

Abstract
In this study, we obtained evidence indicating that annexin 1 is a new target of the p38/MAPKAP kinase-2 pathway and that it regulates endothelial cell migration in response to vascular endothelial growth factor (VEGF). These conclusions are supported by a series of substantiating experiments. First, by two-dimensional gel electrophoresis and mass spectrometry, we identified annexin 1 as a protein whose phosphorylation is induced by VEGF and is impaired by inhibiting p38. Second, using in vitro kinase assays and in vivo phosphorylation assays, we found that VEGF-mediated activation of LIM kinase 1 downstream of the p38 pathway triggers the phosphorylation of annexin 1. Third, VEGF-induced cell migration and tube formation in Matrigel are inhibited following small interfering RNA-mediated knockdown of annexin 1. Fourth, both processes are rescued in cells expressing an annexin 1 construct insensitive to the small interfering RNA knockdown. Finally, the VEGF/annexin 1-mediated cell migration is impaired by inhibiting p38. We therefore conclude that phosphorylation of annexin 1 regulates the angiogenic effect that is associated with the activation of the p38/LIM kinase 1 axis by VEGF.
Keywords: Actin, Annexin, Cell Migration, Endothelium, p38 MAPK, Angiogenesis, VEGF
Introduction
Angiogenesis, the sprouting of new capillaries from pre-existing blood vessels, contributes to the expansion of the vascular network in a number of physiological and pathological situations (1). Physiological angiogenesis is a fundamental process that is highly regulated during development and wound repair (2). However, persistent dysregulated angiogenesis is a common denominator and often a causal factor of several diseases, including proliferative retinopathy, rheumatoid arthritis, tumor progression, and metastasis (3, 4). Angiogenesis is a multistep process that involves proteolytic degradation of the extracellular matrix, followed by migration and proliferation of capillary endothelial cells, pericyte recruitment, and assembly of the mature vessel (5). The angiogenic process is regulated by a tight balance between pro- and antiangiogenic agents. For instance, endostatin is a typical antiangiogenic agent, whereas vascular endothelial growth factor (VEGF)3 is the major promoter of both physiological and pathological angiogenesis (5).
VEGFs encompass a family of six structurally related proteins: VEGF-A, VEGF-B, VEGF-C, VEGF-D, VEGF-E, and placenta-derived growth factor (6). Human VEGF-A monomers exist as five different isoforms, of which VEGF165, generally referred to as VEGF, is the most abundant and biological active form (5, 6). VEGF-A165 (herein called VEGF) binds two tyrosine kinase receptors on blood vessel endothelial cells, VEGF receptor-1 (VEGFR-1/Flt-1) and VEGF receptor-2 (VEGFR-2/KDR/Flk-1). Knock-out of both VEGFRs are embryonic lethal, which indicates that both receptors are essential for developmental neovascularization (7, 8). VEGF also binds to neuropilin, a co-receptor for VEGFR-2 that increases the binding affinity of VEGF for VEGFR-2 (9).
VEGFR-2 is the major signaling endothelial cell receptor for VEGF in adults. By activating several kinase pathways, it regulates all the major steps of angiogenesis including endothelial cell proliferation and migration (5, 6). In particular, as previously reported, activation of the stress-activated protein kinase-2/p38 MAP kinase (p38) pathway is a major regulator of actin remodeling in response to VEGF. By contributing to the phosphorylation of heat-shock protein 27 (HSP27), we have suggested that this pathway mediates actin-based motility by regulating cell contractility (10–12). A recent study suggests that phosphorylation of HSP27 downstream of protein kinase D may also contribute to endothelial cell migration in response to VEGF (13).
Annexin 1 (lipocortin 1, calpactin II herein abbreviated as ANXA1) is one of the 13 members of the annexin superfamily, which consists of closely related calcium- and phospholipid-binding proteins expressed in most eukaryotic cell types (14). The members of the annexin family are characterized by a highly conserved protein core domain that harbors the Ca2+ and phospholipid-binding sites. ANXA1 was initially described as a steroid-regulated protein that is involved in transducing the glucocorticoid anti-inflammatory action by inhibiting phospholipase A2 (15). It has also been proposed that exposure of innate immune cells, such as neutrophils, monocytes, macrophages, and mast cells to glucocorticoids induces the release of ANXA1. The latter acts in a paracrine or autocrine manner to maintain the level and duration of cell activation. The final outcome is that the process could promote the resolution of the inflammatory reaction by actively maintaining homeostatic regulation of innate immune cell functions (16). In line with these findings, ANXA1−/− mice show a partial or complete resistance to the anti-inflammatory effects of glucocorticoids (17). ANXA1 further regulates the inflammatory response by mediating the responsiveness of neutrophils to fMet-Leu-Phe (18). On the other hand, several studies have shown that ANXA1, as a target of EGF and hepatocyte growth factor receptor tyrosine kinases and through its implication in the phosphoinositide 3-kinase and ERK MAP kinase glucocorticoid signaling pathway, plays important signaling functions in cell proliferation, differentiation, and apoptosis (14, 19–21). A role of “signal amplifier” has also been attributed to ANXA1 because it promotes the release of second messengers that affect cell proliferation and migration (21). Furthermore, there is a growing body of evidence indicating that ANXA1 may interact with cytoskeletal proteins such as tubulin and actin (22, 23). In addition, the cell invasion potential of the intestinal epithelial cell line SKCO-15 is increased by 2-fold in the presence of full-length ANXA1 and the N-terminal-derived ANXA1 peptide mimetic Ac2-26. This effect seems to be mediated via autocrine/paracrine signaling involving formyl peptide receptors (24). This role of ANXA1 in tumor invasion is consistent with previous reports showing that ANXA1 is associated with metastasis in several invasive malignancies (25–28).
The LIM kinase (Lin-11/Isl-1/Mec-3 domain-containing protein kinase) family contains two members: LIM kinase 1 (LIMK1) and LIM kinase 2 (LIMK2). These serine/threonine kinases are regulated by several signaling pathways downstream of Rho GTPases. In particular, LIMK1 is a target of Rho kinase, p21-activated kinase, and MAPKAP kinase-2 (29, 30, 31). Following its activation, LIMK1 phosphorylates cofilin and destrin to influence the architecture of the actin cytoskeleton (32). In accordance, LIMK1−/− mice show a decrease of cofilin phosphorylation and abnormalities in the actin cytoskeleton that impairs growth cone motility (33, 34). Additionally, LIMK1 regulates the urokinase-type plasminogen activator/urokinase-type plasminogen activator receptor system, which modulates tumor-cell invasion and angiogenesis in breast cancer (35, 36).
In the present study, we have identified ANXA1 as a new target that is phosphorylated downstream of p38 and LIMK1 in endothelial cells activated by VEGF. We further found that migration of endothelial cells and tube formation in Matrigel are significantly inhibited following the siRNA-mediated knockdown of ANXA1. We therefore conclude that ANXA1, by mediating cell migration, is an important modulator of the p38-mediated angiogenic effect of VEGF. These novel findings are consistent with a recent study showing that ANXA1−/− mice are unable to heal the damage associated with indomethacin-induced gastric ulcers (37).
EXPERIMENTAL PROCEDURES
Chemicals
The p38 inhibitor SB 203580 was purchased from Calbiochem (Mississauga, ON, Canada). Protein A-Sepharose and protein G-Sepharose were obtained from GE Healthcare. VEGF was obtained from Sigma-Aldrich (Saint-Louis, MS) and R&D Systems (Minneapolis, MN).
