Berillon L.; Graul K.; Marzenell K.; Friebe W. G.; Thomas U.; Schumacher R.; Huber R.; Engh R. A.; Masjost B. Structure-based optimization of novel azepane derivatives as PKB inhibitors. J. Med. Chem. 2004, 47, 1375–1390. [DOI] [PubMed] [Google Scholar]; b Breitenlechner C. B.; Friebe W. G.; Brunet E.; Werner G.; Graul K.; Thomas U.; Kunkele K. P.; Schafer W.; Gassel M.; Bossemeyer D.; Huber R.; Engh R. A.; Masjost B. Design and crystal structures of protein kinase B-selective inhibitors in complex with protein kinase A and mutants. J. Med. Chem. 2005, 48, 163–170. [DOI] [PubMed] [Google Scholar]
- Ko J. H.; Yeon S. W.; Ryu J. S.; Kim T. Y.; Song E. H.; You H. J.; Park R. E.; Ryu C. K. Synthesis and biological evaluation of 5-arylamino-6-chloro-1H-indazole-4,7-diones as inhibitors of protein kinase B/Akt. Bioorg. Med. Chem. Lett. 2006, 16, 6001–6005. [DOI] [PubMed] [Google Scholar]
- a Collins I.; Caldwell J.; Fonseca T.; Donald A.; Bavetsias V.; Rowlands M. G.; Hunter L.-J. K.; Garrett M. D.; Davies T. G.; Berdini V.; Woodhead S.; Davis D.; Seavers L. C. A.; Wyatt P. G.; McDonald E. Structure-based design of isoquinoline-5-sulfonamide inhibitors of protein kinase B. Bioorg. Med. Chem. 2006, 14, 1255–1273. [DOI] [PubMed] [Google Scholar]; b Reuveni H.; Livnah N.; Geiger T.; Klein S.; Ohne O.; Cohen I.; Benhar M.; Gellerman G.; Levitski A. Toward a PKB inhibitor: modification of a selective PKA inhibitor by rational design. Biochemistry 2002, 41, 10304–10314. [DOI] [PubMed] [Google Scholar]
- Donald A.; McHardy T.; Rowlands M. G.; Hunter L-J. K.; Davies T. G.; Berdini V.; Boyle R. G.; Aherne G. W.; Garrett M. D.; Collins I. Rapid evolution of 6-phenylpurine inhibitors of protein kinase B through structure-based design. J. Med. Chem. 2007, 50, 2289–2292. [DOI] [PubMed] [Google Scholar]
- Saxty G.; Woodhead S. J.; Berdini V.; Davies T. G.; Verdonk M. L.; Wyatt P. G.; Boyle R. G.; Barford D.; Downham R.; Garrett M. D.; Carr R. A. Identification of novel inhibitors of protein kinase B using fragment-based lead discovery. J. Med. Chem. 2007, 50, 2293–2296. [DOI] [PubMed] [Google Scholar]
- Caldwell J. J.; Davies T. G.; Donald A.; McHardy T.; Rowlands M. G.; Aherne G. W.; Hunter L. K.; Taylor K.; Ruddle R.; Raynaud F. I.; Verdonk M.; Workman P.; Garret M.; Collins I. Identification of 4-(4-Aminopiperidin-1-yl)-7H-pyrrolo[2,3-d]pyrimidines as Selective Inhibitors of Protein Kinase B through Fragment Elaboration. J. Med. Chem. 2008, 51, 2147–2157. [DOI] [PubMed] [Google Scholar]
- Lippa B.; Pan G.; Corbett M.; Li C.; Kauffman G. S.; Pandit J.; Robinson S.; Wei L.; Kozina E.; Marr E. S.; Borzillo G.; Knauth E.; Barbacci-Tobin E. G.; Vincent P.; Troutman M.; Baker D.; Rajamohan F.; Kakar S.; Clark T.; Morris J. Synthesis and structure based optimization of novel Akt inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 3359–3363. [DOI] [PubMed] [Google Scholar]
- a Lin X.; Murray J. M.; Rico A. C.; Wang M. X.; Chu D. T.; Zhou Y.; Del Rosario M.; Kaufman S.; Ma S.; Fang E.; Crawford K.; Jefferson A. B. Discovery of 2-pyrimidyl-5-amidothiophenes as potent inhibitors for AKT: synthesis and SAR studies. Bioorg. Med. Chem. Lett. 2006, 16, 4163–4168. [DOI] [PubMed] [Google Scholar]; b Seefeld M. A.; Rouse M. B.; McNulty K. C.; Sun L.; Wang J.; Yamashita D. S.; Luengo J. I.; Zhang S.; Minthorn E. A.; Concha N. O.; Heerding D. A. Discovery of 5-pyrrolopyridinyl-2-thiophenecarboxamides as potent AKT kinase inhibitors. Bioorg. Med. Chem. Lett. 2009, 19 (8), 2244–2248. [DOI] [PubMed] [Google Scholar]
