

Abstract
Classical Parkinson's disease (PD) is characterized by the appearance of Lewy bodies (LBs) in affected brain regions, showing mostly compact alpha-synuclein deposition, in contrast with punctate or granular deposition, hypothesized to represent early stages of aggregation. Leucine-rich repeat kinase 2 (LRRK2) is the commonest mutated gene in inherited and idiopathic PD. LRRK2 mutation carriers display a diverse neuropathology, including alpha-synuclein and tau inclusions, suggesting an upstream role for LRRK2 in protein aggregation. We studied LRRK2 expression throughout the normal human brain with three different antibodies. We also examined the pattern of LRRK2 expression in relation to alpha-synuclein aggregation and LB formation in the brainstem of sporadic LB disease. Physiological LRRK2 expression was not restricted to regions preferentially affected in PD and LRRK2 often localised to the nuclear envelope in addition to the known cytoplasmic expression. In PD, we were able to consistently detect LRRK2 in the halo of a minority (~10%) of nigral LBs using three different antibodies. Only one antibody detected LRRK2 in the core of ~80% of classic LBs. In the lower brainstem, most notably in the dorsal motor nucleus of the vagus, we found previously unrecognised LRRK2 labelling of complex globular lesions, filled with LB-like-matter showing a punctate or granular staining for alpha-synuclein. This was often accompanied by strong LRRK2 expression within dystrophic neurites. Our findings confirm widespread physiological LRRK2 expression in the human brain and suggest an association of LRRK2 with possible early-stage alpha-synuclein pathology in the brainstem of PD.
Keywords: LRRK2, dardarin, alpha-synuclein, Parkinson's disease, brainstem, Lewy body
Introduction
Parkinson's disease (PD) is a common neurodegenerative disorder affecting the motor system with a prevalence of about 2% in the population >60 years of age. Symptoms mainly arise from the degeneration of the substantia nigra associated with the formation of Lewy bodies (LBs), the characteristic pathological lesion of PD. However, LBs can be found in other regions, such as the dorsal motor nucleus of the vagus (DMNV), recently proposed to be the earliest affected brain region in PD [1], or the olfactory bulb. LBs can also appear in the cortex and may cause dementia (dementia with LB (DLB)). Other pathological lesions also form in PD, such as the LB-like matter and pale bodies (PBs), which are suspected to be precursors of LBs [2-4], and Lewy neurites or axonal spheroids [5, 6]. LB-matter is distinguished from PBs by some authors because of their different staining properties, such as being more eosinophilic than the PBs [4]. LBs are composed mainly of the protein alpha-synuclein [7], which is mutated in a few autosomal dominant forms of PD or DLB [8-10]. There seems to be a continuum in the aggregation morphology of alpha-synuclein within PD-related lesions: from granular or punctate forms, observed only faintly with haematoxylin-eosin (H-E), or associated with structures such as PBs, to a more compact form, such as in LBs. Thus, granular alpha-synuclein and PBs are hypothesised to precede the formation of LBs, in a compaction process that recruits ubiquitin (Ub) and the protein p62. The latter is absent in pure granular alpha-synuclein lesions, shows a punctate pattern in PBs and a compact pattern in LBs [3, 11, 12].
In addition to the alpha-synuclein gene, mutations in at least five genes have been clearly implicated in various inherited forms of PD. The most common to date is LRRK2 (dardarin), which mutations cause autosomal dominant PD associated with the PARK8 locus [13, 14]. More importantly, LRRK2 is the first familial PD gene frequently mutated in apparently sporadic forms of the disease. In Western countries the most frequent LRRK2 mutation (G2019S) may account for up to ~7% of familial and up to 3% of sporadic PD [15]. In North African Arabs and Ashkenazi Jewish populations these numbers could be as high as ~40% and ~30%, respectively [15].
Although LRRK2 mutations usually lead to typical PD, the pathological findings are pleomorphic in some cases, even within the same family [14, 16, 17]. Most commonly, LRRK2 mutation carriers display typical LB neuropathology (both brainstem-confined and diffuse). Other pathologies which may be present include tau tangles resembling those found in progressive supranuclear palsy (PSP), motoneuron disease features, frontotemporal lobar degeneration and pure nigral degeneration [14, 17]. It has been suggested, therefore, that LRRK2 may have a central role across the spectrum of neurodegenerative disease and lies upstream of the pathogenic deposition of alpha-synuclein and tau [18].
