

Abstract
The relationship between the enzymatic and the transcriptional activity of the bifunctional protein pterin-4a-carbinolamine dehydratase/dimerization cofactor for hepatocyte nuclear factor 1 (DCoH) has been elucidated by site-directed mutagenesis. DCoH dimers harbor a binding site for hepatocyte nuclear factor 1 (HNF1), two active centers that bind pterins, and a saddle-shaped surface that resembles nucleic acid binding domains. Two domains of the protein have been selectively targeted to determine if a change in one activity affects the other. No strong correlation has been found, supporting the idea that carbinolamine dehydratase activity is not required for HNF1 binding in vitro or transcriptional coactivation in vivo. Double mutations in the active center, however, influence the in vivo transcriptional activity but not HNF1 binding. This finding suggests that some active center residues also are used during transcription, possibly for binding of another (macro)molecule. Several mutations in the saddle led to a surprising increase in transcription, therefore linking this domain to transcriptional regulation as well. The transcriptional function of DCoH therefore is composed of two parts, HNF1 binding and another contributing effect that involves the active site and, indirectly, the saddle.
Pterin-4a-carbinolamine dehydratase (EC 4.2.1.-) is a protein associated with aromatic amino acid hydroxylation (1). It stimulates phenylalanine hydroxylase (PAH, EC 1.14.16.1) by catalyzing a dehydration step in the regeneration of the essential cofactor tetrahydrobiopterin (BH4), thereby preventing the formation of 7-BH4, an inhibitory isomeric form of BH4 (2–5). Mutations in the human dehydratase gene lead to a form of hyperphenylalaninemia (6), and a lack of dehydratase activity in the skin has been linked to the depigmentation disorder vitiligo (7). Moreover, the discovery that pterin-4a-carbinolamine dehydratase is identical to DCoH, the dimerization cofactor for hepatocyte nuclear factor 1 (HNF1) suggested the possibility that the dehydratase also may regulate PAH activity at the nucleic acid level (8, 9). HNF1 is a tissue-specific transcription factor in mammals and amphibians, regulating the expression of typical genes of liver, pancreas, kidney, and other organs (10–12). A knockout mouse model for HNF1 and analysis of the PAH promotor/enhancer revealed that the PAH gene is regulated by HNF1; indeed, the regulation is even more pronounced than it is with most other known targets of this transcription factor (13, 14). Another important gene regulated by HNF1 is that of insulin. Defects in the HNF1 gene have been linked to a variant of inherited juvenile diabetes (15). Dimerization of HNF1 is required for its binding to DNA. DCoH, which exists in tetrameric form in the cytosol, binds as a dimer to the rather unstable HNF1 dimers. The resulting stable 2:2 complex can be isolated from cell nuclei. However, the stabilization of HNF1 dimers is not considered to be the sole cause of transcriptional activity of DCoH (10, 16). A link between PAH expression and DCoH also had been shown to exist in Pseudomonas aeruginosa where no HNF1 is present. DCoH from Pseudomonas is 33% identical to the human protein and has similar enzymatic properties (17). It exists exclusively as a dimer and does not bind to mammalian HNF1 (18). Inactivation of Pseudomonas DCoH itself completely suppresses the expression of bacterial PAH (17). A restoration of bacterial PAH mRNA levels after complementation with mammalian DCoH has been observed (19). A binding of DCoH to mRNA thus could be a possible scenario to account for that effect. In a recent study, an apparently nonspecific binding between HNF1 and RNA was described. This interaction was prevented by DCoH, which, however, did not bind to the selected RNAs itself (20).
The overall shape of DCoH dimers (Fig. 1) resemble that of the TATA binding protein (21). A β-sheet forms the concave part of a saddle structure, including “stirrups,” with α-helices covering the convex side of the structure. In each subunit, two loops between the sheet and the helices form part of the active center. The saddle appears to be too small to accommodate B-DNA but shows some features common to RNA-binding proteins. A putative binding site for HNF1 is a helical domain at the top of the saddle structure, which also serves as the site where two DCoH dimers assemble to the cytosolic tetramer (19, 22, 23). The developmental pattern of nuclear localization during Xenopus embryogenesis suggests that DCoH is not solely a coactivator of HNF1 but also may interact with other transcription factors (24).
Figure 1.
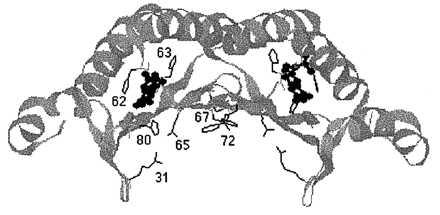
Structure of a DCoH dimer. Shown is a ribbon diagram with only those amino acid residues displayed that had been targeted for mutagenesis. The residues of the left monomer are also numbered. Bound to the active sites is the product analogue 7,8-BH2. The coordinates were taken from ref. 23 and converted with RasMac v2.6 (written by R. Sayle).
Despite solving the three-dimensional structure of DCoH representing a milestone in the study of this protein, many questions remain about its mechanism of action and its function: How can a small tetrameric protein of 12-kDa/subunit perform at least two functions? Are the functions mediated on spatially separated domains, and if they are, do they influence each other, or does the protein even use the same molecular functionalities to perform different tasks? We focused our investigations on the question if dehydratase activity is required for the transcriptional function.