- Rouse M. B.; Seefeld M. A.; Leber J. D.; McNulty K. C.; Sun L.; Miller W. H.; Zhang S.; Minthorn E. A.; Concha N. O.; Choudhry A. E.; Schaber M. D.; Heerding D. A. Aminofurazans as potent inhibitors of AKT kinase. Bioorg. Med. Chem. Lett. 2009, 19 (5), 1508–1511. [DOI] [PubMed] [Google Scholar]
- Heerding D. A.; Rhodes N.; Leber J. D.; Clark T. J.; Keenan R. M.; Lafrance L. V.; Li M.; Safonov I. G.; Takata D. T.; Venslavsky J. W.; Yamashita D. S.; Choudhry A. E.; Copeland R. A.; Lai Z.; Schaber M. D.; Tummino P. J.; Strum S. L.; Wood E. R.; Duckett D. R.; Eberwein D.; Knick V. B.; Lansing T. J.; McConnell R. T.; Zhang S.; Minthorn E. A.; Concha N. O.; Warren G. L.; Kumar R. Identification of 4-(2-(4-amino-1,2,5-oxadiazol-3-yl)-1-ethyl-7-{[(3S)-3-piperidinylmethyl] oxy}-1H-imidazo[4,5-c]pyridin-4-yl)-2-methyl-3-butyn-2-ol (GSK690693), a novel inhibitor of AKT kinase. J. Med. Chem. 2008, 51, 5663–5679. [DOI] [PubMed] [Google Scholar]
- Coffer P. J.; Woodgett J. R. Molecular cloning and characterisation of a novel putative protein-serine kinase related to the cAMP-dependent and protein kinase C families. Eur. J. Biochem. 1991, 201, 475–481. [DOI] [PubMed] [Google Scholar]
- Lindsley C. W.; Zhao Z.; Leister W. H.; Robinson R. G.; Barnett S. F.; Defeo-Jones D.; Jones R. E.; Hartmen G. D.; Huff J. R.; Huber H. E.; Duggan M. E. Allosteric Akt (PKB) inhibitors: discovery and SAR of isoenzyme specificity. Bioorg. Med. Chem. Lett. 2005, 15, 761–764. [DOI] [PubMed] [Google Scholar]
- Barnett S. F.; Defeo-Jones D.; Fu S.; Hancock P. J.; Haskell K. M.; Jones R. E.; Kahana J. A.; Kral A. M.; Leander K.; Lee L. L.; Malinowski J.; McAvoy E. M.; Nahas D. D.; Robinson R. G.; Huber H. E. Identification and characterisation of pleckstrin-homology-domain-dependent and isoenzyme-specific Akt inhibitors. Biochem. J. 2005, 385, 399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolcher A. W.; Yap T. A.; Fearen I.; Taylor A.; Carpenter C.; Brunetto A. T.; Beeram M.; Papadopoulos K.; Yan L.; de Bono J.. A phase I study of MK-2206, an oral potent allosteric Akt inhibitor (Akti), in patients (pts) with advanced solid tumor (ST). J. Clin. Oncol. 2009, 27 (15s), (Suppl. abstr 3503) (Abstract of presentation at ASCO, American Society of Clinical Oncologists, 2009 Annual Meeting, May 29-Jun 2, Orlando, FL). [Google Scholar]
- Davies T. G.; Verdonk M. L.; Graham B.; Saalau-Bethell S.; Hamlett C. C.; McHardy T.; Collins I.; Garrett M. D.; Workman P.; Woodhead S. J.; Jhoti H.; Barford D. A structural comparison of inhibitor binding to PKB, PKA and PKA-PKB chimera. J. Mol. Biol. 2007, 367, 882–894. [DOI] [PubMed] [Google Scholar]
- Kinase selectivity was determined in the SelectScreen assay, Invitrogen Ltd. Compounds were tested at 1 μM, with [ATP] = 10 μM, against the following enzymes: CDK2/cyclinA, CHK1, CHK2, CK2α1, EGFR, FGFR1, FLT3, GSK3β, IGFR1, JAK3, KDR, MAPK1, MAPKAPK2, PKA, PKCα, PKCδ, PKCγ, ROCK2, RSK2, p70S6K, SGK1, SRC.