It is a feature of several neurodegenerative disorders that the mutated protein is deposited in pathological lesions in autosomal dominantly inherited forms of disease, whereas the wild-type protein is found in very similar inclusions in idiopathic disease. Within the lesions such proteins show different morphological and biochemical properties compared to their normal physiological expression. Thus, the objectives of our study were first, to delineate the physiological expression of LRRK2 in control human brain; second, to reveal any disease-specific LRRK2 deposition, in relation to controls, in key affected brainstem regions of idiopathic LB disorders; and, third, to compare co-localisation of LRRK2-positive pathology with alpha-synuclein positive pathology.
Methods
Cases and controls
Brain tissue was obtained from the Thomas Willis Oxford Brain Collection, incorporating the Oxford Project to Investigate Memory and Ageing (OPTIMA) tissue resource. Human tissue was collected with full consent of the patient or next of kin and with approval of the local ethical committee (COREC approval number 1656). The analysis of gene expression undertaken here has also been approved by local research ethics committee review, reference 06/Q1605/8. The characteristics of cases and controls are displayed in Table 1. Cases were selected according to neuropathological diagnosis. Control brain tissue was selected on the basis of showing no evident pathological signs of neurodegenerative disease. None of the cases or controls had a familial history of movement disorders. Formalin-fixed, paraffin-embedded sections and flash frozen samples were used.
Table 1.
Profile of patients and controls used in the study.
| Pathological diagnosis |
Number cases |
Comments | Mean age in years (range) (without the infant) |
Mean post mortem delay in days (range)* |
|
|---|---|---|---|---|---|
| Histological studies |
Control | 5 | 4 adults and 1 infant of 2 months |
54 (31-80) | 2.4 (0.3-7) |
| LB disease | 5 | 2 brainstem confined LB disease and 3 diffuse LB disease |
72 (37-88) | 3.8 (2-4) | |
| Homogenates for western blotting |
Biopsy control |
1 | Temporal lobectomy for hippocampal sclerosis |
22 | 0 |
| Postmortem control |
1 | Frontal cortex | 80 | 1 | |
| Postmortem diffuse LB disease |
2 | Anterior cingulate cortex | 74 (72-76) | 2.3 (1.75- 2.9) |
|
| Postmortem PSP |
1 | Frontal cortex | 77 | 1 |
All bodies were kept at 4°C from the time of death until brain extraction
Histological sections
Regions of normal brains studied are shown in Table 2. In patients, midbrain and medulla sections were studied. In order to estimate the percentage of LBs stained by the anti-LRRK2 antibodies in relation to alpha-synuclein, consecutive sections were stained with each antibody and their number counted in the nigra.
Table 2. Expression of LRRK2 in normal human adult brain.
Semiquantitative analysis of neuronal LRRK2 expression using the C-terminal antibody. Very similar results were observed for the N-terminal and Ev antibodies.