MATERIALS AND METHODS
Materials.
Restriction endonucleases were purchased from New England Biolabs. BH4 and 7,8-dihydrobiopterin (7,8-BH2) were purchased from Schircks Laboratories (Jona, Switzerland). Hepes was obtained from ICN. All other chemicals and proteins were purchased from Sigma.
Site-Directed Mutagenesis and Subcloning.
The CloneAmp pUC19 System (GIBCO) was used to generate H62N/H63L and all single mutants as described before for the C82R mutant (25, 26). The mutagenic oligonucleotides for this PCR approach were synthesized at Lofstrand Labs (Gaithersburg, MD). Our rat cDNA clone, which translates into a protein that is identical to human DCoH, served as template. PCR products were subcloned in pAMP19 (GIBCO). The other double mutants were generated by domain exchange between single mutants, by using the NcoI site and a unique BsrGI restriction site in the coding region. For bacterial expression, the NcoI/BamHI cut inserts were subcloned in pET3d (Novagen). NotI/EcoRI-digested inserts were subcloned in the mammalian expression vector pBJ5 (26, 27). A truncated form of HNF1 that lacks the 345 amino acids of the C-terminal transactivation domain, but retains the fully active dimerization and DNA binding domains, was created by cutting pBJ5-HNF1 with NcoI and religating the fragments with the vector pQE60 (Qiagen) (28). The resulting pQE60-trHNF1 is a vector for expression of a carboxyl-terminally His-tagged HNF1 (the vector was a generous gift of G. Crabtree, Stanford University, Stanford, CA). Plasmids were purified with Qiagen kits. All plasmids used for mammalian transfections were highly purified with EndoFree Plasmid kits (Qiagen). The mutations were confirmed by sequencing with an ALF sequencer (Pharmacia).
Expression and Purification of Proteins.
Wild-type DCoH and all mutants were expressed and purified as described previously (26, 29). The truncated His-tagged form of HNF1 was purified on Ni-NTA resin (Qiagen) according to the manufacturer’s recommendations. Protein concentrations were quantitated with the Bradford and the BCA assays from Pierce. The purity of proteins was assessed by SDS/PAGE as described previously (26). Samples of wild type and mutants were heated for 5 min at 68–70°C in a temperature-controlled water bath to check for heat stability. To confirm the tetrameric state of each mutant, gel filtration on a Superdex 75 HR 10/30 column (Pharmacia) was performed. Proteins were eluted in 20 mM Hepes, pH 7.6/150 mM NaCl with a flow rate of 0.25 ml/min. Protein standards from Pharmacia were used for calibration.
Cell Culture and Transfection.
Chinese hamster ovary cells were grown in Ham’s F-12 medium supplied with 10% fetal calf serum (GIBCO) at 37°C under 5% CO2. Cells were subcultured 22–24 hr before transfection. Transient transfections were performed with 1 μg pSV-β-galactosidase vector (Promega) as internal control, 10 μg (β28)3-Luc (28), 5 ng pBJ5-HNF1 (27), and 1–4 μg pBJ5 (the latter three vectors were a generous gift of G. Crabtree) containing either no insert, wild-type, or mutant insert by using a Gene Pulser apparatus (Bio-Rad) under the following conditions: 960 μF, 270 V, electrode gap 0.4 cm, 107 cells/cuvette (16, 28). Cells were harvested 46 hr after transfection and extracted with reporter lysis buffer (Promega). To confirm the expression of wild-type and mutant DCoH, SDS/PAGE and Western blots were performed as described previously (26, 30).
Enzyme Assays.
The PAH stimulation assay was used to determine dehydratase activities of the mutants as described previously (26, 29). The specific activity of wild-type DCoH determined by this assay was 28.3 μmol/min per mg. The activities of the mutants in Table 1 are expressed as percent of this wild-type activity. The luciferase activity of Chinese hamster ovary cell crude extracts was determined with the Luciferase Assay System from Promega. Measurements were performed with a Packard TRI-CARB scintillation counter in the single photon mode. β-Galactosidase activities were measured spectrophotometrically by using the β-Galactosidase Enzyme Assay System from Promega, and used to normalize for transfection efficiency.
Table 1.
