

Abstract
The IQ-motif protein PEP-19, binds to the C-domain of calmodulin (CaM) with significantly different kon and koff rates in the presence and absence of Ca2+, which could play a role in defining the levels of free CaM during Ca2+ transients. The initial goal of the current study was to determine whether Ca2+ binding to sites III or IV in the C-domain of CaM was responsible for affecting the kinetics of binding PEP-19. EF-hand Ca2+-binding sites were selectively inactivated by the common strategy of changing Asp to Ala at the X-coordination position. Although Ca2+ binding to both sites III and IV appeared necessary for native-like interactions with PEP-19, the data also indicated that the mutations caused undesirable structural alterations as evidenced by significant changes in amide chemical shifts for apoCaM. Mutations in the C-domain also affected chemical shifts in the unmodified N-domain, and altered the Ca2+ binding properties of the N-domain. Conversion of Asp93 to Ala caused the greatest structural perturbations, possibly due to the loss of stabilizing hydrogen bonds between the side chain of Asp93 and backbone amides in apo loop III. Thus, although these mutations inhibit binding of Ca2+, the mutated CaM may not be able to support potentially important native-like activity of the apoprotein. This should be taken into account when designing CaM mutants for expression in cell culture.
Keywords: Biophysics, Calcium, Calcium/Binding Proteins, Calcium/Calmodulin, Methods/NMR, Protein/Conformation, Protein/Ligand Binding, Protein/Metal Ion Interaction
Introduction
PEP-19 is a small (62 amino acids) protein that binds to calmodulin (CaM)2 in the presence or absence of Ca2+ (1) via an IQ CaM binding motif. Although PEP-19 has no known intrinsic activity other than binding to CaM, expression of PEP-19 can protect cells from apoptosis (2, 3), and cell death from Ca2+ overload or excitotoxicity (3). We showed that PEP-19 binds preferentially to the C-domain of CaM and greatly affects the kinetics of Ca2+ binding to the C-domain (4, 5). We also showed that this effect requires the synergistic activities of the IQ motif and an adjacent acidic sequence, and that the rates of association and dissociation of PEP-19 from apoCaM are much slower than for Ca2+-CaM (6). These data support a role for PEP-19 in CaM signaling that involves both modulation of Ca2+ binding to CaM, and inhibition of other proteins that rely on binding to the C-domain of CaM.
The goal of the current study was to use CaM mutants with modified Ca2+-binding loops to determine whether Ca2+ binding to sites III or IV has dominant effects on the properties of PEP-19 binding to CaM. Two strategies have been used to inhibit Ca2+ binding to EF-hand motifs in CaM. One involves conversion of Glu to Gln, Lys, or Ala at position 12 in the Ca2+-binding loops (7, 8) (see Fig. 1 for the consensus Ca2+-binding loop for CaM). The other involves conversion of Asp to Ala at position 1 (9). This latter mutation was used previously to inactivate EF-hand Ca2+-binding sites in cardiac troponin C (10), skeletal troponin C (11), and myosin regulatory light chain (12). Mutation of position 1 has been used extensively to assess the relative functional contribution of Ca2+ binding to the N- and C-domain of CaM by expression of the mutated proteins in cells (13–31). Thus, we elected to convert Asp to Ala in the first ligand position in all CaM Ca2+-binding mutants. The proteins CaM1,2, CaM3, CaM4, and CaM3,4 correspond to mutations: D20A/D56A, D93A, D129A, and D93A/D129A, respectively.
FIGURE 1.

Consensus EF-hand Ca2+-binding loop and mutation strategy. The consensus Ca2+-binding loop for the 4 EF-hand Ca2+-binding motifs of CaM consists of 12 amino acids (32). Positions 1 to 6 adopt a coil secondary structure, 7 to 9 form a short β-strand, and 10 to 12 adopt an α-helix. Ca2+ is coordinated by side chain oxygens at X, Y, and −Z. The carbonyl oxygens of Glu at the −Z provide bidentate coordination. Water provides a hydrogen bond link to oxygens of side chains at Z and −X. Calcium is coordinated by the backbone amide at −Y. An invariant Gly is at position 6. As described in the text, we elected to inactivate Ca2+ binding to CaM by converting Asp to Ala at position Asp93 in site III (CaM3), Asp129 in site IV (CaM4), both 93 and 129 (CaM3,4), or both 20 and 56 (CaM1,2).
Our initial experiments indicated that Ca2+ binding to both sites III and IV in the C-domain of CaM are necessary for native-like interactions with PEP-19. However, the data also pointed to undesirable structural alterations due to mutation of Asp93 and Asp129 to Ala. For example, the affinity of apoCaM and apoCaM3,4 for PEP-19 should be the same if the mutations do not greatly alter the structure of CaM, however, apoCaM3,4 and the other apo mutant CaM proteins had lower affinity for PEP-19 than native apoCaM. Thus, we investigated the structural consequence of these mutations. The data show that, whereas conversion of Asp93 and Asp129 to Ala effectively inhibits Ca2+ binding to sites III and IV, the resulting mutated C-domain exhibits extensive structural perturbations. This has implications for cell expression studies because the C-domain of CaM3,4 may not support important native-like activities of the apo C-domain of CaM.
EXPERIMENTAL PROCEDURES
Materials
[15N]NH4Cl was obtained from Cambridge Isotope Laboratories, Inc. (Andover, MA). Quin-2 (free acid), 1-anilinonaphthalene-8-sulfonic acid (1,8-ANS), and 5-((((2- iodoacetyl)amino)ethyl)amino)naphthalene-1-sulfonic acid (1,5-IAEDANS) were purchased from Invitrogen. N-(4-Dimethylamino-3,5-dinitrophenyl)maleimide (DDPM) was purchased from Alfa Aesar.
Expression and Purification of CaM Proteins
A pET23 expression vector with a mammalian CaM cDNA optimized for bacterial codon usage was kindly provided by Andy Hudmon at the Indiana School of Medicine. QuikChange® II XL Site-directed Mutagenesis Kit (Stratagene) was used to generate all mutant proteins. Calcium binding was inhibited by mutation of the X Ca2+ coordination position (see Fig. 1), which is the first of 12 amino acids in a canonical EF-hand Ca2+-binding loop (32). In addition, a panel of Ca2+-binding mutants with Lys75 converted to Cys as described previously (33) was generated for labeling with fluorescent probes. A cDNA encoding human PEP-19 was synthesized with optimal bacterial codon usage (DNA2.0 Inc.) and subcloned into a pET23 expression vector. PEP-19 with a C-terminal Gly-Cys extension was used for labeling with the FRET acceptor, DDPM as described previously (5). All constructs were transformed into BL21 DE3 or BL21 DE3 pLys-S host cells for expression of protein with or without [15N]NH4Cl and proteins were purified as described previously (4, 33).