| Brain area | LRRK2 |
|---|---|
| Isocortex | + |
| Hippocampal area | |
| Fascia dentata | ++ |
| CA4-CA3 | ++ |
| CA2-CA1 | + |
| Subiculum | ++ |
| Entorhinal cortex | + |
| Transentorhinal | + |
| Cerebral grey nuclei | |
| Striatum (large neurons) | ++ |
| (medium) | + |
| Globus pallidus | ++ |
| Claustrum | + |
| Basal nucleus | ++ |
| Thalamus | ++ |
| Amygdala | ++ |
| Lateral geniculate nucleus | ++ |
| Subthalamus | + |
| Cerebellum | |
| Dentate nucleus | ++ |
| Purkinje cells | + |
| Molecular layer | + |
| Granular layer (Golgi cells) | ++ |
| Midbrain | |
| SN | ++ |
| Red nucleus | ++ |
| Superior colliculus | + |
| Periaqueductal grey | + |
| III nerve nucleus | +++ |
| Pons | |
| Locus caeruleus | ++ |
| Pontine nuclei | ++ |
| Raphe | ++ |
| Medulla | |
| XII nerve nucleus | +++ |
| IX/X dorsal motor nucleus | ++ |
| Nucleus solitarius | + |
| Nucleus cuneatus | +++ |
| Nucleus oblongatae | + |
| Nucleus ambiguus | + |
| Inferior olive | +++ |
| Cord | |
| Motoneurons | +++ |
Scale: + = weak, ++ = moderate, +++ = strong
Antibodies
Immunohistochemistry for LRRK2 protein was performed using the rabbit antibodies NB300-268 (C-Terminal), NB300-267 (N-Terminal) (Novus-Biologicals), and the goat EB06550 (Ev) (Everest Biotech), also corresponding to a C-terminal epitope. All antibodies were supplied affinity purified. To asses staining specificity, immunoabsorption with >10 fold molar ratio peptide/antibody was done for C- and N-terminal antibodies using their specific antigens (NB300-268PEP and NB300-267PEP; Novus-Biologicals) and checked for signal disappearance in both western blotting and immunohistochemistry. In addition we used an anti-alpha-synuclein antibody (LB509, Zymed), an anti-p62 (sequestosome-1) antibody (p62 lck ligand, BD Transduction Laboratories), an anti-ubiquitin antibody (Z0458, Dako), a monoclonal anti-actin antibody (AC-40, Sigma), a monoclonal anti-nucleoporin p62 (np62, Clone 53, BD Biosciences) and a goat anti-GFP (ab5450-25, Abcam).
Immunohistochemistry
Formalin-fixed blocks were cut into sections of 4-6 μm, dewaxed, hydrated, treated with 0.3% H2O2 and microwaved for 10 minutes in citrate buffer pH 6 for antigen retrieval. Some preparations were additionally treated with 0.1% trypsin [19]. Sections were incubated with C- and N-terminal antibodies diluted 1/300 or Ev antibody diluted 1/1000. For the latter antibody, sections were subsequently incubated with 1/200 rabbit anti-goat antibody (Zymed). Detection was done with REAL EnVision Detection System (Peroxidase/DAB+, Rabbit/Mouse; Dako). LB509 and the anti-nucleoporin p62 antibodies were used at 1/200 after microwaving the sections. The anti-p62 (sequestosome-1) antibody was used at 1/800 dilution after microwaving the sections. The anti-Ub antibody was used at a 1/300 dilution. Photomicrographs were taken using an AxioCam MRc camera and KS300 software (Zeiss).
Immunofluorescence
After antigen retrieval, a HRP-goat anti rabbit Tyramide Signal Amplification Kit (Invitrogen) based on Alexa Fluor 594 dye was used according to the manufacturer's instructions. Co-staining was achieved using the C-terminal antibody at a 1/75 dilution and the anti-alpha-synuclein at 1/50 dilution. Fluorescence detection was performed using an Alexa Fluor 405 goat anti mouse antibody (Invitrogen) and the Alexa 594-tyramide solution at a 1/75 dilution and either DAPI or TOPRO3 (Invitrogen). Images were captured with a Zeiss LSM510 META Confocal Imaging System.
Human brain homogenates and western blotting
Frozen human brains were processed to obtain high-salt homogenate extracts according to Giasson et al [20] and run in denaturing conditions in 4-15% Criterion acrylamide gradient gels (Biorad). Portions of some samples were left for 1 or 3 hours at 37°C before processing to study post-mortem LRRK2 degradation. The extracts were electroblotted to a PVDF (Immovilon P, Millipore) membrane, blocked with fat-free milk and incubated over night at 4°C with anti-LRRK2 antibodies diluted at 1/2000 (C-terminal), 1/500 (N-terminal) or 1/10000 (Ev), followed by incubation with HRP conjugated protein A (Amersham). For the Ev antibody a rabbit anti-goat antibody (Zymed) was also used at 1/10000. Chemiluminescence signal was detected using ECL-plus (Amersham) in conjunction with X-ray film (Kodak).