Enzymatic, HNF1-binding, and transcriptional activity of DCoH mutants compared to the wild type
| DCoH mutants | Dehydratase activity, % | Binding to HNF1, % | Transcriptional activity, % |
|---|---|---|---|
| Wild type | 100 ± 5 | 100 ± 6 | 100 ± 16 |
| H62N | 2.6 ± 0.2 | 99 ± 3 | 90 ± 20 |
| H63L | 0.49 ± 0.03 | 106 ± 8 | 91 ± 19 |
| H80L | 20 ± 1 | 82 ± 22 | 98 ± 11 |
| C82R | 49 ± 4 | 93 ± 13 | 97 ± 11 |
| H63L/H80L | 0.15 ± 0.01 | 83 ± 11 | 54 ± 25 |
| H62N/H63L | 0.012 ± 0.002 | 111 ± 10 | 14 ± 10 |
| K72E | 96 ± 3 | 101 ± 7 | 100 ± 12 |
| R31Q | 118 ± 5 | 108 ± 11 | 122 ± 25 |
| R31Q/K72E | 135 ± 8 | 101 ± 10 | 432 ± 53 |
| E65A | 45 ± 2 | 105 ± 13 | 156 ± 46 |
| E65A/K72E | 53 ± 3 | 96 ± 14 | 310 ± 37 |
| F67A | 72 ± 2 | 65 ± 7 | 185 ± 28 |
The dehydratase activities were determined as described in Materials and Methods. The results are the means ± SD of three measurements. The HNF1-binding assays were performed in MaxiSorp 96 well microtiter plates (Nunc) and are based on standard ELISA protocols (37). In each well 400 ng of HNF1 in 40 μl Hepes buffered saline (HBS, 20 mM Hepes pH 7.5/150 mM NaCl) were immobilized. The wells were washed and blocked with HBS/5% milk for 1 hr, then rinsed with HBS/1% BSA. Incubation with wild-type DCoH or its mutants occurred in 40 μl of HBS/1% BSA for 2–4 hr at 22°C. The final concentration of DCoH was 30 μg/ml. After three washes, antibody against DCoH (30), was added and incubation proceeded overnight. The primary antibody was washed away and secondary antibody, anti-rabbit IgG alkaline phosphatase conjugate (Promega), was added for 1 hr at a dilution of 1:4,000. The color reaction was performed with 5 mM p-nitrophenyl-phosphate. Optical densities (405 nm) were measured with a microplate reader (Bio-Tek EL340). The following controls were included in each experiment: minus HNF1 and minus DCoH (Fig. 3). Each value represents the mean ± SD of at least three independent experiments. The antigenicity of each mutant was assessed by immobilizing wild-type and mutant DCoH (0.8–3.2 ng) either on Multisorb plates or nitrocellulose (dot blots) and then probing with antibody. As a control, gel shift assays were done as described (16) with the mutants C82R, H63L, and wild-type DCoH, and gave the same results as the ELISA assay. The determination of the in vivo transcriptional activity is described in Materials and Methods. Transfections were performed with duplicate samples. The results are means ± SD of four independent experiments each and are corrected for transfection efficiency. All listed activities are percentages of the wild-type activity.
RESULTS
Twelve mutant forms of DCoH have been generated, expressed in Escherichia coli, and purified to homogeneity. The selection of the mutated sites originally was based on a comparison of conserved amino acids in the DCoH sequences from different species (17) and the known positions of two naturally occurring mutations that were associated with reduced dehydratase activity (6). After the three-dimensional structure of DCoH became available, a more targeted mutagenesis of the active center as well as of a putative region for transcriptional coactivation, the saddle, was possible. Previously, we and others have described the enzyme kinetics of the naturally occurring mutant C82R (26, 31). This mutant has reduced enzymatic activity but showed normal binding to HNF1, already indicating a separation between the two activities. We also have reported that substitutions of the His residues in positions 62, 63, or 80 led to markedly reduced catalytic activities (26). During our investigations, an enzymatic characterization of several similar DCoH mutants was published (32). Questions regarding the transcriptional activity of DCoH mutants, however, have not been addressed so far. Our objective in the present study was to compare the enzymatic and transcriptional activities of different mutants to determine the degree of correlation or separation of these two activities.
Every mutation has the potential to cause major conformational disturbances. We therefore performed only single and double point mutations and tested each mutant for its degree of aggregation and heat stability. All mutants described here formed only tetramers and were as heat resistant as the wild-type protein. The mutations that targeted the active center of DCoH to knock out dehydratase activity were H62N, H63L, H80L, and the double mutations H62N/H63L and H63L/H80L. The saddle domain of DCoH has been described as a possible binding site for macromolecules like nucleic acids and proteins involved in transcription (19). Binding between proteins and nucleic acids usually is mediated by the sum of numerous single interactions; many are of weaker nature, e.g., van der Waals forces. Stronger interactions exist between a protein’s positively charged lysines or arginines and a nucleic acid’s negative backbone or bases (33–35). To disrupt the binding of a macromolecule, it is expected that more than one point mutation might be necessary to cause a detectable effect. To archive a maximum effect, we targeted the charged residues that likely contribute to stronger interactions. We generated the following substitutions to disrupt potential binding interactions: R31Q, E65A, K72E, and the combinations R31Q/K72E and E65A/K72E. Based on the evidence that with another protein, U1A, two aromatic residues at the center of a β-sheet are crucial for binding to single-stranded RNA through base stacking (34), we mutated Phe-67 at the center of the saddle of DCoH to F67A.