- Graczyk P. P. Gini coefficient: a new way to express selectivity of kinase inhibitors against a family of kinases. J. Med. Chem. 2007, 50, 5773–5779. [DOI] [PubMed] [Google Scholar]
- Manning G.; Whyte D. B.; Martinez R.; Hunter T.; Sudarsanam S.. The protein kinase complement of the human genome. Science 2002, 298 (5600), 1912−1916, 1933−1934. [DOI] [PubMed] [Google Scholar]
- Vlietstra R. J.; van Alewijk D. C.; Hermans K. G.; van Steenbrugge G. J.; Trapman J. Frequent inactivation of PTEN in prostate cancer cell lines and xenografts. Cancer Res. 1998, 58, 2720–2723. [PubMed] [Google Scholar]
- Raynaud F. I.; Eccles S.; Clarke P. A.; Hayes A.; Nutley B.; Alix S.; Henley A.; Di-Stefano F.; Ahmad Z.; Guillard S.; Bjerke L. M.; Kelland L.; Valenti M.; Patterson L.; Gowan S.; de Haven Brandon A.; Hayakawa M.; Kaizawa H.; Koizumi T.; Ohishi T.; Patel S.; Saghir N.; Parker P.; Waterfield M.; Workman P. Pharmacologic characterization of a potent inhibitor of class I phosphatidylinositide 3-kinases. Cancer Res. 2007, 67, 5840–5850. [DOI] [PubMed] [Google Scholar]
- Gowan S. M.; Hardcastle A.; Hallsworth A. E.; Valenti M. R.; Hunter L.-J. K.; de Haven Brandon A. K.; Garrett M. D.; Raynaud F.; Workman P.; Aherne W.; Eccles S. A. Application of mesoscale technology for the measurement of phospho-proteins in human tumour xenografts. Assay Drug Dev. Technol. 2007, 5, 391–401. [DOI] [PubMed] [Google Scholar]
- Skehan P.; Storeng R.; Scudiero D.; Monks A.; McMahon J.; Vistica D.; Warren J. T.; Bokesch H.; Kenney S.; Boyd M. R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [DOI] [PubMed] [Google Scholar]
- Moreno-Farre J.; Workman P.; Raynaud F. I. Analysis of potential drug−drug interactions for anticancer agents in human liver microsomes by high throughput liquid chromatography/mass spectrometry assay. Recent Adv. Res. Updates 2006, 7, 207–224. [Google Scholar]
- a Said S. H.; Karickhoff S. W.; Carreira L. A. A rigorous test for SPARC's chemical reactivity models: estimation of more than 4300 ionization pKa's. Quant. Struct. Act. Rel. 1995, 14, 348–355. [Google Scholar]; b Milletti F.; Storchi L.; Sforna G.; Cruciani G. New and original pKa prediction method using grid molecular interaction fields. J. Chem. Inf. Model. 2007, 47, 2172–2181. [DOI] [PubMed] [Google Scholar]
- Pallai P; Goodman M. Preparation of optically pure monoacyl 2-alkyl gem-diamines from peptide amides. J. Chem. Soc., Chem. Commun. 1982, 280–281. [Google Scholar]
- Caldwell J. J.; Collins I. Rapid synthesis of 4-benzyl-4-aminopiperidines by addition of Grignard reagents to N-(1-Boc-piperidin-4-ylidene)-tert-butanesulfinyl imine. Synlett 2006, 2565–2568. [Google Scholar]
- Caldwell J. J.; Cheung K. M.; Collins I. Synthesis of 4-(cyclic dialkylamino)-7-azaindoles by microwave heating of 4-halo-7-azaindoles and cyclic secondary amines. Tetrahedron Lett. 2007, 48, 1527–1529. [Google Scholar]
- a Matthews T. P.; Klair S.; Burns S.; Boxall K.; Cherry M.; Fisher M.; Westwood I. M.; Walton M. I.; McHardy T.; Cheung K.-M. J.; Van Montfort R.; Williams D.; Aherne G. W.; Garrett M. D.; Reader J.; Collins I. Identification of Inhibitors of Checkpoint Kinase 1 through Template Screening. J. Med. Chem. 2009, 52, 4810–4819. [DOI] [PubMed] [Google Scholar]; b Hoehn H.; Denzel T.. 1H-Pyrazolo[3,4-b]pyridines. Patent Appl. DE2301268. Chem. Abs. 1973, 79, 115578. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.