Results
LRRK2 is extensively expressed in neurons of the human central nervous system
To check specificity, we confirmed that all antibodies recognized LRRK2 by western blot in lysates from HEK cells over-expressing a LRRK2-GFP fusion protein (Fig 1, A). We then investigated detection of the endogenous full length LRRK2 in human brain homogenates by the C- and N-terminal antibodies. A biopsy of temporal cortex (Fig 1, B) and several post-mortem control and diseased brain samples (Fig 1, C) were used. Both the C- and the N-terminal antibodies detected the predicted 286 KDa band (Fig 1, B and C) in the biopsy, and the N-terminal antibody also detected a band of ~125 KDa. Other N-terminal antibodies have been shown to recognise the smaller band [21]. In contrast to the biopsy, in post-mortem samples both C- and N-terminal antibodies detected additional smaller bands of ~140 and ~100 KDa, respectively, in the frontal cortex of an aged control, the anterior cingulate cortex of two DLB cases or even from the frontal cortex of a PSP case. These bands did not show a disease-specific pattern and a representative blot is shown in Fig. 1C (PSP). When a portion of the biopsy was thawed and left for degradation, the post-mortem pattern was reproduced (Fig. 1, C). This result indicates that the antibodies are able to detect their epitopes even if some post-mortem degradation had occurred. This degradation is likely to occur early (< 1 day), since differences only appeared between the biopsy and all the PM samples, regardless of their PM delay (1-2.9 days). Preabsorption of both the C- and the N-terminal antibody by their respective immunogenic peptides completely abolished antibody staining in both western blotting and immunohistochemistry (data not shown).
Fig. 1. LRRK2 detection by immunoblotting.
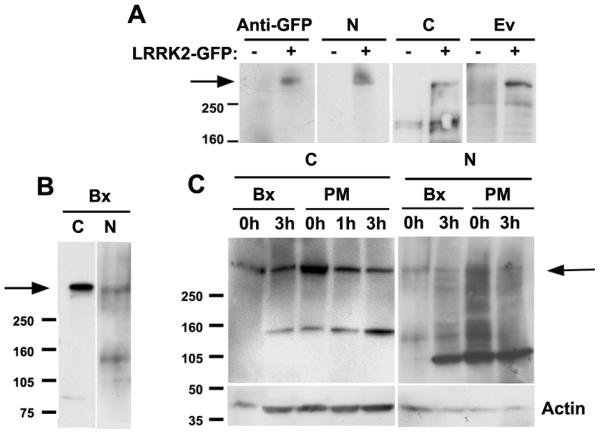
(A) Detection of a LRRK2-GFP reporter gene overexpressed in HEK cells using the three anti-LRRK2 antibodies used in this study. Recognition of the appropriate band is confirmed with an anti-GFP antibody. (B) Detection of the predicted 286 KDa band on a brain homogenate from a temporal lobe biopsy (Bx) by the C- and N-terminal antibodies. (C) LRRK2 detection in human post-mortem samples. Both the C-terminal and the N-terminal antibodies detect the same band of about 286 KDa in human post-mortem brain high salt homogenate extracts from several controls and patients. Since post-mortem (PM) samples from control and diseased brain gave a different pattern to the Bx sample, possibly due to post-mortem degradation, we performed a degradation assay of 1 or 3 hours with portions of the Bx sample and reproduced the same pattern as with the PM samples.
All three antibodies showed a similar widespread cytoplasmic neuronal staining (Fig. 2A-J). A mottled, Nissl-like appearance was seen in cell types with prominent rough endoplasmic reticulum (RER), such as motoneurons (Fig. 2F). Table 2 summarizes the areas studied in control adult brain and provides a semi-quantitative evaluation.
Fig. 2. LRRK2 is extensively expressed in the cytoplasm of neurons of the normal CNS. LRRK2 localises to the nuclear envelope.