Cells
Human umbilical vein endothelial cells (HUVECs) were isolated by collagenase digestion of umbilical veins from undamaged sections of fresh cords (38). Subcultures were maintained in 199 medium containing 20% heat-inactivated fetal bovine serum, endothelial cell growth supplement (60 μg/ml), gelatin, heparin, and antibiotics. Replicated cultures were obtained by trypsinization and were used at passages < 4. Treatments were done on HUVECs cultivated on gelatin and were made quiescent by incubation for 16–20 h in endothelial cell growth supplement-free medium containing 5% heat-inactivated fetal bovine serum.
Antibodies
Anti-ANXA1 monoclonal mouse antibody was purchased from BD Transduction Laboratories. The anti-mouse IgG-horseradish peroxidase was from The Jackson Laboratory (Bar Harbor, ME). Monoclonal anti-α-tubulin, a mouse IgG1 isotype, was purchased from Sigma. The anti-HA tag (clone 12Ca5) is a mouse monoclonal antibody that was purchased from Roche Applied Science. Anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is a mouse monoclonal antibody purchased from Novus Biologicals (Littleton, CO). Anti-p38 and anti-MAP kinase-activated protein kinase-2 are polyclonal antibodies that were raised in rabbits (38, 39). Anti-LIMK1 polyclonal rabbit antibody was obtained from Cell Signaling Technology (Pickering, ON, Canada). Rabbit IgG (NRS1) was obtained from Dr. Jacques Landry (Laval University). Mouse gamma globulin was purchased from The Jackson Laboratory.
Plasmids, Small Interfering RNA (siRNA), and Adenovirus
Validated ANXA1 siRNA 7 and 8 were purchased from Qiagen (Mississauga, ON, Canada), and siRNA 7 was designed to target the mRNA of human ANXA1 (GenBankTM accession no. NM_000700). The target sequence of ANXA1 siRNA 7 is as follows: sense, 5′-ATGCCTCACAGCTATCGTGAA-3′ and antisense, 5′-TTCACGATAGCTGTGAGGCAT-3′. Control siRNA that targets GAPDH mRNA and siRNA that targets mRNA of human MAP kinase-activated protein kinase-2 were purchased from Ambion (Austin, TX). Validated LIMK1 siRNA 7 was purchased from Qiagen and was designed to target the mRNA of human LIMK1 (GenBankTM accession no. NM_002314). The target sequence of LIMK1 siRNA 7 is as follows: sense, 5′-GCATCTAGGAAGTATTAAA-3′ and antisense, 5′-TTTAATACTTCCTAGATGC-3′. Plasmid pCMV-SPORT6 containing human ANXA1 was obtained from ATCC. Human recombinant ANXA1 (rANXA1) construct was created following subcloning from pCMV-SPORT6 to pET-52b(+) (Novagen, Gibbstown, NJ) between sacI-XmaI restriction sites. pIRES-HA-ANXA1 was generated following subcloning from pCMV-SPORT6 to pIRES-hr- GFP-2a between sacII-xhoI restriction sites. The pIRES-HA-ANXA1 construct was rendered insensitive to ANXA1 siRNA 7 by PCR-mediated mutagenesis at the sites indicated in boldface type in the mutagenic oligonucleotides: 5′-GCCTGACAGCCATAGTCAAGTGCGCC-3′ (sense) and 5′-GCACTTGACTATGGCTGTCAGGCATTTCTCAAT-3′ (reverse). pEGFP-N3 ANXA1 wild-type was obtained following subcloning from pCMV-SPORT6 to pEGF-N3 between SST2-bamH1 restriction sites. The adenovirus expressing a dominant negative mutant form of p38α (p38AGF) was described previously (40, 41). pCMV-p38AGF was a generous gift of Dr. Roger Davis (University of Massachusetts). Adenovirus carrying GFP was given by Dr. Josée N. Lavoie (Laval University, Québec, Canada). Gene and siRNA transfers in HUVECs were done by electroporation as reported previously (42). Co-transfection or co-infection with vectors expressing enhanced green fluorescent protein (EGFP) has allowed us to evaluate transfer efficiency as 30 and 90%, respectively.
Immunoprecipitation and Western Blotting
After treatment, cells were washed with phosphate-buffered saline and were lysed in 250 μl of B buffer containing 150 mm NaCl, 50 mm tris-HCl, pH 7.5, 1% Triton X-100, 0.1% sodium deoxycholate, 2 mm EDTA, 2 mm EGTA, 1 mm NA3VO4, 1 mm benzamide, 1 μm leupeptin, 50 mm NaF, and 1 mm phenylmethylsulfonyl fluoride. Cells were centrifuged at 13,000 rpm for 10 min, and proteins were precleared with 15 μl 50% (v/v) protein G-Sepharose for 45 min. Supernatants were incubated on ice for 16 h with appropriate antibodies. Then, 15 μl of 50% (v/v) protein G-Sepharose was added, and the incubation was extended for 45 min on ice with shaking. Antibody-antigen complexes were washed three times with B buffer and then SDS-PAGE loading buffer was added. Proteins were separated by SDS-PAGE, and the gels were transferred onto nitrocellulose membranes for Western blotting. After incubating nitrocellulose membranes with the appropriate primary antibody, antigen-antibody complexes were detected with an anti-IgG antibody coupled to horseradish peroxidase and then revealed using an enhanced chemiluminescence kit. Quantification of the immunoreactive bands was done by densitometric scanning using the ImageJ software.
Immunocomplex Kinase Assays
HUVECs were serum-starved for 16–20 h and then stimulated with 5 ng/ml VEGF for 5 or 15 min. Thereafter, p38 or LIMK1 were immunoprecipitated and used to determine their ability to phosphorylate human rANXA1 in a buffer that contains 50 μm ATP and 4 μCi of [γ-32P]ATP (3000 Ci/mmol). Recombinant GST-ATF2 was used as a control substrate for p38. The kinase assays were performed for 30 min at 30 °C. The activities of p38 and LIMK1 were quantified by measuring the incorporation of radioactivity into specific substrates after SDS-PAGE fractionation using PhosphorImager (GE Healthcare) (38, 43).
In Vitro Kinase Assay
Direct in vitro kinase assays of MAPKAP kinase-2, MAPKAP kinase-3, and MAPKAP kinase-5 (p38-regulated/activated protein kinase) were performed by adding MAPKAP kinase-2/-3/-5 (Upstate) to rANXA1 or to recombinant HSP27 (rHSP27) for 30 min at 30 °C in the presence of 4 μCi [γ-32P]ATP (3000 Ci/mmol) (42). Thereafter, proteins were run through SDS-PAGE and transferred on nitrocellulose membranes. The activity of the kinases was quantified by measuring the incorporation of radioactivity into the specific substrates using PhosphorImager. The purity of all recombinant substrates was >80%.
In Vivo Phosphorylation Assays
HUVECs were serum-starved for 16–20 h before being incubated in phosphate-free medium for 1 h. The cells were then labeled with H332PO4 (125–200 μCi/ml) for 90 min. Cells were treated and were extracted and immunoprecipitated in IEF buffer (7 m urea, 4% CHAPS, 0.5% IPG buffer 3–10, 20 mm dithiothreitol) for two-dimensional separation or in 250 μl of B buffer for one-dimensional separation.