Three assays have been used to characterize the various properties of DCoH: An enzymatic assay that (indirectly) determines the rate of dehydration of 4a-hydroxytetrahydrobiopterin (4a-OH-BH4), a binding assay that measures the ability of DCoH to form a complex with HNF1, and a cotransfection procedure with Chinese hamster ovary cells that determines the stimulation by DCoH of HNF1-mediated transcription of a reporter gene. The enzymatic assay has been described extensively before (26, 29, 30). We used the natural substrate 4a-OH-BH4, synthesized in situ during PAH-mediated hydroxylation of phenylalanine, to characterize the mutants. In Table 1, the specific activities of the mutants are listed, expressed as percentages of the wild-type activity. With 0.49% wild-type activity, H63L has the lowest specific activity of the single mutants, followed by H62N with 2.6%. No stable single mutant could be found that was completely devoid of dehydratase activity. Double substitutions, however, could produce the desired mutants. H63L/H80L has only 0.15% residual activity, and H62N/H63L is essentially inactive. Two other residues located in the saddle domain and relatively close to the active center showed some effects on dehydratase activity. E65A (45%) and F67A (72%) both are in proximity of the loop that carries the important residues His-63 and His-62. Their reduced catalytic activities can be interpreted as being caused by a conformational disturbance of the loop. The activity of K72E is unchanged. The substitution of Lys-72 displays almost no influence in E65A/K72E (53%) either, whereas R31Q (118%) shows a slight increase in activity that is more pronounced in the double mutant R31Q/K72E (135%). At first glance, Arg-31, which is located close to the stirrup, appears to be too far from the active center to mediate any change. A closer look at the three-dimensional structure, however, reveals a salt bridge between Arg-31 and Glu-65. Mutation of Arg-31 disrupts the contact to Glu-65 that might then reorient itself and thereby influence the positions of His-63 and His-62.
We developed a binding assay that allowed us to determine the interaction between DCoH and HNF1. Compared with the usual gel shift assay, no in vitro cotranslation and no addition of (radiolabeled) DNA are necessary (16). The assay is based on the immunochemical detection of DCoH that binds to immobilized HNF1, similar to a blot overlay assay (36). We estimate that, when DCoH is in excess, at least 10% of the immobilized HNF1 forms a complex. This result is in contrast to attempts to form a protein complex by mixing free HNF1 and DCoH in solution, which does not lead to sufficient binding (16, 22). The validity of the assay was confirmed by testing the mutants C82R and H63L with this assay as well as with the regular gel shift assay (16, 26). The new assay can be performed in Western, dot blot, or ELISA formats (37). We preferred the ELISA version, which allows a more convenient quantitation. Recombinant, highly purified HNF1 is bound to 96-well plates. After blocking, the wells are incubated with wild-type or mutant DCoH in the presence of competing protein, e.g., BSA. Excess DCoH is washed away and primary antibody against DCoH is added, which binds only to the wells that contain bound DCoH. An optimal concentration of wild-type DCoH protein for the incubation step with HNF1 was determined first (Fig. 2). With increasing concentrations of DCoH, the formation of the DCoH/HNF1 complex increases until it reaches a plateau. Comparisons of wild type and mutants were performed at a DCoH concentration of 30 μg/ml, which lies within the linear section of the curve. The background is usually low, because DCoH does not bind nonspecifically and HNF1 does not crossreact with anti-DCoH antibody (see Fig. 3). The antigenicity of all mutants was tested in parallel experiments and corrections were made, if necessary. Table 1 summarizes the results. Most mutants show no significant change in binding to HNF1.
Figure 2.
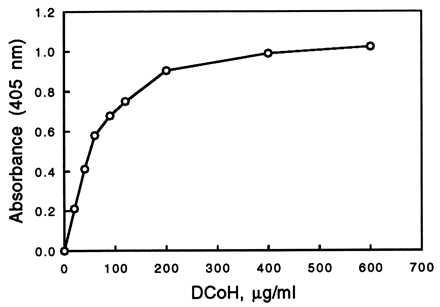
Dependence of the DCoH/HNF1 complex formation, expressed as absorbance, on the concentration of wild-type DCoH. The experimental conditions are the same as described in the legend to Table 1, except that the concentration of DCoH was varied. Absorbance was measured at 405 nm.
Figure 3.
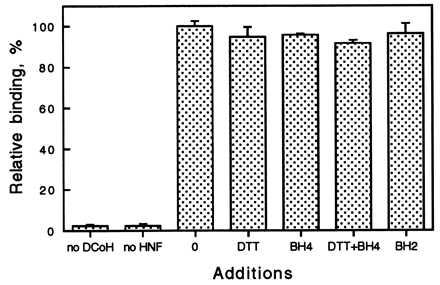
The effect of pterins on the formation of the DCoH/HNF1 complex, expressed as relative binding activity. The conditions are the same as described in the legend to Table 1. Measurements were done in duplicate. The first two bars are negative controls, where either DCoH or HNF1 had been left out. Additions to the incubation step are as follows: 0, no addition; DTT, 1 mM DTT; BH4, 100 μM BH4; BH4+DTT, 100 μM BH4/1 mM DTT (DTT was added to keep BH4 reduced); BH2, 25 μM 7,8-BH2.