All images from adult control tissue unless stated otherwise. (A) Substantia nigra. (B) Hippocampus. (C) Substantia nigra (infant control). (D) Dentate granule cells. (E) Striatum, showing large (arrow) and medium neurons (arrowhead). (F) Anterior horn of the spinal cord (infant control). (G) CA2 sector of the hippocampus. (H) CA4 sector of the hippocampus. (I) III nerve nucleus. (J) Red nucleus (PD case). (K) CA4 sector of the hippocampus. (L) CA4 sector of the hippocampus. Single slide from a z-stack three dimensional reconstruction. LRRK2 (red colour) in an apparent intranuclear rod is actually at the nuclear membrane. In G-L, arrows point at the nuclear membrane enhancement of staining. Arrowheads point at nuclear indentations and rods. The antibody used is shown in each image. Slides were counterstained with haematoxylin/DAPI (L). Scale bars: 100 μm in A and B, rest, 10 μm.
LRRK2 localizes to the nuclear envelope
All three LRRK2 antibodies stained the nuclear envelope in neurons across the control and LB/DLB brains. However, the staining intensity was different depending on neuroanatomical region and age (Fig. 2, G-J and L). Staining was more intense in the red nucleus (Fig. 2, J), the amygdala, the CA4 sector of the Ammon's horn (Fig. 2, H), or the somatic motor nuclei of the brainstem (Fig. 2, I). Less intense staining was seen in the neonate.
Within the nucleus, staining appeared to be confined to the nuclear envelope; however, indentations forming intranuclear bodies or rods were also frequently noted (Fig. 2, H, L). We therefore compared LRRK2 staining to that of nucleoporin p62, a component of the nuclear pore complex, and observed a very similar pattern to that of LRRK2. In addition, we performed three dimensional reconstructions based on a Z-stack multiple image acquisition, of which one section is shown in Fig. 2, L, to demonstrate the nuclear envelope origin of these rods and indentations.
LRRK2 presence in LBs
We conducted a detailed study of midbrain and medulla, two regions consistently affected by disease, from PD/DLB patients to evaluate any disease-specific LRRK2 presence compared to controls. All three LRRK2 antibodies stained the halo of a minority of perikaryal LBs(Fig. 3, A-D), consistently labelling about 10% of those stained by alpha-synuclein in the nigra. Using fluorescence double-labelling and confocal microscopy we observed that the LRRK2 halo could either be outside (Figure 3, E-G) or inside the alpha-synuclein ring (Fig. 3, H-J), with only partial overlapping between them. Only the NB300-268 C-terminal antibody stained the core of perikaryal LBs (Fig. 3, K, L), most notably in ~77% of nigral LBs. Staining of alpha-synuclein in the halo and LRRK2 in the core is shown in Fig. 3, M-O. No difference was seen between PD and DLB cases. LBs commonly adopt the form of intraneuritic LBs in the DMNV, frequently with a tubular morphology. They were frequently stained with the N-terminal antibody and, although less frequently, with the Ev and the C-terminal antibodies as well. In perikaryal LBs in the DMNV we could rarely detect LRRK2 in either halo or core (Fig. 3, D).
Fig. 3. Association of LRRK2 with brainstem lesions in PD/DLB.

All images are from PD/DLB cases. (A-D) LB halo staining in the midbrain (A-C) or medulla (D). In D note the concomitant staining of the core; this was rare. (E-J) LB halo staining with immunofluorescence double labelling in the midbrain. In E, a faint inner ring was also present (arrowhead). In J, the green arrow points to the alpha-synuclein-positive ring. Red arrow points to the LRRK2-positive ring. (K and L) LB core staining in the midbrain. (M-O) LB core staining with immunofluorescence double labelling in the midbrain. (P-V) LRRK2-positive lesions in the lower brainstem associated with granular alpha-synuclein staining. Alpha-synuclein staining was stronger in the periphery. Note in R that the dense peripheral structures are associated with a halo-like compact alpha-synuclein staining. U and V show weak or absent staining, respectively, of the body of these LRRK2-positive lesions for Ub and p62, with a potential “aggregation centre” localized in the periphery. P-R and S-V are thin consecutive sections. (W-Y) LRRK2-positive neurites in the lower brainstem. X and Y are thin consecutive sections. No difference between PD and DLB was observed. The antibody used is shown in each image. Slides were counterstained with haematoxylin/TOPRO3. Scale bars: 10 μm.