Two-dimensional Gel Electrophoresis
After in vivo phosphorylation, cells extracted in IEF buffer were run for 16 h with a 18-cm Immobilon Drystrip pH 3–10 on an IPGPhor (GE Healthcare). Thereafter, gels were incubated in 1% dithiothreitol/SDS equilibration buffer (50 mm Tris-HCl, pH 8.8, 6 m urea, 30% glycerol, 2% SDS, 1% dithiothreitol) for 15 min and were incubated for another 15 min in 2.5% iodoacetamide SDS equilibration buffer (50 mm Tris-HCl, pH 8.8, 6 m urea, 30% glycerol, 2% SDS, 2.5% iodoacetamide). Gels were run into 11% SDS-PAGE and then dried and exposed for imaging with PhosphorImager. Control gels that were run similarly, but not dried, were used to cut off the spots of interest of the exposed gels.
Mass Spectrometry Analysis
Protein extracts of cells treated or not treated by VEGF in the presence or not of SB203580 or p38AGF were separated by two-dimensional gel electrophoresis as described above. The spots of interest were excised and digested with trypsin. Tryptic peptides were analyzed on an Elite STR matrix-assisted laser desorption time-of-flight (MALDI-TOF) mass spectrometer (Applied Biosystems, Foster City, CA) with saturated α-cyanocinnamic acid as the matrix. The mass spectrum was acquired in the reflector mode and was internally mass calibrated. The tryptic peptide ions obtained were scanned against the Swiss-Prot and Genpep databases by using the MS-FIT program of Protein Prospector developed by Karl Clauser and Peter Baker.
Cell Migration Assays
HUVECs were electroporated with an siRNA that targets LIMK1 mRNA or with siRNA 7 or 8 that target ANXA1 mRNA in the presence or not in the presence of HA-tagged wild-type ANXA1 not targeted by ANXA1 siRNAs. An siRNA that specifically targets human GAPDH mRNA was used as a control. Co-electroporation of EGFP-expressing plasmid has also been done to evaluate migration of electroporated cells. Forty-eight h later, the cells were made quiescent by serum starvation before use a day later. Then, cells were harvested with trypsin, counted, centrifuged, and resuspended at 1.5 × 106 cells/ml in migration buffer (199 medium, 10 mm HEPES, pH 7.4, 1 mm MgCl2, 0.5% bovine serum albumin). Cells (1.5 × 105) were added on the upper part of 8.0-μm pore size gelatin-coated FluoroBlock migration chambers (Becton Dickinson Labware, Franklin Lakes, NJ), separating the upper and lower chambers of a 6.5-mm transwell as described previously (44). Cells were left to adhere for 1 h. Then, 5 ng/ml VEGF was added in the lower chamber, whereas 1 μm of SB203580 was added or not in the lower and the upper chambers. Four h later, the sum of fluorescent cells that crossed the membrane of a Boyden chamber was calculated from five fields and was used as the measure of migration. All experiments were performed at least three times in duplicates. In each experiment, the log2 of the sum of migrating cells in each condition was divided by the sum of migrating cells in the control condition without VEGF. Student's t test was performed on all of the log2 ratios, and a p value < 0.05 was considered significant. All statistical analyses were performed using R software.
Tubulogenesis in Matrigel
Tube formation assays were performed with commercial growth factor reduced (GFR) Matrigel Matrix obtained from BD Biosciences. HUVECs were electroporated with or without 20 μg HA-tagged wild-type ANXA1 not targeted by ANXA1 siRNA 7 together with 400 pmol of siRNAs that specifically target human GAPDH mRNA or human ANXA1 mRNA (siRNA 7; Qiagen). Co-electroporation of EGFP-expressing plasmid has also been done to evaluate migration of control electroporated cells. Thirty-six h later, the cells were serum-starved for 8 h before being evaluated in the tube formation assay. Briefly, 24-well plates were coated with 200 μl of GFR Matrigel and incubated at 37 °C for 30 min to promote polymerization. Quiescent HUVECs in serum-starved medium, without or without VEGF (5 ng/ml), were added to each well (5 × 104 cells/well). After 8 h of incubation, a wide field, representing an area of 9.4 mm2, was examined for each sample and photographed using contrast microscopy with a 4× objective. Pictures were captured as 16-bit TIFF files and were processed using ImageJ software.
Live Cell Microscopy
HUVECs were electroporated with EGFP-ANXA1 wild-type and were plated on gelatin-coated 35-mm glass bottom microwell dishes (MatTek Corp., Ashland, MA). Twenty-four h later, the cells were made quiescent by serum starvation for 16 h. They were then placed in an incubator of the VivaView LCV110U. Images of green cells were taken every 4 min during 6 h using the time-lapse imaging VivaView LCV110U system equipped with a 40× objective UPLSAPO40X (NA0.95,WD0.18 mm). At the beginning, cells were left untreated for 3 h before the addition of 5 ng/ml VEGF. Images were captured as 16-bit TIFF files, and the supplemental Movie was created using MetaMorph software.
RESULTS
Mass Spectrometry Identification of Annexin 1 (ANXA1) as a Downstream Target of p38 Activated by VEGF
Phosphorylation of HSP27 downstream of the VEGFR2-p38-MAPKAP kinase-2 axis triggers actin polymerization and is an important determinant of cytoskeletal remodeling in endothelial cells (10, 44). To examine whether other targets of this axis contribute to modulating actin remodeling and actin-based motility in endothelial cells, we decided to identify the proteins that were phosphorylated downstream of p38 following activation of primary cultures of HUVECs by VEGF. HUVECs labeled with H332PO4 were pretreated with or without SB203580 (1 μm for 15 min) or infected with adenovirus carrying a dominant negative form of p38α (p38AGF) to inhibit p38 activity and were treated with or without VEGF (5 ng/ml for 15 min). Following two-dimensional gel separation of [32P]-labeled proteins, gels were stained to visualize the spots and were dried and exposed for autoradiography or excised for mass spectrometry analysis. Six proteins were phosphorylated by VEGF in a p38-dependent manner. One protein, with a pI of ∼6.6 and an Mr of ∼38,000, shows a marked increase in phosphorylation level (Fig. 1, A and B). This spot was analyzed by mass spectrometry. Sequence analysis and data base searching identified this protein as ANXA1 with 12 matching peptides representing 36% of the sequence coverage with a total ion score of 102 (Fig. 1C). Interestingly, the inhibition of the ERK pathway was not associated with an inhibition of ANXA1 phosphorylation in response to VEGF (data not shown).
FIGURE 1.

MALDI-TOF identification of annexin 1 as a downstream target of p38 activated in response to VEGF. A, HUVECs from exponentially growing cultures were plated in gelatin-coated Petri dishes before being infected with adenovirus (Adeno) carrying GFP or with adenovirus carrying the dominant negative form of p38 (p38AGF). Forty-eight h later, the quiescent cells were incubated in phosphate-free culture medium in the presence of H332PO4 and were treated or not treated with 5 ng/ml of VEGF for 15 min. Then, proteins were extracted and run into IEF (pH 3–10) gels and in a second dimension into 11% SDS-PAGE prior to analysis by PhosphorImager. B, quiescent HUVECs were labeled with H332PO4 as in A and were pretreated for 15 min with vehicle (dimethyl sulfoxide, 0.01%) or with the p38 inhibitor SB203580 (SB, 1 μm). HUVECs were then treated or not with 5 ng/ml of VEGF for 15 min. Proteins were extracted and separated by two-dimensional gel electrophoresis as described in A. Representative autoradiograms from four separate experiments are shown. The regions from 31 kDa to 45 kDa and from pI ∼ 5.0 to 7.0 in A and B are enlarged below each panel to facilitate visualization of the phosphorylated spot. Representative autoradiograms from four separate experiments are shown. C, in three separate experiments, spot circled in A or B was excised and processed for MALDI-TOF analysis. Data base searching identified the protein as ANXA1 with 12 matching peptides that represent 36% sequence coverage and a total ion score of 102. The identified peptides within ANXA1 sequence are in boldface black letters.