Equilibrium Binding and Koff of CaM Mutants with PEP-19
A FRET-based assay described previously (5) was used to measure equilibrium binding of CaM to PEP-19. Typically, a solution of 1.0 μm 1,5-IAEDANS-labeled CaM(K75C) with 10.0 μm DDPM-labeled PEP-19 was prepared in a base buffer containing 20 mm MOPS, pH 7.5, 100 mm KCl, with either 2 mm CaCl2 or 1 mm EGTA. A concentrated stock solution of unlabeled native CaM (control) or CaM mutant in the same solution was used to titrate the complex. Apparent IC50 values were derived by plotting the fluorescence response versus log(competitor) and fitting to the following equation.
 |
Where Fmin and Fmax are fluorescence in the absence or large excess of competitor.
Stopped-flow fluorescence was used to determine the dissociation rate (Koff) of the CaM mutants from PEP-19. Briefly, syringe A contained a solution of 1 μm IAEDANS-labeled CaM mutant and 10 μm DDPM-labeled PEP-19 in base buffer. Syringe B contained buffer only. The final concentration of these reagents in the optical chamber was one-half of these values resulting from the 1:1 mixing ratio.
Measuring Calcium Binding Affinity of CaM Mutants
The relative equilibrium Ca2+ binding affinities of the various CaM mutants were determined using Ca2+-dependent Tyr fluorescence, and the dye 1,8-ANS, which fluoresces upon binding to hydrophobic surfaces exposed by Ca2+ binding to CaM. Briefly, solutions contained 4 μm CaM, 50 mm MOPS, pH 7.5, 100 mm KCl, 1 mm EGTA, 1 mm HEDTA, and 1 mm NTA with or without 4 μm 1,8-ANS. The level of total Ca2+ needed to achieve a desired free Ca2+ in the presence of the above chelators was calculated using WEBMAXC EXTENDED. Calcium was added from a concentrated stock made in the above buffer such that CaM and 1,8-ANS were not diluted during the titration. Temperature was set at 23 °C. Fluorescence data were collected with a QuantaMaster fluorimeter (Photon Technology International) using excitation wavelengths of 276 for Tyr and 370 for 1,8-ANS. Emission was integrated from 290 to 320 nm for Tyr, and from 430 to 520 nm for 1,8-ANS. Data were fit to a Hill equation with one or two binding components as follows.
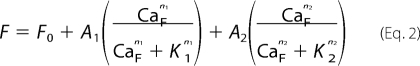 |
Where F0 is the fluorescence without Ca2+, CaF is the free Ca2+ level, A is the amplitude of change due to a given binding event, n is the Hill coefficient, and K is the KCa50.
Stopped-flow Measurements
Experiments were performed at 23 °C using an Applied Photophysics Ltd. (Leatherhead, UK) model SX20 MV sequential stopped-flow spectrofluorimeter with a 150 watt Xe/Hg lamp. The system was configured with two 3-ml syringes (A and B) and has a dead time of 1.7 ms, all solutions contained a base buffer of 20 mm MOPS, pH 7.5, 100 mm KCl. Three independent fluorescence indicators were used to measure the rate of dissociation (koff) of Ca2+ from CaM or CaM mutants; intrinsic tyrosine, 1,8-ANS, and Quin-2. For Tyr fluorescence, syringe A contained 4 μm CaM and 0.1 mm CaCl2, whereas syringe B contained either 10 mm EGTA or 0.1 mm CaCl2 as a negative control for each CaM. The optical/mixing chamber was excited at 276 nm and fluorescence was collected using Oriel filter 51662. Changes in fluorescence obtained with Ca2+ in syringe B were subtracted from data obtained with EGTA in the syringe. Experiments using 1,8-ANS were performed as for Tyr, but with 4 μm 1,8-ANS included in syringe A. 1,8-ANS was excited at 366 nm and fluorescence was collected using a 430-nm cutoff filter (Oriel 51282). Measurements using Quin-2 included 4 μm CaM and 30–60 μm CaCl2 in syringe A, and 200–400 μm Quin-2 in syringe B. The samples were excited at 334.5 nm and fluorescence was collected using a 430-nm cutoff filter (Oriel 51282). Data collected using all three fluorescent markers fit well to an equation with a single exponential rate constant. Fluorescence from Quin-2 was calibrated by replacing CaM in syringe A with 2, 4, 6, 8, or 10 μm EGTA. The amplitude of the slow rate of release of Ca2+ from EGTA (koff = 0.7 s−1) could be readily used to calibrate the fluorescence response.
NMR Methodology
All NMR experiments were collected on a DRX 600 MHz spectrometer instrument using a 5-mm TXI Cryoprobe at 298 K for apo samples and 310 K for Ca2+ samples. The data were processed using FELIX 2007 software. Resonance assignments were made by comparison to wild-type CaM. For experiments requiring decalcified proteins, proteins were decalcified by adding 1 to 5 mm 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid followed by desalting on a Bio-Gel P-10 column equilibrated in buffers that were decalcified by passage over a Calcium Sponge column (Molecular Probes). The decalcified proteins were lyophilized and resuspended in decalcified buffer containing 10 mm imidazole, 100 mm KCl, 5% D2O at pH 6.3. Calcium was added to 20 mm to the [15N]apoCaM mutants and the pH adjusted accordingly. Chemical shift perturbation was calculated using the following equation.
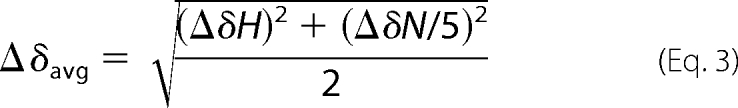 |
Where ΔδH is the change in 1H chemical shift and ΔδN is the change in 15N chemical shift.
RESULTS
Mutation at Ca2+-binding Sites Alters Binding of PEP-19 to CaM
Fig. 1 shows the consensus sequence of a canonical EF-hand Ca2+-binding loop (32), which consists of 12 amino acids that provide 7 coordination positions for Ca2+. All mutated CaMs used in this study have this Asp converted to Ala at position 1 as indicated by the arrowhead. Nomenclature for mutant CaM proteins was adopted from the literature (34). For example, CaM3,4 indicates that Asp93 and Asp129 were changed to Ala at the X coordination position of Ca2+-binding loops III and IV, respectively.