LRRK2 presence in brainstem lesions associated with a granular form of alpha-synuclein
We found strong LRRK2 staining of large lesions in the DMNV and dorsal raphe nuclei in the medulla of PD/DLB patients (Fig. 3, Q, S). To characterize these lesions further, we stained consecutive thin sections with H-E, anti-LRRK2, anti-alpha-synuclein, anti-ubiquitin and anti-p62 (sequestosome-1) antibodies. In H-E (Fig. 3, P) the lesions appeared as ellipsoidal structures of ~50 μm or larger. The structures were smooth, slightly/moderately eosinophilic, without a nucleus, and commonly associated with peripheral, smaller, round, dense and intensely eosinophilic ~10 μm structures. The N-terminal and the Ev antibodies consistently showed strong staining for these lesions (Fig. 3, Q and S), although the N-terminal antibody required an additional epitope retrieval step through trypsin proteolysis, which reduced the physiological staining concomitantly. The peripheral dense material, when present, was negative for anti-LRRK2 antibodies, but was surrounded by strong halo-like staining. With alpha-synuclein staining the whole lesion showed a light granular staining, usually more intense in the periphery (Fig. 3, R and T). The halo around the dense peripheral material was also typically intensely stained with alpha-synuclein. The staining for Ub of the ellipsoidal lesions was weak, although with frequent small areas of greater intensity in the periphery (Fig. 3 U). These small peripheral areas were the only positive structures for p62 (sequestosome-1) immunoreactivity (Fig. 3 V). Notably, similar large, ellipsoidal structures were rarely observed in the nigra. No differences in staining were attributable to variation in the PM delay.
LRRK2 is found in alpha-synuclein-positive neurites in the brainstem
Alpha-synuclein-positive neurites in the DMNV and the intramedullary fascicles of the IX/X cranial nerve were frequently and strongly stained with both the N-terminal (after trypsin treatment) and the Ev antibodies. Such neurites were ≥3-5 μm thick and sometimes several millimetres long (Fig. 3, W and X), with occasional swellings at varying intervals. Neurites on consecutive thin sections were shown to stain for both alpha-synuclein and LRRK2 (Fig. 3, X and Y).
Discussion
In the present study we describe several aspects of LRRK2 expression in human brain both in controls and LB disease cases: a semiquantitative neuroanatomical evaluation of physiological LRRK2 expression; the presence of LRRK2 in the nuclear envelope; the common co-localisation of LRRK2 with granular alpha-synuclein deposits and neurites in the brainstem of PD; and the detection of a minority of classical perikaryal LBs by three different LRRK2 antibodies.
Several studies have already described the cytoplasmic occurrence of LRRK2 and some describe its presence in different brain regions [22, 23]. However, a semi-quantitative evaluation in human brain was previously lacking. We show here that LRRK2 is broadly present in the cytoplasm of human brain neurons including those affected by neurodegeneration. However, there are significant differences in the detected level of protein expression across neuroanatomical regions, with affected areas in LB disease, such as the nigra, only harbouring moderate LRRK2 expression. In comparison to previous studies, mostly done in rodents using mRNA detection [24-26], the widespread expression of LRRK2 in neurons is becoming apparent. However, mRNA levels in rodent seem to be higher in the striatum, piriform cortex and fascia dentata, while protein levels in humans are greater in motoneurons, certain brainstem nuclei and fascia dentata, perhaps reflecting species-specific differences. We detected, however, the presence of LRRK2 in the nigra and in both large and medium spiny neurons in the striatum, in contrast to an mRNA detection study in humans [26].
LRRK2 and the nuclear envelope
We describe for the first time the localization of LRRK2 to the nuclear envelope. Interestingly, our finding agrees with a reported association of LRRK2 to membranous structures [21, 27-29] and is consistent with the occasional unexplained presence of LRRK2 in nuclear biochemical fractions [28]. The nuclear membrane is composed of the outer nuclear membrane (ONM) and the inner nuclear membrane (INM) which join together at the nuclear pores. The ONM is very similar to the RER, to which it is connected, whereas the INM has a different composition. It is possible that LRRK2 associates with the RER, and then to the ONM. We have detected an RER-like staining in some neurons (Fig. 2,F) and LRRK2 is associated to membranous fractions including those rich in RER [27]. Furthermore, at least three proteins involved in translation (a process that occurs at the RER) have been immunoprecipitated with LRRK2 [30], suggesting a role of LRRK2 in translation.