We next proceeded to transfection experiments to further ascertain that ANXA1 was phosphorylated downstream of p38. HUVECs expressing or not expressing wild-type HA-tagged ANXA1 (HA-ANXA1) were incubated with H332PO4 and were either pretreated or not pretreated with SB203580 (1 μm for 15 min) or transfected with p38AGF before being treated by VEGF (5 ng/ml for 15 min). Thereafter, HA-ANXA1 was immunoprecipitated from the [32P]-labeled protein extracts before being run through SDS-PAGE. Results showed that VEGF induced the phosphorylation of HA-ANXA1. Incidentally, we found in five different experiments that VEGF increased the phosphorylation level of ANXA1 by an average of 1.8-fold. The phosphorylation was sensitive to p38 inhibition by SB203580 and by p38AGF, thereby further confirming that ANXA1 was phosphorylated downstream of p38 (Fig. 2, A and B). Interestingly, HA-ANXA1 was not immunoprecipitated by irrelevant antibodies, which supports the specificity of the ANXA1 immunoprecipitation experiments. Overall, these results indicate that ANXA1 is phosphorylated by VEGF downstream of p38 MAP kinase.
FIGURE 2.
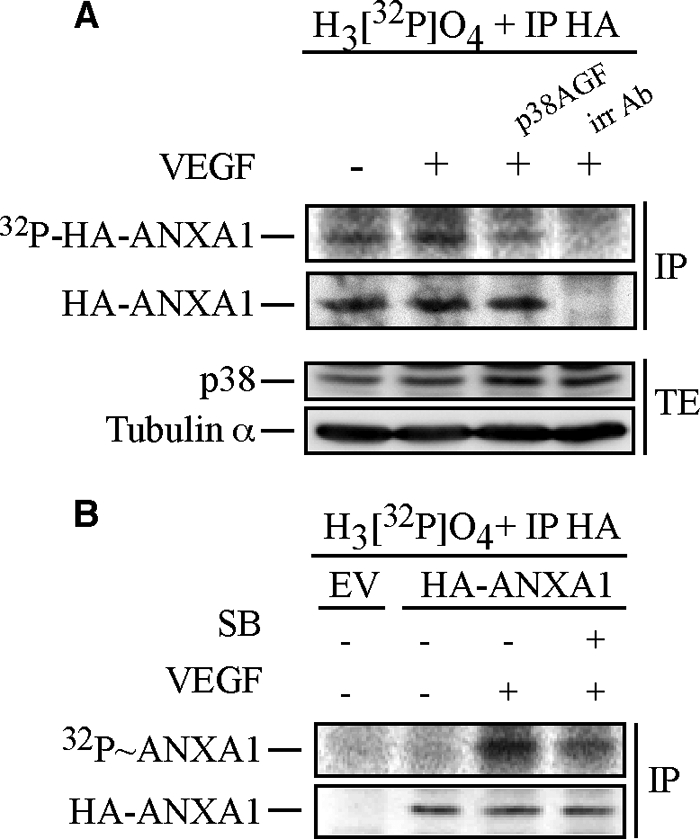
Annexin 1 is phosphorylated via the p38 pathway in response to VEGF. A, HUVECs were co-electroporated with pIRES-hrGFP-2a containing HA-tagged wild-type ANXA1 together with empty vector (EV) or pCMV carrying p38AGF. After 24 h, cells were serum-starved for 16–20 h. Then, quiescent HUVECs were treated or not treated with 5 ng/ml of VEGF for 15 min. Cells were extracted as described under “Experimental Procedures”. The HA-tagged proteins were immunoprecipitated (IP) using anti-HA mouse antibody. Immunoprecipitation using a mouse γ globulin irrelevant antibody (irr Ab) shows the specificity of the anti-HA antibody. Proteins were separated by SDS-PAGE and transferred to a nitrocellulose membrane, and the HA-ANXA1 band was analyzed using PhosphorImager. Thereafter, the membrane was processed for immunodetection of immunoprecipitated HA-ANXA1. Total proteins were kept before immunoprecipitation to monitor p38AGF expression. Tubulin α is also shown as a loading control. B, quiescent HUVECs transiently expressing an empty vector (EV) or HA-tagged wild-type ANXA1 were processed and treated as described in Fig. 1B. Cells were extracted as described under “Experimental Procedures.” After extraction, the HA-tagged proteins were immunoprecipitated using anti-HA mouse antibody. Proteins were separated by SDS-PAGE and transferred to a nitrocellulose membrane. The membrane was then exposed for autoradiography, analyzed using PhosphorImager, and processed for immunodetection of immunoprecipitated HA-ANXA1. Representative blots of at least two separate experiments are shown in A and B. SB, SB203580. TE, total extract.
LIM Kinase 1 Phosphorylates Annexin 1 in Response to VEGF
To determine whether ANXA1 was phosphorylated by p38, HUVECs treated or not treated with 5 ng/ml of VEGF for 5 min were extracted. Thereafter, p38 was immunoprecipitated and incubated in vitro with [γ-32P]ATP in the presence of rANXA1 or of GST-ATF2, a known substrate of p38 (38, 45) (Fig. 3A). Results showed that p38 immunoprecipitated from VEGF-stimulated cells did phosphorylate GST-ATF2 but not rANXA1. These results suggest that p38 does not phosphorylate rANXA1 directly. To search for the ANXA1 kinase downstream of p38, we investigated the potential of MAPKAP kinase-2, MAPKAP kinase-3, and p38-regulated/activated protein kinase (MAPKAP kinase-5), three p38-targeted kinases, to phosphorylate rANXA1. rANXA1 and rHSP27 were incubated with [γ-32P]ATP in the presence or not of purified recombinant active form of the three kinases. As expected, we found that no [32P] was incorporated in rHSP27 or rANXA in the absence of the kinases. However, we found that [32P] incorporation into rHSP27 was induced in the presence of the three kinases (Fig. 3B). In contrast, none of the kinases induced the [32P] incorporation into rANXA1 indicating that they are not ANXA1 kinases (Fig. 3B). Given that LIMK1 has been reported to be a target of MAPKAP kinase-2 in response to VEGF (31), we next investigated whether LIMK1 could phosphorylate ANXA1. HUVECs were treated or not treated with VEGF in the presence or not of SB203580 to inhibit p38 activity. Thereafter, LIMK1 was immunoprecipitated and allowed to react with rANXA1 in the presence of [γ-32P]-ATP. We found that LIMK1 immunoprecipitated from VEGF-treated cells phosphorylates rANXA1 in an SB203580-sensitive manner, suggesting that it was an ANXA1 kinase downstream of p38 (Fig. 4A, left and right panels). Of note, an isotypic irrelevant antibody fails to precipitate LIMK1, which supports the specificity of the anti-LIMK antibody used to precipitate LIMK1 (Fig. 4A, left panel). We next confirmed the role of LIMK1 as an ANXA1 kinase in vivo by showing that the siRNA-mediated knockdown of the kinase (40%) was associated with an inhibition of ANXA1 phosphorylation in response to VEGF (Fig. 4B). As expected, the siRNA-mediated knockdown of MAPKAP kinase-2, which activates LIMK1 in response to VEGF, also inhibits ANXA1 phosphorylation (Fig. 4C) (31). Overall, these findings suggest that LIMK1 phosphorylates ANXA1 in response to VEGF and suggest that the phosphorylation is downstream of the p38/MAPKAP-kinase-2 axis.
FIGURE 3.