The in vivo reporter gene assay is a slightly modified version of the procedure described by Mendel et al. (16). A construct consisting of three copies of the β-fibrinogen HNF1 binding sequence in front of a TATA box promotor and the gene of firefly luciferase [(β28)3-Luc] served as the reporter (28). Cotransfections included the construct, an expression vector for HNF1 (pBJ5-HNF1), and either the parent vector pBJ5 or pBJ5-DCoH. The amount of HNF1 vector used was 5 ng, which is almost saturating (data not shown). The stimulation of the basal luciferase activity by HNF1 alone was about 60-fold; the enhancement of this HNF1 effect by DCoH was another 2-fold. Under these conditions the reported 200-fold enhancement of HNF1 could not be observed, probably because the stabilizing influence of DCoH on dimerization is most effective at very low cellular concentrations of HNF1 (16). The advantage of using higher amounts of HNF1 is better reproducibility. It also allows the dissection of the HNF1 binding effect from another possible mechanism that might contribute to the DCoH coactivator function in vivo. The results are summarized in Table 1. The amount of pBJ5-DCoH used in each experiment depended on the expression level of the corresponding mutant, and varied between 1 and 4 μg, which is in excess and produces an amount of DCoH protein that can be monitored in Coomassie-stained SDS/PAGE gels (data not shown). The stimulation of HNF1 by increasing amounts of pBJ5-DCoH is shown in Fig. 4. Saturation already is reached with 40–60 ng of pBJ5-DCoH. Consequently, we also performed cotransfections with lower amounts (20–40 ng) of expression vector. The active center mutants gave similar results as those listed in Table 1, whereas the saddle mutants that are more active than the wild type lost their excess activity with decreasing amounts of expression vector, as shown for R31Q/K72E in Fig. 4.
Figure 4.
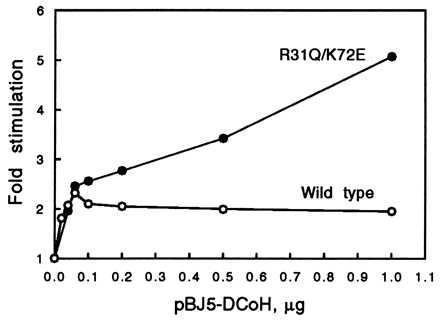
Dependence of the in vivo stimulation of HNF1 by wild-type DCoH and the mutant R31Q/K72E on the concentration of the expression vector pBJ5-DCoH. The basal HNF1 activity is set to 1. The experimental conditions are the same as described in Materials and Methods, except that the amount of vector was varied. The curves represent a typical experiment. Measurements were performed in duplicate.
DISCUSSION
We succeeded in generating mutants that have, to various degrees, reduced dehydratase activities, including the essentially inactive double mutant H62N/H63L (Table 1). The mutations confirmed the localization of the active center (23, 32) and provided us with a tool to probe the effects of enzymatic inactivation on transcriptional activity. Mutations in the saddle region of DCoH had a moderate impact, negative as well as positive, on the enzymatic activity. This result implies that binding of a molecule to the saddle might influence the characteristics of the active center.
We developed a convenient binding assay, which allowed us to quantitate the ability of each mutant to bind to HNF1. Despite the large range in their enzymatic activities, none of the 12 mutants showed any significant change in their ability to bind to HNF1. Only F67A led to a more pronounced reduction (about 35%), which could be explained by the location of Phe-67 at the monomer-monomer interface within the saddle. A change in the contact angle between the monomers most likely also would affect the contact between two DCoH dimers or a dimer and HNF1. The other saddle mutants displayed no change in their binding capability. We also tested the effect of pterins on DCoH/HNF1 binding. Addition of BH4 or 7,8-BH2 to the binding reaction did not affect the complex formation (Fig. 3). All binding results are consistent with the interpretation of crystal structure data, which favors binding of dimeric HNF1 not to the concave saddle surface but to the two helices at the top of a DCoH dimer, forming a mixed four-helix bundle (18, 19, 22). This helical site is spatially separated from the active center and is not affected by a conformational change on binding of the product analogue 7,8-BH2 to the active site (23). In the reciprocal experiment, it was found that binding of HNF1 to DCoH also does not influence dehydratase activity (20). Biopterins seem not to play a role in the formation of the DCoH/HNF1 complex, but a role in transcriptional coactivation in vivo is still possible. However, preliminary results from transfection experiments in NIH 3T3 cells, which do not synthesize pterins, show the same DCoH-mediated stimulation of HNF1 as in the BH4-synthesizing Chinese hamster ovary cells (unpublished work). Recently, it had been reported that the mutant C82R binds to HNF1 in the yeast two-hybrid system (38). Because this mutant still has significant (49%) dehydratase activity and the two-hybrid system cannot be quantitated precisely enough, these results are difficult to interpret.
The mutants H62N and H63L have severely reduced dehydratase activity. In contrast, their transcriptional function remains essentially unchanged (Table 1). The partially active mutants H80L and C82R also are not affected in their transcriptional activity. It has to be noted, however, that the in vivo transcriptional assay is not accurate enough to allow a clear statement for mutants that have considerable residual enzymatic activities. Nevertheless, the pterin-4a-carbinolamine dehydratase function can clearly be separated from the transcriptional function. Moreover, the cytosolic substrate for DCoH, 4a-OH-BH4, is very unstable and therefore unlikely to occur in considerable amounts in the nucleus. Furthermore, there is no evidence that the phenylalanine hydroxylating system is present in the nucleus. In contrast to single substitutions, double mutations in the active center of DCoH not only destroy the catalytic activity but also reduce its transcriptional activity. The active center—or at least some of its residues—contributes to the transcriptional function. Because the single mutations already exclude the dehydration of 4a-OH-BH4, the dehydration of another substrate or a completely different mechanism has to be involved in the coactivator function. An alternative to an enzymatic mechanism would be the binding of a macromolecule, like a protein of the transcriptional machinery or a nucleic acid. A bridging function, where DCoH acts as an adapter between HNF1 and other components of the transcriptional apparatus, had been proposed before (10, 19). DCoH, while bound to the dimerization domain, also could interact with other parts of the HNF1 protein itself. The putative substrate could bind to one or both active centers of a DCoH dimer; it might involve additional domains of DCoH, including the saddle. The present results support the idea that binding to HNF1 is not the only cause of DCoH’s transcriptional activity but one aspect of a bipartite function. Despite their reduced activity in the in vivo assay, the HNF1 binding function of H62N/H63L and H63L/H80L is not diminished. The low in vivo activity of H62N/H63L speaks for a synergistic role of the two partial functions, as one would expect for a bridging function of DCoH, or it even suggests that the contribution of HNF1 binding to the total transcriptional effect is minor. However, our in vivo assay uses an amount of HNF1 vector that produces saturating HNF1 protein levels, which would minimize the dimerization/stabilization effect of DCoH.