We first determined the relative affinity of PEP-19 for native CaM and the CaM mutant proteins, with a focus on CaM3, CaM4, and CaM3,4 because PEP-19 binds selectively to the C-domain of CaM. CaM with Lys75 changed to Cys was labeled with the FRET donor 1,5-IAEDANS (CaMD), and PEP-19 with a C-terminal Gly-Cys extension was labeled with the FRET acceptor DDPM (PEP-19A). The relative affinity of the CaM mutants for PEP-19 in the presence or absence of Ca2+ was determined by their ability to compete with CaMD for binding to PEP-19A. Because PEP-19 binds to CaM with similar affinity in the presence or absence of Ca2+ (6), we anticipated that inactivation of Ca2+-binding sites would have little effect on binding affinity. However, Fig. 2 shows that only Ca2+-CaM3 had similar affinity for PEP-19 as native Ca2+-CaM (see Fig. 2A), all other mutants had lower affinity in either the presence or absence of Ca2+ as indicated by a rightward shift in the binding curves. Results shown in Fig. 2B in the absence of Ca2+ are particularly interesting. If the mutations inhibit or prevent Ca2+ binding to a given EF-hand in CaM, but do not greatly alter the structure of CaM in the absence of Ca2+, then all mutant proteins should have similar affinity for PEP-19 in the apo forms. In contrast, the apo forms of all mutant proteins have lower affinity for PEP-19 relative to native apoCaM.
FIGURE 2.

Relative affinity of PEP-19 for native CaM and Ca2+-binding mutants. The relative affinity of CaMs for PEP-19 was determined using a competition FRET assay in which IAEDANS-labeled native CaM (donor) bound to DDPM-labeled PEP-19 (acceptor) was displaced by increasing concentrations of the indicated unlabeled CaM or the indicated mutant proteins. Under these conditions, effective competition with IAEDANS-labeled CaM results in an increase in fluorescence. In the presence of Ca2+ (panel A), only CaM3 has an affinity for PEP-19 that is comparable with that of native CaM. In the absence of Ca2+ (panel B), CaM3, CaM4, and CaM3,4 all show lower affinity for PEP-19 than native CaM. Sample conditions for all assays included 1 μm IAEDANS-labeled CaM, 10 μm DDPM-labeled PEP-19, in 20 mm MOPS, pH 7.5, 100 mm KCl with 1 mm EGTA or 2 mm CaCl2. The lines indicate fits to the equations described under “Experimental Procedures” to derive IC50 values.
We showed previously that the kon and koff rates for binding PEP-19 to Ca2+-CaM are too rapid to measure using a stopped-flow fluorimeter (6). Fig. 3A shows that mutation of sites III, IV, or both III and IV greatly decrease the rate of dissociation of PEP-19 from CaM in the presence of Ca2+. This indicates that Ca2+ binding to both sites III and IV are necessary to allow a fast rate of dissociation of PEP-19 from CaM.
FIGURE 3.

Rate of dissociation of PEP-19 from native CaM versus Ca2+-binding mutants. Dissociation of CaM from PEP-19 was determined using a stopped-flow fluorimeter as described under “Experimental Procedures.” Essentially, donor-labeled CaM (1 μm) bound to acceptor-labeled PEP-19 (10 μm) was rapidly diluted to achieve partial dissociation of the complex, and an increase in fluorescence. Syringe A contained labeled CaM (1 μm) and labeled PEP-19 (10 μm) with 1 mm EGTA or 2 mm CaCl2. Syringe B contained 1 mm EGTA or 2 mm CaCl2, but no proteins. Panel A shows the dissociation in the presence of Ca2+ (2 mm CaCl2). Dissociation from native Ca2+-CaM is too fast to detect using a stopped flow with a 1.7-ms dead time. Panel B shows dissociation in the absence of Ca2+ (1 mm +EGTA).
In contrast to Ca2+-CaM, both kon and koff rates for binding PEP-19 to apoCaM are slow and can be easily measured. Rates for mutant proteins should be identical to CaM if mutation of the Ca2+-binding loops does not alter the structure of CaM, however, Fig. 3B shows this is true only for CaM4. The koff rates for CaM3 and CaM3,4 are 3- to 4-fold greater than native CaM. Based on the data in Figs. 2B and 3B, the kon rates (μm−1 s−1) for binding PEP-19 are: CaM = 0.36; CaM3 = 0.45; CaM4 = 0.15; CaM3,4 = 0.59. Thus, differences in affinity of binding PEP-19 to the mutant proteins can result from changes in kon or koff.
Mutations in the C-domain Affect Ca2+ Binding to the N-domain
The data in Figs. 2 and 3 suggest that conversion of Asp93 and/or Asp129 to Ala causes undesirable structural perturbations in CaM. Thus, we performed a series of experiments to characterize the mutant proteins, especially CaM3,4. We first compared the Ca2+ dependence of fluorescence from Tyr99 and Tyr138 in the C-domain of CaM. Fig. 4A shows that CaM and CaM1,2 respond similarly to Ca2+, indicating that mutations in the N-domain do not greatly affect Ca2+-dependent Tyr fluorescence from the C-domain. Interestingly, Tyr fluorescence from both CaM3,4 and CaM3 showed little response to Ca2+, but had increased basal Tyr fluorescence in the absence of Ca2+. This suggests a more hydrophobic environment for Tyr98 and Tyr138 in the apo forms of CaM3 and CaM3,4 relative to native apoCaM.
FIGURE 4.

Calcium-binding mutations alter intrinsic Tyr fluorescence and 1,8-ANS binding. Panel A shows Tyr fluorescence in the presence and absence of Ca2+. Solutions contained 4 μm CaM, 50 mm MOPS, pH 7.5, 100 mm KCl, 1 mm EGTA or 1 mm CaCl2. Panel B shows fluorescence from 1,8-ANS, which fluoresces upon binding to hydrophobic surfaces. Conditions were the same as in panel A but with 4 μm 1,8-ANS.
We next compared exposure of hydrophobic surfaces in CaM and mutant proteins using 1,8-ANS, which fluoresces upon binding to hydrophobic surfaces. Fig. 4B shows much greater fluorescence from 1,8-ANS in the presence of Ca2+-CaM versus apoCaM. In contrast, there is little change in 1,8-ANS fluorescence when Ca2+ is added to CaM1,2, indicating that the increase in 1,8-ANS fluorescence in the presence of native CaM is primarily due to Ca2+ binding to sites I and II in the N-domain. This is confirmed by CaM3,4, which does not bind Ca2+ to sites III and IV based on Tyr fluorescence (see Fig. 4A), but has a robust Ca2+-dependent increase in ANS fluorescence upon binding Ca2+. The increase is much greater than observed with native CaM, suggesting that mutation of sites III and IV in the C-domain alters exposure of hydrophobic surfaces in response to Ca2+ binding to sites I and II in the N-domain. This is also observed for CaM3, but to a much lesser extent with CaM4.