p38 and its downstream kinases, MAPKAP kinase-2, MAPKAP kinase-3, and MAPKAP kinase-5, do not phosphorylate annexin 1 in response to VEGF. A, quiescent HUVECs were left untreated or stimulated with VEGF (5 ng/ml for 5 min). After treatments, cells were lysed, and p38 was immunoprecipitated (IP) using rabbit polyclonal antibody and subjected to an in vitro kinase assay. Reaction mixtures for kinase assay were put in the presence of [γ-32P]ATP. p38 activity was determined in immunocomplex assays using 2 μg recombinant GST-ATF2 as control substrate or 5 μg rANXA1. Protein mixtures were separated through SDS-PAGE and were transferred to a nitrocellulose membrane. Kinase activity was then quantified by autoradiography using the PhosphorImager system by measuring [32P] incorporation into the specific substrates. Membrane was also processed for immunodetection of immunoprecipitated p38. Representative results from three distinct experiments, each realized in duplicates, are shown. Total recombinant GST-ATF2 and total rANXA1 are shown as internal controls for the amount of added substrates. B, commercial purified and activated recombinant MAPKAP kinase-2 (rMK2), MAPKAP kinase-3 (rMK3) and MAPKAP kinase-5 (rMK5) were used in an in vitro kinase assay. As in A, reaction mixtures for kinase assays were put in the presence of [γ-32P]ATP, and kinase activities were determined using 1 μg of rHSP27 as a control substrate and 5 μg rANXA1. Protein mixtures were separated through SDS-PAGE. Kinase activities were then quantified by autoradiography using PhosphorImager. Total rHSP27 and total rANXA1 are shown as internal controls for the amount of added substrates. Control tracks containing all kinase assay compounds, except activated kinases, are also shown. Representative results of three separate experiments are shown.
FIGURE 4.

Immunoprecipitated LIMK1, activated in response to VEGF, phosphorylates annexin 1 downstream of MAPKAP kinase-2. A, quiescent HUVECs were pretreated or not pretreated for 1 h with the p38 inhibitor SB203580 (SB, 1 μm). HUVECs were then treated or not treated with VEGF (5 ng/ml for 15 min). After treatments, cells were lysed, and LIMK1 was immunoprecipitated (IP) using rabbit polyclonal antibody and was subjected to an in vitro kinase assay. Reaction mixtures for kinase assays were put in the presence of [γ-32P]ATP, and LIMK1 activity was determined in immunocomplex assays using 5 μg rANXA1. Protein mixtures were separated through SDS-PAGE and were transferred to a nitrocellulose membrane. Kinase activity was then quantified by autoradiography using the PhosphorImager system by measuring [32P] incorporation into rANXA1. Membrane was also processed for immunodetection of immunoprecipitated LIMK1. Total rANXA1 is shown as an internal control for the amount of added substrate. A control track containing all kinase assay compounds except immunoprecipitated kinase is shown (Control). Immunoprecipitation using a rabbit isotype irrelevant antibody (Irr Ab) shows the specificity of the anti-LIMK1 antibody. In vitro phosphorylation of ANXA1 by LIMK1 was obtained in five distinct experiments. Representative results of those experiments are shown. In B, HUVECs were co-electroporated with siRNAs that target human GAPDH or LIMK1 mRNAs (200 pmol of siRNA) together with pIRES-hrGFP-2a containing HA-tagged wild-type ANXA1 construct. In C, HUVECs were co-electroporated with siRNAs that target human GAPDH or MAPKAP kinase-2 (MK2) mRNAs (80 pmol of siRNA) along with pIRES-hrGFP-2a containing HA-tagged wild-type ANXA1 construct. After 48 h, in B and C, cells were serum-starved for 16–20 h before being assayed for in vivo phosphorylation, as described under “Experimental Procedures.” Cells were treated or not treated with VEGF (5 ng/ml for 15 min). Thereafter, proteins were extracted, and the HA-tagged proteins were immunoprecipitated and processed as described in Fig. 2B. Incorporation of [32P] into HA-ANXA1 (upper panel) and immunoprecipitated HA-ANXA1 (second panel) are shown. The efficiency of the siRNA knockdown of LIMK1 and GAPDH (B) or MAPKAP kinase-2 and GAPDH (C) was determined (lower two panels) in Western blotting as described above using the total cell extracts. Representative results of at least three separate experiments are shown in B and C. MK2, MAPKAP kinase-2.
Phosphorylation of Annexin 1 Regulates Endothelial Cell Migration and Tubulogenesis in Matrigel
Given the determinant role played by the p38 pathway in VEGF-induced endothelial cell migration, we next examined the role of ANXA1 in mediating endothelial cell migration in response to VEGF. HUVECs were electroporated or not with siRNAs (7 and 8 from Qiagen) that knock down the expression of ANXA1 and with or without a HA-tagged ANXA1 construct insensitive to the siRNA (Fig. 5). Thereafter, cells were put in the upper part of a Boyden chamber, and VEGF was added to the lower part. In some experiments, SB203580 was added to both chambers to inhibit p38 activity. After 4 h, the number of electroporated cells that have migrated across the polycarbonate membranes separating the two chambers was counted. Results showed that VEGF increased by 2.5-fold the migration of control HUVECs. This increase was completely abolished in cells in which the expression of ANXA1 had been knocked down (Fig. 5), indicating that ANXA1 was instrumental in regulating endothelial cell migration. In accordance, the VEGF-induced increase in cell migration was maintained in HUVECs expressing an HA-tagged version of ANXA1 that was rendered insensitive to the siRNA directed against ANXA1 mRNA. In this latter case, the rescued cell migration was impaired by SB203580 suggesting that ANXA1 required phosphorylation downstream of p38 to drive cell migration. We next verified whether it regulates endothelial cell migration in response to VEGF. HUVECs were electroporated or not with siRNA that knocks down the expression of LIMK1. Thereafter, cell migration was evaluated in a Boyden chamber as above. Consistent with our result showing that LIMK1 is an ANXA1 kinase, we found that VEGF-induced cell migration was abolished in cells in which the expression of LIMK1 has been knocked down by siRNA (Fig. 6).
FIGURE 5.

Annexin 1 is required for p38-mediated endothelial cell migration in response to VEGF. HUVECs were co-electroporated, with siRNA to knock down human GAPDH or ANXA1 mRNAs (400 pmol of siRNA) along with pEGFP-C1 and pIRES-hrGFP-2a containing HA-tagged wild-type ANXA1 not targeted by ANXA1 siRNA. After 48 h, cells were serum-starved for 16–20 h before being treated or not treated with SB203580 (1 μm) and treated or not treated with VEGF (5 ng/ml for 4 h) Thereafter, cell migration was assayed in a modified Boyden chamber, as described under “Experimental Procedures.” The efficiency of siRNA knockdown was determined in Western blotting of the total cell extract (three right panels). The results of cell migration are expressed as the ratio of the number of fluorescent endothelial cells that have crossed the membrane in each experimental condition over the number of fluorescent cells that have crossed the membrane in untreated conditions (mean number = 23 cells). Data points represent the mean ratio with 95% confidence intervals. p was determined by Student's t test. Representative results of three separate experiments are shown. SB, SB203580.
FIGURE 6.