How do we picture the second aspect of the bipartite transcriptional function? If we assume that a nucleic acid would be a binding partner for DCoH, double-stranded DNA has to be excluded (19). This assumption still would leave RNA and single-stranded DNA as possible candidates. The purine bases of nucleic acids, especially guanine with its pyrimidine moiety, resemble biopterin. Binding studies with several different pterins have demonstrated that DCoH can bind a variety of related structures (31). Even a crystal structure with a bound product analogue has been published (23). Contrary to those results, however, with the exception of the unstable quinonoid dihydrobiopterin, none of these compounds was able to compete with the natural substrate 4a-OH-BH4 in the enzymatic assay (39). Coherent with the latter observations, neither free guanosine nor free GMP were able to inhibit the dehydratase (unpublished data). Still, we can imagine a nucleic acid molecule “wrapped” around DCoH or spanning through the saddle, with two unpaired guanine residues in different positions of the sequence, each binding to an active center adjacent to the sides of the saddle. The bases (or backbone) between these two residues could make additional contacts to the saddle or the outer helices. The concerted interaction of the guanines and other parts of the nucleic acid should provide enough binding energy to compete with the natural substrate or other pterins—in case these molecules should be present in the nucleus.
In our experiments, most single mutations in the saddle domain of DCoH had no significant effect and even the removal of four positive charges (two per subunit) did not reduce transcriptional activity. Instead, a remarkable increase of more than 400% with the mutant R31Q/K72E was observed. E65A/K72E has a similar positive effect on transcription. An increase simply could mean that the mechanism of coactivation had been improved and nucleic acids either do not bind to DCoH or bind in a different way. A reason for the unexpected increase could be that the aforementioned salt bridge between Arg-31 and Glu-65 might influence the geometry of the saddle domain. If interrupted by mutating either one of the two residues, the positions of the stirrups could change, possible moving outward and thereby widening the opening of the saddle. This change, in turn, should make it easier for a macromolecule to bind to the saddle. Unfortunately, this model does not explain the synergistic role of K72E or why the single mutants R31Q and E65A do not cause an enhancement similar to the double mutants. Nor does it explain the considerable effect of the single mutant F67A, whose transcriptional activity is increased to 185% despite its reduced HNF1 binding and dehydratase activity. In that context, it should be noted that there is no correlation between the saddle mutants’ increase in transcriptional activity and their dehydratase function. Most mutants show a loss in dehydratase activity; only R31Q/K72E exhibits an increase in both carbinolamine dehydratase and transcriptional activity. This loss again is evidence for a separation of the two functions.
Another explanation for the activity-enhanced mutants could be that the mutated saddle residues are not the actual mediators of transcriptional coactivation but are rather involved in down-regulating this function. Because it is easier to destroy than to improve a function, it appears to be more likely that the different mutations in R31Q/K72E, E65A/K72E, and F67A did not enhance the transcriptional mechanism directly. For example, some mutagenic improvements of enzyme catalysis turned out to result from a removal of an (allosteric) inhibition (40, 41). Thus, mutations in the DCoH saddle might disrupt the binding of an inhibitory protein, nucleic acid, or other (macro)molecule. Some support for this model can be derived from crystal structure studies. In the crystal, individual tetramers of DCoH are associated with each other. A helix of one tetramer interacts with the saddle of another tetramer (22, 23), suggesting that DCoH is blocking one of its own functional domains. A similar interaction has been reported for TATA binding protein dimers (42). For the saddle mutants, the oligomerization of DCoH tetramers might be weakened or are no longer possible, therefore making the saddle more readily accessible to other macromolecules. However, gel filtration experiments with wild-type DCoH have never shown any evidence for (noncovalent) higher aggregates in solution (19, 29).