Relative Ca2+ binding affinities were determined by monitoring Tyr or 1,8-ANS fluorescence during titration of CaM and mutant derivatives with Ca2+. The results shown in Fig. 5 and Table 1 can be interpreted with respect to binding Ca2+ to the C- and N-lobes with KCa50 values of around 2 and 13 μm, respectively (5, 35, 36). Tyr fluorescence detects a single class of Ca2+-binding sites in native CaM with KCa50 of 2.5 μm, which corresponds with Ca2+ binding to the C-domain. Fluorescence data for native CaM collected using 1,8-ANS fit well to a single class of binding sites with a KCa50 of 5.2 μm. We interpret this KCa50 to reflect binding to sites I and II because Fig. 4 shows that 1,8-ANS is most sensitive to Ca2+ binding to the N-domain. The lower KCa50 relative to the native CaM may be due to a contribution to fluorescence from the C-domain, or an increase in Ca2+ binding affinity of the N-domain due to 1,8-ANS binding.
FIGURE 5.

Equilibrium Ca2+ binding to native and mutant CaM. Fluorescence from Tyr (panel A) and 1,8-ANS (panel B) was used to measure equilibrium Ca2+ binding to CaM. Solutions contained 4 μm CaM, 50 mm MOPS, pH 7.5, 100 mm KCl, 1 mm EGTA, 1 mm HEDTA, 1 mm NTA with or without 4 μm 1,8-ANS. The level of total Ca2+ needed to achieve a desired free Ca2+ in the presence of the above chelators was calculated using WEBMAXC EXTENDED. Data are expressed as a percent of maximal fluorescence for each protein to normalize for differences in absolute maximal fluorescence shown in Fig. 4. Lines represent fits of the data to a Hill equation with one or two binding components as described under “Experimental Procedures.”
TABLE 1.
Equilibrium calcium binding
Data such as that shown in Fig. 5 were fit to Hill equations with one or two Ca2+-binding components. All values are the average ± S.D. of at least three independent titrations. S.D. values of less than 0.1 are reported as 0.1.
| Method |
KCa50 (μm) and Hill coefficient (n) |
||||
|---|---|---|---|---|---|
| K1 | n1 | K2 | n2 | ||
| CaM | Tyr | 2.5 ± 0.7 | 2.1 ± 0.1 | —a | — |
| ANS | 5.2 ± 0.5 | 1.5 ± 0.1 | — | — | |
| CaM1,2 | Tyr | 2.4 ± 0.5 | 2.1 ± 0.1 | — | — |
| ANS | 1.9 ± 0.1 | 1.9 ± 0.1 | — | — | |
| CaM3,4 | Tyr | — | — | — | — |
| ANS | 2.1 ± 0.1 | 2.3 ± 0.1 | — | — | |
| CaM3 | Tyr | 1.0 ± 0.1 | 2.0 ± 0.1 | 33 ± 7 | 1.3 ± 0.1 |
| ANS | 2.2 ± 0.1 | 2.0 ± 0.1 | 30 ± 5 | 1.4 ± 0.1 | |
| CaM4 | Tyr | 10 ± 2 | 2.2 ± 0.4 | 44 ± 10 | 1.3 ± 0.2 |
| ANS | 4.1 ± 0.1 | 2.2 ± 0.1 | 53 ± 3 | 1.5 ± 0.1 | |
a Indicates either no detectable signal or that the data fit well to an equation with a single binding component.
Both Tyr and 1,8-ANS fluorescence detect a single class of Ca2+-binding sites in CaM1,2 with a KCa50 of about 2 μm, which is consistent with Ca2+ binding to the C-domain. Although CaM3,4 shows no Ca2+-dependent change in Tyr fluorescence, 1,8-ANS detects a single binding component with a KCa50 of 2.1 μm. This is due to Ca2+ binding to the N-domain because NMR data show that Ca2+ does not bind to the C-domain of CaM3,4. The KCa50 of 2.1 μm is less than for the N-domain of native CaM by about 11 μm (5, 35, 36). Thus, mutation of Ca2+ binding sites III and IV increases the affinity of Ca2+-binding to sites I and II in the N-domain.
Fig. 5B and Table 1 show that CaM3 and CaM4 have biphasic Ca2+ binding curves. Although values derived using Tyr and 1,8-ANS fluorescence are not identical, the trends are clear. Barring some unusual cooperative interactions between N- and C-domain EF-hands, the simplest interpretation of the data is that the higher affinity site(s) associated with a large increase in 1,8-ANS fluorescence is due to binding Ca2+ at sites I and II in the N-domain of CaM3 and CaM4 because the Hill coefficient is close to 2. The lower affinity class of binding sites has a KCa50 of 30–50 μm, and Hill coefficient of less than 2. This is consistent with Ca2+ binding to the remaining active site in the C-domain of CaM3 and CaM4, which would have lower affinity due to the absence of cooperativity (6).
Regardless of the microscopic assignment of Ca2+-binding sites, Fig. 5 and Table 1 demonstrate interdomain effects of mutations. First, fluorescence from Tyr99 and Tyr138 in the C-domain, which is insensitive to Ca2+ binding to the N-domain of native CaM, responds to Ca2+ binding to the N-domain of CaM3 and CaM4 because biphasic Ca2+ binding curves are observed. Second, the apparent affinity of Ca2+ binding to sites I and II is increased for CaM3 and CaM3,4 relative to native CaM.
Effect of Ca2+-binding Mutations on Rates of Ca2+ Dissociation
We next determined the effect of mutations on the rates of Ca2+ dissociation (koff) using stopped-flow techniques to monitor fluorescence from 1,8-ANS, Tyr, and Quin-2. Data obtained with 1,8-ANS are shown in Fig. 6, and rates obtained with all methods are shown in Table 2.
FIGURE 6.

Effect of mutations on the rates of Ca2+ dissociation. Dissociation of Ca2+ (koff) from the various CaMs was measured using fluorescence from 1,8-ANS, Tyr, and Quin-2 as described under “Experimental Procedures.” All fluorescent markers gave very similar koff values as shown in Table 2. The arrow in panel A indicates very rapid release of Ca2+ from CaM, which occurs in the dead-time of the stop flow, indicating a koff of >500.
TABLE 2.
Calcium dissociation rate constants
All values are the average ± S.D. of at least three independent data sets. S.D. values of less than 0.1 are reported at 0.1. Moles of Ca2+ released per mol of protein determined using Quin-2 were based on calibrating the fluorescence response with Ca2+ release from EGTA.
| Method | koff | Ca2+/protein | |
|---|---|---|---|
| s−1 | |||
| CaM | Quin-2 | 9.5 ± 0.2 | 2.0 ± 0.1 |
| 1,8-ANS | 10.5 ± 0.1 | ||
| Tyr | 10.3 ± 0.4 | ||
| CaM1,2 | Quin-2 | 9.7 ± 0.1 | 2.0 ± 0.1 |
| 1,8-ANS | 12.5 ± 0.2 | ||
| Tyr | 9.8 ± 1.1 | ||
| CaM3,4 | Quin-2 | 296 ± 1 | 1.9 ± 0.1 |
| 1,8-ANS | 270 ± 9 | ||
| Tyr | NDa | ||
| CaM3 | Quin-2 | 282 ± 11 | 1.9 ± 0.1 |
| 1,8-ANS | 217 ± 6 | ||
| Tyr | ND | ||
| CaM4 | Quin-2 | 59 ± 3 | 1.6 ± 0.1 |
| 1,8-ANS | 52 ± 2 | ||
| Tyr | 40 ± 2 |
a ND indicates no detectable change in Tyr fluorescence.