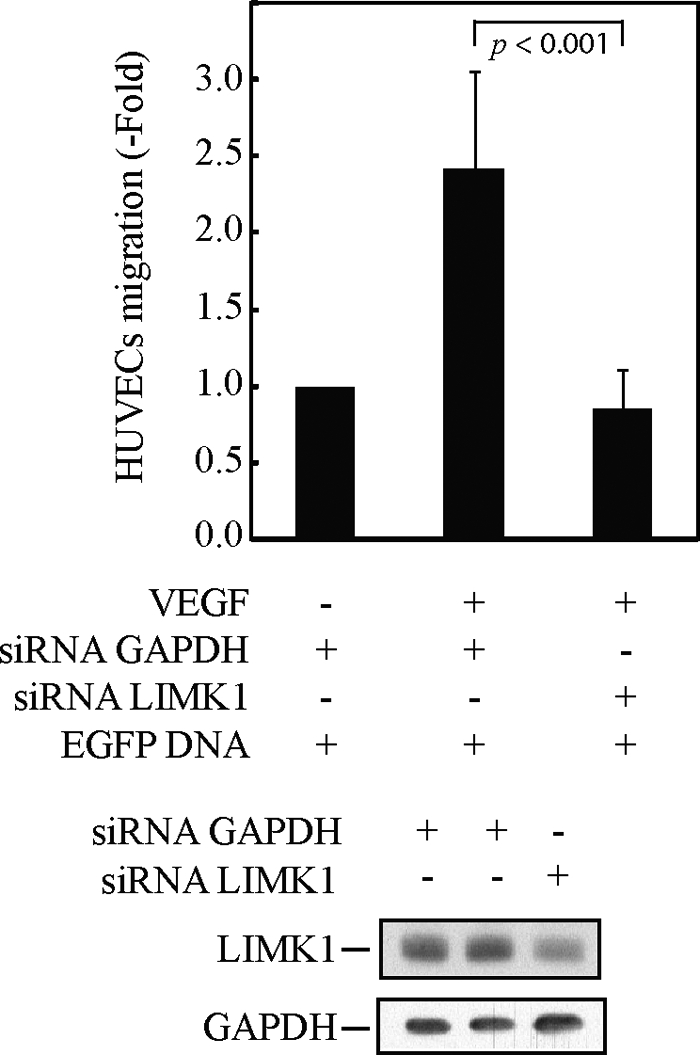
LIMK1 is required for p38-mediated endothelial cell migration in response to VEGF. HUVECs were co-electroporated, with siRNA to knock down human GAPDH or LIMK1 mRNAs (200 pmol of siRNA) along with pEGFP-C1. After 48 h, cells were serum-starved for 16–20 h before being treated or not treated with VEGF (5 ng/ml for 4 h) and being assayed for migration in a modified Boyden chamber, as described under “Experimental Procedures.” The efficiency of siRNA knockdown was determined in Western blotting using the total cell extracts (lower two panels).The results of cell migration are expressed as the ratio of the number of fluorescent endothelial cells that have crossed the membrane in each experimental condition over the number of fluorescent cells that have crossed the membrane in untreated conditions (mean number = 12 cells). Data points represent the mean ratio with 95% confidence intervals. p was determined by Student's t test. Representative results of three separate experiments are shown.
To further ascertain the involvement of ANXA1 in endothelial cell migration and angiogenesis, we examined its role in the formation of capillary-like tubes by endothelial cells in Matrigel (5). HUVECs were electroporated or not with siRNA that knocks down the expression of ANXA1 and with or without an HA-tagged ANXA-1 construct insensitive to the siRNA. Thirty-six h later, the cells were serum-starved for 8 h. Then, the quiescent cells were embedded in Matrigel and were treated or not treated for 8 h with VEGF. Thereafter, the formation of tubes was examined under phase contrast microscopy. Results show that VEGF triggers the formation of a dense network of tubules when added to control HUVEC in Matrigel (Fig. 7, A and B). This effect was markedly repressed in cells in which the expression of ANXA1 has been knocked down (Fig. 7, C and D). Importantly, the increase in tube formation was maintained in cells that expressed an ANXA1 construct insensitive to siRNA knockdown (Fig. 7, E and F). Incidentally, in line with the role of LIMK1 as an ANXA1 kinase, it has previously been reported that inhibiting LIMK1 with a dominant negative form impairs VEGF-induced tube formation (31). Taken together, these results suggest that phosphorylation of ANXA1 is required to sustain endothelial cell migration and angiogenesis.
FIGURE 7.

Expression of annexin 1 is required for tubulogenesis in Matrigel. Tube formation assays were performed with commercial growth factor-reduced (GFR) Matrigel Matrix. HUVECs were electroporated with 400 pmol siRNAs that specifically target human GAPDH mRNA (A and B) or with 400 pmol siRNAs that specifically target human ANXA1 mRNA (C and D). In E and F, HUVECs were co-electroporated with 20 μg HA-tagged wild-type (WT) ANXA1 insensitive to ANXA1 siRNA together with siRNAs that specifically target human ANXA1 mRNA. A schematic representation of the formed tubules is shown in the middle panels to facilitate interpretation. Tubulogenesis assays were performed as described under “Experimental Procedures.” Fields from each sample were photographed using a contrast microscopy with a 4× objective. Representative results of three separate experiments are shown.
DISCUSSION
Chemotactic endothelial cell migration is a major process by which VEGF regulates angiogenesis. It involves the binding of VEGF to VEGFR-2 and the downstream activation of several signaling pathways that converge on actin remodeling (5). In the present study, we show for the first time that ANXA1 is phosphorylated downstream of the VEGFR-2/p38/MAPKAP kinase-2/LIMK1 pathway, which in turn is required to sustain endothelial cell migration and capillary-like tube formation.
The identification of ANXA1 as a target of the p38 pathway has been obtained by mass spectrometry analysis of two-dimensional gel electrophoresis spots that were phosphorylated by VEGF in control HUVECs but not in HUVECs treated with SB203580 or infected with adenovirus carrying p38AGF to inhibit p38α activity. Thereafter, we obtained further experimental evidence supporting the point that ANXA1 was phosphorylated downstream of p38 by showing that the in vivo phosphorylation of transfected HA-tagged ANXA1 in response to VEGF was inhibited by blocking the activity of p38 with SB203580 or p38AGF. Interestingly, previous studies have indicated that ANXA1 is phosphorylated on Tyr and Ser/Thr residues by a variety of kinases including EGF and hepatocyte growth factor receptor tyrosine kinases and protein kinase C (14, 19–21). The involvement of p38 as an ANXA1 kinase has also been suggested because SB203580 inhibits the protective function of the N-terminal fragment of ANXA1 (Anx-1-(2–26)) against metabolic injury (46). However, we found that immunoprecipitated p38 from VEGF-activated HUVECs does not phosphorylate ANXA1 in vitro in contrast to ATF2. Hence, this indicates that p38 is not the kinase that directly phosphorylates ANXA1 in response to VEGF in vivo.
One of the major finding of our study is the evidence we have provided indicating that LIMK1 is an ANXA1 kinase downstream of p38. It is supported by key experimental observations. First, none of the direct p38 targets (MAPKAP kinase-2/-3/-5) could directly phosphorylate rANXA1 in vitro, whereas they all phosphorylated their known substrate HSP27 (38, 47, 48). Conversely, immunoprecipitated LIMK1 from VEGF-treated-HUVECs phosphorylates rANXA1, and the activation is sensitive to SB203580. Second, in vivo phosphorylation of ANXA1 is abolished in cells in which the expression of LIMK1 is knocked down by siRNA directed against human LIMK1 mRNA. Incidentally, this proves the in vivo relevance of LIMK1 in phosphorylating ANXA1. Third, phosphorylation of ANXA1 is inhibited by knocking down MAPKAP kinase-2, a known activator of LIMK1 (31).