Besides general reasons like higher flexibility, what would be the biological meaning of an inhibitory modulation of the transcriptional function? DCoH is able to translocate into the nucleus on its own (38). In the liver, DCoH has to perform its two different functions within the same cell type. The cellular concentration of the protein, estimated to be 6 μM (39), is relatively high. This amount is sufficient to prevent the formation of the potentially harmful (4, 5) 7-BH4 in the cytosol, but might be in excess for subtle regulatory functions in the nucleus. In that case, it can be speculated that a compensatory feedback mechanism would be necessary. For example, inhibition of the transcriptional, but not the enzymatic, activity of DCoH by its own mRNA, by the DCoH protein itself, or by a still unknown factor that is released by the action of DCoH could occur through binding to the saddle domain. The dose–response behavior of wild-type DCoH and the mutant R31Q/K72E in transfection experiments points in that direction (Fig. 4). The wild-type protein already reaches its maximum stimulation of HNF1 with about 50 ng of expression vector, whereas the activity of R31Q/K72E continues to increase until its effect levels off around 1 μg at a much higher stimulation. A self-inhibition of DCoH protein expression was not observed in this experiment: in Western blots the DCoH protein band increased with the amount of vector (data not shown).
In our study we presented evidence for an involvement of the active center and the saddle domain of DCoH in transcriptional coactivation. Further studies are necessary to determine the mechanism and regulation of this complex function. A future direction might include the isolation of the potential macromolecular binding partner(s) of DCoH, in addition to the development of an in vitro transcriptional assay.
Acknowledgments
We are grateful to L. Hansen, W. Wang, and G. Crabtree for helpful discussions and for providing the vectors for the transcriptional assays. We are also very grateful to J. Cronk, J. Endrizzi, and T. Alber for giving us access to x-ray structure data before publication. We thank A.-M. Vasquez and L. Lee for their skillful help in performing the tissue culture experiments, and C. Falke for help with sequencing.
ABBREVIATIONS
- BH4
tetrahydrobiopterin
- 4a-OH-BH4
4a-hydroxytetrahydrobiopterin
- 7
8-BH2, 7,8-dihydrobiopterin
- DCoH
dimerization cofactor for HNF1
- HNF1
hepatocyte nuclear factor 1
- PAH
phenylalanine hydroxylase
References
- 1.Kaufman S. J Biol Chem. 1970;245:4751–4759. [PubMed] [Google Scholar]
- 2.Lazarus R A, Benkovic S J, Kaufman S. J Biol Chem. 1983;258:10960–10962. [Google Scholar]
- 3.Davis M D, Kaufman S, Milstien S. Proc Natl Acad Sci USA. 1991;88:385–389. doi: 10.1073/pnas.88.2.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davis M D, Ribeiro P, Tipper J, Kaufman S. Proc Natl Acad Sci USA. 1992;89:10109–10113. doi: 10.1073/pnas.89.21.10109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Adler C, Ghisla S, Rebrin I, Haavik J, Heizmann C W, Blau N, Kuster T, Curtius H C. Eur J Biochem. 1992;208:139–144. doi: 10.1111/j.1432-1033.1992.tb17167.x. [DOI] [PubMed] [Google Scholar]
- 6.Citron B A, Kaufman S, Milstien S, Naylor E W, Greene C L, Davis M D. Am J Hum Genet. 1993;53:768–774. [PMC free article] [PubMed] [Google Scholar]
- 7.Schallreuter K U, Wood J M, Pittelkow M R, Gütlich M, Lemke K R, Rodl W, Swanson N N, Hitzemann K, Ziegler I. Science. 1994;263:1444–1446. doi: 10.1126/science.8128228. [DOI] [PubMed] [Google Scholar]
- 8.Citron B A, Davis M D, Milstien S, Gutierrez J, Mendel D B, Crabtree G R, Kaufman S. Proc Natl Acad Sci USA. 1992;89:11891–11894. doi: 10.1073/pnas.89.24.11891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hauer C R, Rebrin I, Thöny B, Neuheiser F, Curtius H C, Hunziker P, Blau N, Ghisla S, Heizmann C W. J Biol Chem. 1993;268:4828–4831. [PubMed] [Google Scholar]
- 10.Hansen L P, Crabtree G R. Curr Opin Genet Dev. 1993;3:246–253. doi: 10.1016/0959-437x(93)90030-s. [DOI] [PubMed] [Google Scholar]
- 11.Bartkowski S, Zapp D, Weber H, Eberle G, Zoidl C, Senkel S, Klein-Hitpass L, Ryffel G U. Mol Cell Biol. 1993;13:421–431. doi: 10.1128/mcb.13.1.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cereghini S. FASEB J. 1996;10:267–282. [PubMed] [Google Scholar]
- 13.Pontoglio M, Barra J, Hadchouel M, Doyen A, Kress C, Bach J P, Babinet C, Yaniv M. Cell. 1996;84:575–585. doi: 10.1016/s0092-8674(00)81033-8. [DOI] [PubMed] [Google Scholar]