The vertical arrow in Fig. 6A shows a rapid decrease of fluorescence from 1,8-ANS during the dead time of the stopped-flow fluorimeter when Ca2+-CaM is rapidly mixed with EGTA. This is consistent with rapid dissociation of Ca2+ from the N-domain of CaM, which is too fast to be detected by stopped-flow methods. This fast phase is not seen for CaM1,2, because both N-domain sites are inactivated. Both CaM and CaM1,2 show a slower dissociation of 2 Ca2+/mol of protein with similar rates of 10–12 s−1, which is consistent with release of Ca2+ from sites III and IV in the C-domain. These data, together with Table 1, show that mutation of sites I and II do not affect Ca2+ binding to sites III and IV.
Very similar data were obtained using CaM3,4 and CaM3. Neither protein showed a detectable change in Tyr fluorescence, indicating an insensitivity to Ca2+ release, or that the change occurs too rapidly to measure using stopped-flow techniques. Data collected with Quin-2 and 1,8-ANS fit single exponential decays with rates of between 200 and 300 s−1, and stoichiometry of 2 Ca2+/mol of protein. The simplest explanation for these data is that Quin-2 and 1,8-ANS detect dissociation of Ca2+ from the N-domain of CaM. This would mean that mutation of C-domain Ca2+-binding sites decreases the rate of Ca2+ dissociation from the N-domain, and would account for the increase in affinity of Ca2+ binding to the N-domain in CaM3 and CaM3,4 shown in Table 1.
Dissociation of Ca2+ from CaM4 shows a single rate of 40 to 60 s−1. There are two possibilities for this result. One is that the observed rate is associated with Ca2+ release from the active site III in CaM4, and that release of Ca2+ from sites I and II in the N-lobe is too rapid to detect. The other possibility is that release of Ca2+ from the active site III is too fast to be observed, and that the observed rate is associated with dissociation of Ca2+ from the N-domain, which is sensed by Tyr residues due to mutation of site IV in CaM4. The latter seems more plausible because: 1) low affinity binding of Ca2+ to site III would likely be associated with a rapid rate of dissociation; 2) rapid release of Ca2+ from the N-lobe would be detected as a large decrease of 1,8-ANS fluorescence during the dead time of the stopped-flow fluorimeter, but this is not observed; and 3) the stoichiometry of Ca2+ release is consistent with release of 2 Ca2+ from the N-lobe rather than 1 Ca2+ from site III.
Together, the data in Figs. 5 and 6 and Tables 1 and 2 indicate that mutation of Ca2+-binding sites III and IV in the C-domain can affect Ca2+ binding to sites I and II in the N-domain. This is most evident for CaM3,4 because it does not bind Ca2+ to the C-domain, and exhibits Ca2+ binding to the N-domain with higher affinity and slower koff rate than native CaM.
NMR Spectra Indicate Altered Apo Structure in CaM3,4 and Interdomain Effects of Mutations
NMR was used to assess potential structural changes due to conversion of Asp93 and Asp129 to Ala. Fig. 7A compares a region of the 1H-15N HSQC spectra for CaM (gray) and CaM3,4 (black) collected in the absence of Ca2+ (see supplemental Fig. S1 for complete spectra). Cross-peaks for amides in the N-domain of CaM3,4 overlap with those of CaM, but amides in the C-domain (residues 76–148) of CaM3,4 do not overlap with those of native CaM due to severe exchange broadening or large differences in chemical shifts. Fig. 7B shows that major differences are also seen in the crowded, central region of the spectra. Almost all C-domain resonances are significantly affected by changing Asp93 and Asp129 to Ala, and could not be assigned by comparison to amide chemical shifts for native CaM.
FIGURE 7.

Mutation of Asp93 and Asp129 destabilizes the C-domain of CaM. Fig. 7 compares selected regions of 1H-15N HSQC spectra collected for native CaM (gray) and CaM3,4 (black) in the absence of Ca2+. The upper panel shows that amide resonances for residues in the N-domain of CaM3,4 (residues 1–75) are very similar to those of CaM, but amides for residues in the C-domain (residues 76–148) are greatly affected by the mutations. The lower panel shows that major differences are also seen in the crowded central region of the spectra.
Overall, C-terminal amide resonances in apoCaM3,4 show a loss of dispersion that is consistent with a loss of tertiary structure, or increased disorder in the C-domain due to the mutations. An immediate question is whether conversion of Asp93 or Asp129 to Ala is most responsible for this structural change? A quantitative answer would require extensive heteronuclear experiments for backbone assignments. Instead, we compared 15N HSQC spectra for apo-native CaM, CaM3, and CaM4, and focused on C-domain amides that are in well resolved regions of the spectra. This type of analysis shows that mutation of Asp93 in site III causes the greatest structural change because amide cross-peaks for all of the following residues overlap in the spectra for apoCaM and apoCaM4, but are significantly shifted for apoCaM3: 91, 92, 95, 96, 97, 100, 101, 102, 109, 110, 113, 117, 118, 119, 127, 139, 146, 147, and 148. The “Discussion” will provide a plausible explanation for the greater structural perturbation due to mutation of Asp93 in site III.
The majority of N-lobe amide assignments for the mutant proteins could be made by comparison with native CaM, however, resonances in the N-domain were affected by mutations in the C-domain. Fig. 8 shows chemical shift differences between native apoCaM and the apo forms of CaM3, CaM4, and CaM3,4. Chemical shifts for amides in site II are most affected by mutations in sites III and IV, especially Asn60 and Gly61 in CaM3, CaM4, and CaM3,4, and Asp64 in CaM3,4 shown by the gray bars. These data are consistent with Figs. 4–6, which show that Ca2+ binding to the N-domain is altered as a consequence of mutations in the C-domain.
FIGURE 8.

Mutation of Asp93 and Asp129 affects the conformation of the apo N-domain. The majority resonances for backbone amides in the N-domain of the mutant CaM proteins could be assigned by comparison of 1H-15N HSQC spectra of mutant proteins to that of native CaM. Panels A–C compare chemical shift differences between native apoCaM versus apoCaM3, -CaM4, and -CaM3,4. The differences are small but measurable, and the greatest differences are localized to Ca2+-binding loops I and II. The insets indicate positions of residues in the solution structure of the apo N-domain that have amide chemical shift differences greater than 0.02.