Very little is known concerning the role of phosphorylated ANXA1. However, previous studies have shown that its phosphorylation by the tyrosine kinase activity of EGFR regulates ERK activation and cell proliferation (19). Phosphorylation of ANXA1 has also been proposed to regulate the G2/M transition (49). In addition, its phosphorylation by kinases, including protein kinase C, is essential for its role in regulating the anti-inflammatory effects of glucocorticoids (14). Moreover, as reported above, the phosphorylation of ANXA1 within its N-terminal domain confers protective functions against metabolic injury in cardiomyocytes (46). Interestingly, EGF-induced phosphorylation of ANXA1 triggers its colocalization with F-actin in the lamellipodia (50). Given the important role that lamellipodia play in conferring forward movements to endothelial cells during their migration (5), this observation is consistent with our finding that ANXA1 mediates VEGF-induced endothelial cell migration. Incidentally, we also found that, in both immunofluorescence and live cell microscopy, ANXA1 colocalized with F-actin in the lamellipodia in basal and VEGF-induced conditions (data not shown and supplemental data). On the other hand, the inhibition of cell migration associated with the knockdown of ANXA1 can be rescued in a p38-dependent manner by expressing an ANXA1 form insensitive to the knockdown. Taken together, these findings are consistent with the interpretation that ANXA1 should be present in the lamellipodia to sustain cell migration but that the triggering event is induced by its phosphorylation. The identification of the serine/threonine sites phosphorylated by LIMK1 within ANXA1 should allow us to address this question by using an Ala mutant of the site(s).
Intriguingly, we previously reported that p38-MAPKAP kinase-2 may regulate VEGF-induced endothelial cell migration by stimulating HSP27 phosphorylation and formation of stress fibers necessary to throw the rear of the cells toward its front (5, 42–44). Our present findings thus suggest that p38 has a dual complementary function in endothelial cell migration. By contributing to the phosphorylation of HSP27 via MAPKAP-kinase 2, p38 mediates the formation of stress fibers and cell contractility that throws the rear of the cell to the front. By contributing to the phosphorylation of ANXA1 via LIMK1, p38 allows lamellipodia-dependent swimming of the cell forward. Interestingly, a recent study has identified ANXA1 as a possible target of hypoxia-inducible factor-1 that confers migration and invasion to cancer cells (51). Consistent with the role of ANXA1 in endothelial cell migration, we obtained evidence indicating that the expression of ANXA1 is required for tubulogenesis in Matrigel because the knockdown of ANXA1 is associated with a decrease in tube formation in response to VEGF. This finding is further strengthened by the fact that the siRNA used to target ANXA1 mRNA does not inhibit the formation of tubes when expressed together with an HA-ANXA1 construct that is insensitive to its action. It has been demonstrated that the tubular structures formed by endothelial cells in this model are vascular-like structures containing lumens (52). Hence, our findings suggest that LIMK1-mediated phosphorylation of ANXA1 is an important determinant of angiogenesis. Intriguingly, null mice for MAPKAP kinases-2/-3/-5 and ANXA1 develop normally and do not show apparent defect in vasculature (17, 53). This indicate that the p38 pathway may not be required for developmental angiogenesis. Yet, our results suggest that phosphorylation of ANXA1 by LIMK1 may regulate pathological angiogenesis. This possibility is consistent with a recent study showing that ANXA1−/− mice are unable to heal the damage associated with indomethacin-induced gastric ulcers (37).
Overall, we report in this study that ANXA1 is a new target of the p38/MAPKAP kinase-2/LIMK1 axis activated in response to VEGF and that its phosphorylation triggers endothelial cell migration and tubulogenesis in Matrigel. These findings are important because they highlight a novel function of ANXA1 as a potential regulator of pathological angiogenesis.
Supplementary Material
Acknowledgments
We thank Dr. Nick Morrice from Dundee for helpful contribution to the mass spectrometry analysis. We also thank Dr. Jacques Landry for providing anti-p38 and anti-MAPKAP kinase-2 antibodies. We are indebted to Dr. Kristopher Valerie for giving adenoviral constructs containing CMV p38AGF. We thank Dr. Pierre-Luc Tremblay for having amplified the adenoviral constructs containing GFP and p38AGF, and Dr. Josée N. Lavoie for providing adenovirus carrying GFP. We also thank Carl St-Pierre for help with microscopy, Jean-Yves Masson for giving the pET52b(+) vector, and Éric Paquet for the statistical analysis. We also thank Samantha Benlolo for help in the English language editing of this manuscript.
This work was supported by grants from The Canadian Institutes of Health Research and the Heart Stroke Foundation of Canada (Qc).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Movie 1 and data.
- VEGF
- vascular endothelial growth factor
- ANXA1
- annexin A1
- EGF
- epidermal growth factor
- ERK
- extracellular signal-regulated kinase
- GAPDH
- glyceraldehyde-3-phosphate-dehydrogenase
- HSP27
- Heat-shock protein 27
- HA
- hemagglutinin
- HUVEC
- human umbilical vein endothelial cell
- MAP kinase
- mitogen-activated protein kinase
- rANXA1
- recombinant ANXA1
- VEGFR-2
- vascular endothelial growth factor receptor-2
- EGFP
- enhanced green fluorescent protein
- CHAPS
- 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid
- MALDI-TOF
- matrix-assisted laser desorption time-of-flight
- GST
- glutathione S-transferase
- MAPKAP kinase-2
- MAPK-activated protein kinase-2
- hr
- human recombinant.