- 14.Faust D M, Catherin A M, Barbaux S, Belkadi L, Imaizumi-Scherrer T, Weiss M C. Mol Cell Biol. 1996;16:3125–3137. doi: 10.1128/mcb.16.6.3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamagata K, Oda N, Kaisaki P J, Menzel S, Furuta H, Vaxillaire M, Southam L, Cox R D, Lathrop G M, Boriraj V V, Chen X, Cox N J, Oda Y, Yano H, Le Beau M M, Yamada S, Nishigori H, Takeda J, Bell G I. Nature (London) 1996;384:455–458. doi: 10.1038/384455a0. [DOI] [PubMed] [Google Scholar]
- 16.Mendel D B, Khavari P A, Conley P B, Graves M K, Hansen L P, Admon A, Crabtree G R. Science. 1991;254:1762–1767. doi: 10.1126/science.1763325. [DOI] [PubMed] [Google Scholar]
- 17.Zhao G, Xia T, Song J, Jensen R A. Proc Natl Acad Sci USA. 1994;91:1366–1370. doi: 10.1073/pnas.91.4.1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suck D, Ficner R. FEBS Lett. 1996;389:35–39. doi: 10.1016/0014-5793(96)00573-x. [DOI] [PubMed] [Google Scholar]
- 19.Endrizzi J A, Cronk J D, Wang W, Crabtree G R, Alber T. Science. 1995;268:556–559. doi: 10.1126/science.7725101. [DOI] [PubMed] [Google Scholar]
- 20.Rhee K H, Stier G, Becker P B, Suck D, Sandaltzopoulos R. J Mol Biol. 1997;265:20–29. doi: 10.1006/jmbi.1996.0708. [DOI] [PubMed] [Google Scholar]
- 21.Kim J L, Burley S K. Structure. 1995;3:531–534. doi: 10.1016/S0969-2126(01)00186-1. [DOI] [PubMed] [Google Scholar]
- 22.Ficner R, Sauer U H, Stier G, Suck D. EMBO J. 1995;14:2034–2042. doi: 10.1002/j.1460-2075.1995.tb07195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cronk J D, Endrizzi J A, Alber T. Protein Sci. 1996;5:1963–1972. doi: 10.1002/pro.5560051002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pogge von Strandmann E, Ryffel G U. Development (Cambridge, UK) 1995;121:1217–1226. doi: 10.1242/dev.121.4.1217. [DOI] [PubMed] [Google Scholar]
- 25.Rashtchian A, Thornton C G, Heidecker G. PCR Methods Appl. 1992;2:124–130. doi: 10.1101/gr.2.2.124. [DOI] [PubMed] [Google Scholar]
- 26.Johnen G, Kowlessur D, Citron B A, Kaufman S. Proc Natl Acad Sci USA. 1995;92:12384–12388. doi: 10.1073/pnas.92.26.12384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuo C J, Conley P B, Hsieh C L, Francke U, Crabtree G R. Proc Natl Acad Sci USA. 1990;87:9838–9842. doi: 10.1073/pnas.87.24.9838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hansen L P. Ph.D. thesis. Stanford: Stanford University; 1994. [Google Scholar]
- 29.Huang C Y, Max E E, Kaufman S. J Biol Chem. 1973;248:4235–4241. [PubMed] [Google Scholar]
- 30.Davis M D, Kaufman S, Milstien S. FEBS Lett. 1992;302:73–76. doi: 10.1016/0014-5793(92)80288-r. [DOI] [PubMed] [Google Scholar]
- 31.Köster S, Thöny B, Macheroux P, Curtius H C, Heizmann C W, Pfleiderer W, Ghisla S. Eur J Biochem. 1995;231:414–423. doi: 10.1111/j.1432-1033.1995.tb20714.x. [DOI] [PubMed] [Google Scholar]
- 32.Köster S, Stier G, Ficner R, Holzer M, Curtius H C, Suck D, Ghisla S. Eur J Biochem. 1996;241:858–864. doi: 10.1111/j.1432-1033.1996.00858.x. [DOI] [PubMed] [Google Scholar]
- 33.Nagai K, Oubridge C, Jessen T H, Li J, Evans P R. Nature (London) 1990;348:515–520. doi: 10.1038/348515a0. [DOI] [PubMed] [Google Scholar]
- 34.Oubridge C, Ito N, Evans P R, Teo C H, Nagai K. Nature (London) 1994;372:432–438. doi: 10.1038/372432a0. [DOI] [PubMed] [Google Scholar]
- 35.Burd C G, Dreyfuss G. Science. 1994;265:615–621. doi: 10.1126/science.8036511. [DOI] [PubMed] [Google Scholar]
- 36.Kremer L, Dominguez J E, Avila J. Anal Biochem. 1988;175:91–95. doi: 10.1016/0003-2697(88)90365-x. [DOI] [PubMed] [Google Scholar]
- 37.Ausubel F M, Brent R, Kingston R E, Moore D, Seidman J G, Smith J A, Struhl K, editors. Current Protocols in Molecular Biology. New York: Wiley; 1997. [Google Scholar]
- 38.Sourdive D J, Transy C, Garbay S, Yaniv M. Nucleic Acids Res. 1997;25:1476–1484. doi: 10.1093/nar/25.8.1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rebrin I, Bailey S W, Boerth S R, Ardell M D, Ayling J E. Biochemistry. 1995;34:5801–5810. doi: 10.1021/bi00017a011. [DOI] [PubMed] [Google Scholar]
- 40.Lau F T, Fersht A R. Nature (London) 1987;326:811–812. doi: 10.1038/326811a0. [DOI] [PubMed] [Google Scholar]
- 41.Kowlessur D, Yang X J, Kaufman S. Proc Natl Acad Sci USA. 1995;92:4743–4747. doi: 10.1073/pnas.92.11.4743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Taggart A K, Pugh B F. Science. 1996;272:1331–1333. doi: 10.1126/science.272.5266.1331. [DOI] [PubMed] [Google Scholar]