Fig. 6 suggests that mutation of sites III and IV in the C-domain of CaM affects the structure of the Ca2+-bound N-domain because the rate of dissociation of Ca2+ from the N- domain of CaM3,4 is significantly slower than for native CaM. To further investigate this, the amide chemical shifts of CaM3,4 were followed during titration with Ca2+ and compared with native CaM. Fig. 9 shows chemical shift differences for residues that could be confidently assigned in the Ca2+-bound N-domain of CaM3,4 versus native CaM. Observed differences are greater in magnitude than for the apoproteins shown in Fig. 8, and the greatest changes are clustered in the hydrophobic pocket as shown by the inset. A change in exposure of hydrophobic surfaces in the Ca2+-bound N-domain due to mutations in the C-domain is consistent with the increased ANS fluorescence from CaM3,4 shown in Fig. 4B.
FIGURE 9.

Interdomain effects of Ca2+ binding to CaM3,4. Amide chemical shifts differences are shown for the N-domain of CaM3,4 versus native CaM in the presence of Ca2+. The insets indicate positions of residues in the solution structure of the N-domain that have amide chemical shift differences greater than 0.08 in CaM3,4 relative to native Ca2+-CaM. The greatest differences are localized to the hydrophobic pocket.
DISCUSSION
The original goal of this study was to determine whether Ca2+ binding to sites III or IV in the C-domain of CaM has dominant effects on the characteristics of binding PEP-19. The two most common strategies to inhibit Ca2+ binding to EF-hand motifs are: 1) convert Glu at loop position 12 to Gln, Lys, or Ala (7, 8) (see Fig. 1); and 2) convert Asp at loop position 1 to Ala (9). The latter mutation was used previously to inactivate Ca2+ binding to cardiac troponin C (10), skeletal troponin C (11), myosin regulatory light chain (12), and CaM (9). Mutation at loop position 1 in CaM has been used extensively to assess the relative functional contribution of Ca2+ binding to the N- and C-domains of CaM when expressed in cell culture (13–31). Thus, we elected to convert Asp to Ala in the first ligand position in all Ca2+-binding mutants. Although the data in Figs. 2 and 3 indicate that Ca2+ binding to both sites III and IV of CaM is important for native-like interactions with PEP-19, apparent structural perturbations induced by the mutations complicated interpretation of the data. This led us to characterize the effect of these mutations on the Ca2+ binding properties and structure of CaM.
Data in Figs. 5 and 6 and Tables 1 and 2 show that mutation of Ca2+-binding sites in the C-domain increases the Ca2+ binding affinity of the N-domain by decreasing the rate of Ca2+ dissociation, but that mutation of sites in the N-domain has no apparent effect on the C-domain. This interdomain effect is consistent with other studies showing that mutation of the 1st or 12th ligand positions in sites III and IV decreased the rate of Ca2+ dissociation from the N-domain (37, 38), and that mutation of Val136 to Gly in site IV increases the Ca2+ binding affinity of the N-domain (39).
NMR analyses in Figs. 7–9 show that mutation of Ca2+-binding sites in the C-domain causes significant changes in amide chemical shifts that are consistent with a loss of structure. In addition, mutations in the C-domain affect amide chemical shifts in the N-domain. There are at least two general mechanisms for this observation. One involves a specific interaction between domains that are disrupted by mutations. A recent crystal structure of Ca2+-CaM shows a collapsed structure in which helix A in the N-domain makes significant H-bonding interactions with helices G and H in the C-domain (40). Such a conformation would undoubtedly be a minor species in solution. Moreover, the apparent KCa50 for Ca2+ binding to isolated N and C domains of CaM are very similar (<1.5-fold different) to those determined for the intact protein (35, 36, 41, 42). This would not be expected if the Ca2+ binding properties of the N- and C-domains of intact CaM experience an intrinsic effect of domain-domain interactions.
A second mechanism for interdomain effects of mutations involves relatively nonspecific interactions between the N-domain and the mutated C-domain. Even though a highly flexible tether links these domains, it is reasonable that random arcing motions would allow nonspecific collisions. Changes in the surface chemistry due to mutations could lead to enhanced interactions between domains. Fig. 9 shows that residues in the hydrophobic pocket of the Ca2+-bound N-domain are most affected by mutation of Asp93 and Asp129 in the C-domain. This supports a model in which exposed surfaces in the mutated C-domain associate with the N-terminal hydrophobic pocket in the presence of Ca2+ to stabilize the Ca2+ bound form, and increase Ca2+ binding affinity by decreasing the koff.
Interestingly, inactivation of sites I and II in CaM1,2 does not affect Ca2+ binding to the C-domain. Because the overall affinity of Ca2+ binding to the C-domain (kIIIkIV) is about 30-fold stronger than the N-domain (kIkII) (5, 35, 36, 42, 43), Ca2+ binds sequentially first to the C-domain, whereas the N-domain is predominately in the apo state. Thus, inhibition of Ca2+ binding to the N-domain would not be expected to affect binding to the C-domain. The relative structural stability of the N- and C-domains may also contribute to interdomain effects of mutations. The apo N-domain has a defined hydrophobic core and a single conformation, but the apo C-domain has an ill-defined aromatic hydrophobic core (44, 45), multiple thermal melting transitions (46), with regions of intrinsic disorder (45, 47), and a high degree of backbone conformational entropy that allows global conformation exchange between at least two conformations (45, 48, 49). Thus, mutation in the C-domain may have a greater effect on structural stability than similar mutations in the N-domain.
Comparison of 15N HSQC spectra for native apoCaM, CaM3, and CaM4 shows that conversion of Asp93 to Ala in CaM3 has the greatest effect on structure of the C-domain. Structures of apo Ca2+-binding loops in Fig. 10 provide a mechanistic basis for this observation. The side chains of Asp129 in loop IV, and the corresponding Asp56 in loop II (not shown) are both solvent exposed. Thus, significant structural effects would not be predicted from mutation of these residues. In contrast, Fig. 10 shows that the side chain carboxyl group of Asp93 points toward the backbone of other residues in the Ca2+-binding loop of EF-hand III and is 2–3 Å from backbone amides of Asp95, Gly96, Asn97, and Gly98 as shown by the yellow lines. A similar conformation is seen for Asp20 of loop I in the N-domain. This suggests that the apo forms of loops I and III are stabilized by hydrogen bonds between side chains of Asp at position 1, and backbone amides of residues at positions 3 to 6. Conversion of Asp93 or Asp20 to Ala would eliminate these hydrogen bonds. The extent of structural perturbation in the N- and C-domains may depend on differences in stability in the apo state as summarized above. Specifically, the apo C-domain is less stable than the N-domain and may be more sensitive to mutations.