REFERENCES
- 1.Breier G., Risau W. (1996) Trends Cell Biol. 6, 454–456 [DOI] [PubMed] [Google Scholar]
- 2.Folkman J., Shing Y. (1992) J. Biol. Chem. 267, 10931–10934 [PubMed] [Google Scholar]
- 3.Risau W. (1997) Nature 386, 671–674 [DOI] [PubMed] [Google Scholar]
- 4.Otrock Z. K., Mahfouz R. A., Makarem J. A., Shamseddine A. I. (2007) Blood Cells Mol. Dis. 39, 212–220 [DOI] [PubMed] [Google Scholar]
- 5.Lamalice L., Le Boeuf F., Huot J. (2007) Circ. Res. 100, 782–794 [DOI] [PubMed] [Google Scholar]
- 6.Olsson A. K., Dimberg A., Kreuger J., Claesson-Welsh L. (2006) Nat. Rev. Mol. Cell Biol. 7, 359–371 [DOI] [PubMed] [Google Scholar]
- 7.Fong G. H., Rossant J., Gertsenstein M., Breitman M. L. (1995) Nature 376, 66–70 [DOI] [PubMed] [Google Scholar]
- 8.Shalaby F., Rossant J., Yamaguchi T. P., Gertsenstein M., Wu X. F., Breitman M. L., Schuh A. C. (1995) Nature 376, 62–66 [DOI] [PubMed] [Google Scholar]
- 9.Soker S., Takashima S., Miao H. Q., Neufeld G., Klagsbrun M. (1998) Cell 92, 735–745 [DOI] [PubMed] [Google Scholar]
- 10.Rousseau S., Houle F., Landry J., Huot J. (1997) Oncogene 15, 2169–2177 [DOI] [PubMed] [Google Scholar]
- 11.Rousseau S., Dolado I., Beardmore V., Shpiro N., Marquez R., Nebreda A. R., Arthur J. S., Case L. M., Tessier-Lavigne M., Gaestel M., Cuenda A., Cohen P. (2006) Cell Signal. 18, 1897–1905 [DOI] [PubMed] [Google Scholar]
- 12.Lamalice L., Houle F., Jourdan G., Huot J. (2004) Oncogene 23, 434–445 [DOI] [PubMed] [Google Scholar]
- 13.Evans I. M., Britton G., Zachary I. C. (2008) Cell Signal 20, 1375–1384 [DOI] [PubMed] [Google Scholar]
- 14.Lim L. H., Pervaiz S. (2007) FASEB J. 21, 968–975 [DOI] [PubMed] [Google Scholar]
- 15.Flower R. J., Rothwell N. J. (1994) Trends Pharmacol. Sci. 15, 71–76 [DOI] [PubMed] [Google Scholar]
- 16.Perretti M., D'Acquisto F. (2009) Nat. Rev. Immunol. 9, 62–70 [DOI] [PubMed] [Google Scholar]
- 17.Roviezzo F., Getting S. J., Paul-Clark M. J., Yona S., Gavins F. N., Perretti M., Hannon R., Croxtall J. D., Buckingham J. C., Flower R. J. (2002) J. Physiol. Pharmacol. 53, 541–553 [PubMed] [Google Scholar]
- 18.Rescher U., Danielczyk A., Markoff A., Gerke V. (2002) J. Immunol. 169, 1500–1504 [DOI] [PubMed] [Google Scholar]
- 19.Alldridge L. C., Harris H. J., Plevin R., Hannon R., Bryant C. E. (1999) J. Biol. Chem. 274, 37620–37628 [DOI] [PubMed] [Google Scholar]
- 20.Solito E., Mulla A., Morris J. F., Christian H. C., Flower R. J., Buckingham J. C. (2003) Endocrinology 144, 1164–1174 [DOI] [PubMed] [Google Scholar]
- 21.Skouteris G. G., Schröder C. H. (1996) J. Biol. Chem. 271, 27266–27273 [DOI] [PubMed] [Google Scholar]
- 22.Hayes M. J., Rescher U., Gerke V., Moss S. E. (2004) Traffic 5, 571–576 [DOI] [PubMed] [Google Scholar]
- 23.Traverso V., Morris J. F., Flower R. J., Buckingham J. (1998) J. Cell Sci. 111, 1405–1418 [DOI] [PubMed] [Google Scholar]
- 24.Babbin B. A., Lee W. Y., Parkos C. A., Winfree L. M., Akyildiz A., Perretti M., Nusrat A. (2006) J. Biol. Chem. 281, 19588–19599 [DOI] [PubMed] [Google Scholar]
- 25.Wu W., Tang X., Hu W., Lotan R., Hong W. K., Mao L. (2002) Clin. Exp. Metastasis 19, 319–326 [DOI] [PubMed] [Google Scholar]
- 26.Jiang D., Ying W., Lu Y., Wan J., Zhai Y., Liu W., Zhu Y., Qiu Z., Qian X., He F. (2003) Proteomics 3, 724–737 [DOI] [PubMed] [Google Scholar]
- 27.Bai X. F., Ni X. G., Zhao P., Liu S. M., Wang H. X., Guo B., Zhou L. P., Liu F., Zhang J. S., Wang K., Xie Y. Q., Shao Y. F., Zhao X. H. (2004) World J. Gastroenterol. 10, 1466–1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cicek M., Samant R. S., Kinter M., Welch D. R., Casey G. (2004) Clin. Exp. Metastasis 21, 149–157 [DOI] [PubMed] [Google Scholar]
- 29.Ding Y., Milosavljevic T., Alahari S. K. (2008) Mol. Cell. Biol. 28, 3742–3756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dohn M. R., Brown M. V., Reynolds A. B. (2009) J. Cell Biol. 184, 437–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kobayashi M., Nishita M., Mishima T., Ohashi K., Mizuno K. (2006) EMBO J. 25, 713–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scott R. W., Olson M. F. (2007) J. Mol. Med. 85, 555–568 [DOI] [PubMed] [Google Scholar]
- 33.Meng Y., Zhang Y., Tregoubov V., Janus C., Cruz L., Jackson M., Lu W. Y., MacDonald J. F., Wang J. Y., Falls D. L., Jia Z. (2002) Neuron 35, 121–133 [DOI] [PubMed] [Google Scholar]
- 34.Endo M., Ohashi K., Sasaki Y., Goshima Y., Niwa R., Uemura T., Mizuno K. (2003) J. Neurosci. 23, 2527–2537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bagheri-Yarmand R., Mazumdar A., Sahin A. A., Kumar R. (2006) Int. J. Cancer 118, 2703–2710 [DOI] [PubMed] [Google Scholar]
- 36.Yoshioka K., Foletta V., Bernard O., Itoh K. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 7247–7252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martin G. R., Perretti M., Flower R. J., Wallace J. L. (2008) Am. J. Physiol. Gastrointest. Liver Physiol. 294, G764–G769 [DOI] [PubMed] [Google Scholar]
- 38.Huot J., Houle F., Marceau F., Landry J. (1997) Circ. Res. 80, 383–392 [DOI] [PubMed] [Google Scholar]
- 39.Huot J., Lambert H., Lavoie J. N., Guimond A., Houle F., Landry J. (1995) Eur. J. Biochem. 227, 416–427 [DOI] [PubMed] [Google Scholar]
- 40.Taher M. M., Oakley J. D., Hershey C., Valerie K. (2000) Biochemistry 39, 1709–1715 [DOI] [PubMed] [Google Scholar]
- 41.Chouinard N., Valerie K., Rouabhia M., Huot J. (2002) Biochem. J. 365, 133–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Le Boeuf F., Houle F., Sussman M., Huot J. (2006) Mol. Biol. Cell 17, 3508–3520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rousseau S., Houle F., Kotanides H., Witte L., Waltenberger J., Landry J., Huot J. (2000) J. Biol. Chem. 275, 10661–10672 [DOI] [PubMed] [Google Scholar]
- 44.Rousseau S., Houle F., Huot J. (2000) Trends Cardiovasc. Med. 10, 321–327 [DOI] [PubMed] [Google Scholar]
- 45.Raingeaud J., Gupta S., Rogers J. S., Dickens M., Han J., Ulevitch R. J., Davis R. J. (1995) J. Biol. Chem. 270, 7420–7426 [DOI] [PubMed] [Google Scholar]
- 46.Ritchie R. H., Gordon J. M., Woodman O. L., Cao A. H., Dusting G. J. (2005) Br. J. Pharmacol. 145, 495–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.New L., Jiang Y., Zhao M., Liu K., Zhu W., Flood L. J., Kato Y., Parry G. C., Han J. (1998) EMBO J. 17, 3372–3384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stokoe D., Caudwell B., Cohen P. T., Cohen P. (1993) Biochem. J. 296, 843–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alldridge L. C., Bryant C. E. (2003) Exp. Cell Res. 290, 93–107 [DOI] [PubMed] [Google Scholar]
- 50.Campos-Gonzalez R., Kanemitsu M., Boynton A. L. (1990) Cell Motil. Cytoskeleton 15, 34–40 [DOI] [PubMed] [Google Scholar]
- 51.Liao S. H., Zhao X. Y., Han Y. H., Zhang J., Wang L. S., Xia L., Zhao K. W., Zheng Y., Guo M., Chen G. Q. (2009) Proteomics 9, 3901–3912 [DOI] [PubMed] [Google Scholar]
- 52.Tong Q., Zheng L., Li B., Wang D., Huang C., Matuschak G. M., Li D. (2006) Exp. Cell Res. 312, 3559–3569 [DOI] [PubMed] [Google Scholar]
- 53.Gaestel M. (2006) Nat. Rev. Mol. Cell Biol. 7, 120–130 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.