FIGURE 10.

NMR solution structures for apoCa2+-binding loops III and VI. Solution structure coordinates were taken from Kuboniwa et al. (45). Asp93 and Asp129 are the first amino acids in loops III and IV, respectively, and provide the X Ca2+ coordination ligand. Glu104 and Glu140 are the 12 amino acids in the loops and provide the −Z coordination position. Yellow lines indicate backbone amide hydrogens that are within 2 to 3 Å of carboxyl side chain of Asp93.
This raises the question of what is the best way to mutate Ca2+-binding sites of EF-hand proteins for in vivo and in vitro studies? Mutation strategies must inhibit Ca2+ binding, but should also maintain structural integrity of the apo form to allow productive interactions with target proteins that require the apo state. The data presented here argue that mutation of the first ligand is not the best choice for inactivation of EF-hands III and IV in the C-domain of CaM. However, similar mutations may be suitable for the N-domain, which has greater intrinsic stability in the apo form.
Mutation of Glu at loop position 12 (see Figs. 1 and 10) has been used to inhibit Ca2+ binding to CaM (7, 8), because its side chain carboxyl group provides bidentate coordination of Ca2+ (32) (see Fig. 1) and it is solvent exposed in the apo form (44, 45). Mutation of Glu104 or Glu140 to Gln in sites III and IV, respectively, does not abolish Ca2+ binding, but greatly reduces affinity (50, 51). This is consistent with an analogous mutation in site I of skeletal troponin C, which allows weak Ca2+ binding via the first 5 coordination positions, but the EF-hand does not undergo a conformational change (52). Relative to CaM3 or CaM4, published NMR spectra of CaM with E104Q or E140Q mutations show fewer and less dramatic changes in amide chemical shifts in the C-domain (50, 51). Although this is desirable, it was shown that CaM E140Q regained Ca2+ binding activity in the presence of a high affinity CaM binding peptide (53). We see no evidence for recovery of Ca2+ binding to mutated sites in CaM1,2 or CaM3,4 upon association with a high affinity CaM binding peptide from CaM kinase II, CaMKII-(293–312) (data not shown).
The potential to recover Ca2+ binding at mutated EF-hands is an important consideration, and this potential will likely depend on the nature of the mutation, the EF-hand protein being studied and its putative target proteins. For example, conversion of Asp to Ala at the Y coordination position in sites II, III, or IV of cardiac troponin C inhibited Ca2+ binding to the free protein, however, Ca2+ binding activity was recovered at sites III and IV, but not site II, when the mutant proteins were incorporated into a troponin complex (54).
In summary, mutation strategies for inhibition of Ca2+ binding to EF-hands must balance retention of structural integrity against the potential for recovery of Ca2+ binding activity upon interaction with native protein binding partners. This is especially problematic if the mutated proteins are to be expressed in cell culture where there is the potential for interaction with multiple binding proteins.
Supplementary Material
Acknowledgments
We thank Dr. Andy Hudmon for expression vectors for CaM1,2 and CaM3,4 and Dr. Neal Waxham for helpful discussions.
This work was supported, in whole or in part, by National Institutes of Health Grants GM069611 and NS038310, American Heart Association Grant 09GRATN2280427, and Robert A. Welch Foundation Grant AU1144.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
- CaM
- calmodulin
- 1,8-ANS
- 1-anilinonaphthalene-8-sulfonic acid
- 1,5-IAEDANS
- 5-((((2-iodoacetyl)amino)ethyl)amino)naphthalene-1-sulfonic acid
- DDPM
- N-(4-dimethylamino-3,5-dinitrophenyl)maleimide
- HEDTA
- N-(2-hydroxyethyl)ethylenedinitrilotriacetic acid
- NTA
- nitrilo-2,2′,2″-triacetic acid
- HSQC
- heteronuclear single quantum coherence
- FRET
- fluorescence resonance energy transfer
- MOPS
- 4-morpholinepropanesulfonic acid.
REFERENCES
- 1.Slemmon J. R., Morgan J. I., Fullerton S. M., Danho W., Hilbush B. S., Wengenack T. M. (1996) J. Biol. Chem. 271, 15911–15917 [DOI] [PubMed] [Google Scholar]
- 2.Erhardt J. A., Legos J. J., Johanson R. A., Slemmon J. R., Wang X. (2000) Neuroreport 11, 3719–3723 [DOI] [PubMed] [Google Scholar]
- 3.Kanazawa Y., Makino M., Morishima Y., Yamada K., Nabeshima T., Shirasaki Y. (2008) Neuroscience 154, 473–481 [DOI] [PubMed] [Google Scholar]
- 4.Putkey J. A., Kleerekoper Q., Gaertner T. R., Waxham M. N. (2003) J. Biol. Chem. 278, 49667–49670 [DOI] [PubMed] [Google Scholar]
- 5.Putkey J. A., Waxham M. N., Gaertner T. R., Brewer K. J., Goldsmith M., Kubota Y., Kleerekoper Q. K. (2008) J. Biol. Chem. 283, 1401–1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kleerekoper Q. K., Putkey J. A. (2009) J. Biol. Chem. 284, 7455–7464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maune J. F., Klee C. B., Beckingham K. (1992) J. Biol. Chem. 267, 5286–5295 [PubMed] [Google Scholar]
- 8.Leachman S. A., Gallagher P. J., Herring B. P., McPhaul M. J., Stull J. T. (1992) J. Biol. Chem. 267, 4930–4938 [PMC free article] [PubMed] [Google Scholar]
- 9.Xia X. M., Fakler B., Rivard A., Wayman G., Johnson-Pais T., Keen J. E., Ishii T., Hirschberg B., Bond C. T., Lutsenko S., Maylie J., Adelman J. P. (1998) Nature 395, 503–507 [DOI] [PubMed] [Google Scholar]
- 10.Putkey J. A., Sweeney H. L., Campbell S. T. (1989) J. Biol. Chem. 264, 12370–12378 [PubMed] [Google Scholar]
- 11.Sheng Z., Strauss W. L., Francois J. M., Potter J. D. (1990) J. Biol. Chem. 265, 21554–21560 [PubMed] [Google Scholar]
- 12.Reinach F. C., Nagai K., Kendrick-Jones J. (1986) Nature 322, 80–83 [DOI] [PubMed] [Google Scholar]
- 13.DeMaria C. D., Soong T. W., Alseikhan B. A., Alvania R. S., Yue D. T. (2001) Nature 411, 484–489 [DOI] [PubMed] [Google Scholar]
- 14.Deschênes I., Neyroud N., DiSilvestre D., Marbán E., Yue D. T., Tomaselli G. F. (2002) Circ. Res. 90, E49–E57 [DOI] [PubMed] [Google Scholar]
- 15.Leung L. L. (1984) J. Clin. Invest. 74, 1764–1772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Joseph J. D., Means A. R. (2002) Eukaryot. Cell 1, 119–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kasri N. N., Sienaert I., Parys J. B., Callewaert G., Missiaen L., Jeromin A., De Smedt H. (2003) J. Biol. Chem. 278, 27548–27555 [DOI] [PubMed] [Google Scholar]
- 18.Lambers T. T., Weidema A. F., Nilius B., Hoenderop J. G., Bindels R. J. (2004) J. Biol. Chem. 279, 28855–28861 [DOI] [PubMed] [Google Scholar]
- 19.Kasri N. N., Bultynck G., Smyth J., Szlufcik K., Parys J. B., Callewaert G., Missiaen L., Fissore R. A., Mikoshiba K., de Smedt H. (2004) Mol. Pharmacol. 66, 276–284 [DOI] [PubMed] [Google Scholar]
- 20.Vermassen E., Fissore R. A., Nadif Kasri N., Vanderheyden V., Callewaert G., Missiaen L., Parys J. B., De Smedt H. (2004) Biochem. Biophys. Res. Commun. 319, 888–893 [DOI] [PubMed] [Google Scholar]
- 21.Nadif Kasri N., Bultynck G., Parys J. B., Callewaert G., Missiaen L., De Smedt H. (2005) Mol. Pharmacol. 68, 241–250 [DOI] [PubMed] [Google Scholar]
- 22.Li L., Li Z., Sacks D. B. (2005) J. Biol. Chem. 280, 13097–13104 [DOI] [PubMed] [Google Scholar]
- 23.Kasri N. N., Török K., Galione A., Garnham C., Callewaert G., Missiaen L., Parys J. B., De Smedt H. (2006) J. Biol. Chem. 281, 8332–8338 [DOI] [PubMed] [Google Scholar]
- 24.Spratt D. E., Taiakina V., Guillemette J. G. (2007) Biochim. Biophys. Acta 1774, 1351–1358 [DOI] [PubMed] [Google Scholar]
- 25.Stroffekova K. (2008) Pflugers Arch. 455, 873–884 [DOI] [PubMed] [Google Scholar]
- 26.Mahajan A., Sato D., Shiferaw Y., Baher A., Xie L. H., Peralta R., Olcese R., Garfinkel A., Qu Z., Weiss J. N. (2008) Biophys. J. 94, 411–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ravindran A., Lao Q. Z., Harry J. B., Abrahimi P., Kobrinsky E., Soldatov N. M. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 8154–8159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ravindran A., Kobrinsky E., Lao Q. Z., Soldatov N. M. (2009) Channels 3, 25–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wei F., Xia X. M., Tang J., Ao H., Ko S., Liauw J., Qiu C. S., Zhuo M. (2003) J. Neurosci. 23, 8402–8409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee A., Zhou H., Scheuer T., Catterall W. A. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 16059–16064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dick I. E., Tadross M. R., Liang H., Tay L. H., Yang W., Yue D. T. (2008) Nature 451, 830–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Strynadka N. C., James M. N. (1989) Annu. Rev. Biochem. 58, 951–998 [DOI] [PubMed] [Google Scholar]
- 33.Putkey J. A., Waxham M. N. (1996) J. Biol. Chem. 271, 29619–29623 [DOI] [PubMed] [Google Scholar]
- 34.Pitt G. S., Zühlke R. D., Hudmon A., Schulman H., Reuter H., Tsien R. W. (2001) J. Biol. Chem. 276, 30794–30802 [DOI] [PubMed] [Google Scholar]
- 35.Linse S., Helmersson A., Forsén S. (1991) J. Biol. Chem. 266, 8050–8054 [PubMed] [Google Scholar]
- 36.Bayley P. M., Findlay W. A., Martin S. R. (1996) Protein Sci. 5, 1215–1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martin S. R., Maune J. F., Beckingham K., Bayley P. M. (1992) Eur. J. Biochem. 205, 1107–1114 [DOI] [PubMed] [Google Scholar]
- 38.Dodd R., Peracchia C., Stolady D., Török K. (2008) J. Biol. Chem. 283, 26911–26920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fefeu S., Biekofsky R. R., McCormick J. E., Martin S. R., Bayley P. M., Feeney J. (2000) Biochemistry 39, 15920–15931 [DOI] [PubMed] [Google Scholar]
- 40.Fallon J. L., Quiocho F. A. (2003) Structure 11, 1303–1307 [DOI] [PubMed] [Google Scholar]
- 41.Evans T. I., Shea M. A. (2009) Proteins 76, 47–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.VanScyoc W. S., Sorensen B. R., Rusinova E., Laws W. R., Ross J. B., Shea M. A. (2002) Biophys J. 83, 2767–2780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Renner M., Danielson M. A., Falke J. J. (1993) Proc. Natl. Acad. Sci. U.S.A. 90, 6493–6497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang M., Tanaka T., Ikura M. (1995) Nat. Struct. Biol. 2, 758–767 [DOI] [PubMed] [Google Scholar]
- 45.Kuboniwa H., Tjandra N., Grzesiek S., Ren H., Klee C. B., Bax A. (1995) Nat. Struct. Biol. 2, 768–776 [DOI] [PubMed] [Google Scholar]
- 46.Tsalkova T. N., Privalov P. L. (1985) J. Mol. Biol. 181, 533–544 [DOI] [PubMed] [Google Scholar]
- 47.Lundström P., Mulder F. A., Akke M. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 16984–16989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rabl C. R., Martin S. R., Neumann E., Bayley P. M. (2002) Biophys. Chem. 101–102, 553–564 [DOI] [PubMed] [Google Scholar]
- 49.Chen Y. G., Hummer G. (2007) J. Am. Chem. Soc. 129, 2414–2415 [DOI] [PubMed] [Google Scholar]
- 50.Evenäs J., Malmendal A., Thulin E., Carlström G., Forsén S. (1998) Biochemistry 37, 13744–13754 [DOI] [PubMed] [Google Scholar]
- 51.Evenäs J., Thulin E., Malmendal A., Forsén S., Carlström G. (1997) Biochemistry 36, 3448–3457 [DOI] [PubMed] [Google Scholar]
- 52.Gagné S. M., Li M. X., Sykes B. D. (1997) Biochemistry 36, 4386–4392 [DOI] [PubMed] [Google Scholar]
- 53.Haiech J., Kilhoffer M. C., Lukas T. J., Craig T. A., Roberts D. M., Watterson D. M. (1991) J. Biol. Chem. 266, 3427–3431 [PubMed] [Google Scholar]
- 54.Dotson D. G., Putkey J. A. (1993) J. Biol. Chem. 268, 24067–24073 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.