LRRK2 and Lewy bodies
In our study we help to resolve a recent controversy surrounding the presence of LRRK2 in LBs [23, 31-33], by showing LB labelling with three different anti-LRRK2 antibodies. LRRK2 was present in the halo of a minority (~10%) of nigral LBs consistent with previous reports [31] although not in agreement with a study by Covy et al [33], that denies any presence of LRRK2 in LBs. In addition, we help to characterize anti-LRRK2 antibodies by western blot, and our data are in agreement with other studies [21, 23, 31, 32, 34]. By immunofluorescence double-staining of the halo of LBs we observe that the circle of LRRK2 could be found inside or outside the ring of alpha-synuclein, usually only partially overlapping. While the presence of LRRK2 in LB halos as a result of passive entrapment remains possible, its occasional location within the alpha-synuclein ring appears to make this explanation less likely. Interestingly, the C-terminal antibody also detected LRRK2 in the core of ~80% of nigral LBs (Fig. 3, K and L) where parkin, one of LRRK2's interacting proteins potentially relevant for the pathogenesis of nigral degeneration, is also localised [35]. However, unlike our other findings, we note that this staining pattern was only detected with one of three LRRK2 antibodies and should be interpreted with caution until confirmed by other LRRK2 antibodies.
LRRK2 association with granular alpha-synuclein and neurites
We report the new finding of intense staining with anti-LRRK2 antibodies of pathological lesions in the lower brainstem of PD/DLB patients in association with alpha-synuclein granular staining. It is likely that similar structures were identified by Lewy himself when he drew some of the most complex lesions he found in the DMNV of PD cases [36]. The deposits may correspond to PBs, although perhaps match better the description of “LB-like matter” by Gibb and colleagues [4]. LB-like matter is associated with dense structures contiguous to intraneuritic LBs and may be the true precursor of LBs. The alpha-synuclein aggregation process has been recently reported to have a central role in neurotoxicity in vivo [37], in which protofibrils, non-fibrillar small aggregates, or related oligomeric species, which may correspond to the granular staining [38 ], may be the toxic species [39]. Several post-mortem and in vivo studies have proposed alpha-synuclein granular staining (also described as punctate, cloud-like or dust-like) to precede the compact deposition in the aggregation process [3, 11, 12, 38, 40, 41]. This process may start with small aggregates forming across the somatic cytoplasm and proximal neurites, later fusing to form a fibrillar inclusion body in the perikaryon, possibly as an attempt by the cell to protect itself against the toxic protein species [38]. In agreement with this idea, we found that the LRRK2-positive large globular lesions in the lower brainstem associated with granular alpha-synuclein, were negative or weak for p62 and Ub, respectively, except in small peripheral compaction foci that may represent “aggregation centres” as in the morphogenetic study of Kuusisto et al [3]. Aside from any potential role of the enzymatic activity of LRRK2 in cases of PD, there seems to be a morphological link between LRRK2 and alpha-synuclein, whereby LRRK2 is abundant in lesions where alpha-synuclein deposition begins, but appears to be relatively excluded from compact (end-stage) alpha-synuclein deposits.
In addition, we found that LRRK2 staining was associated with dystrophic neuronal processes in the lower brainstem of PD/DLB cases. Some lesions displayed periodic swellings and are likely to be abnormal axons, similar to those described by Braak et al. for alpha-synuclein staining [5]. Pathology of neurites, which can be extensive [5, 40], seems to occur early in the disease process both in PD patients where alpha-synuclein deposition starts in the DMNV [1], and in transgenic mouse models [40, 42]. Their formation need not be preceded, however, by granular alpha-synuclein deposits [3].
In summary, the most intensely and consistently LRRK2-expressing lesions (LB-matter and neurites) in the lower brain stem of PD can be hypothesized to represent early pathological events. Our findings, taken together with the observation that somatic mutations in LRRK2 are sufficient to cause LB disease, would therefore support placing LRRK2 upstream in the pathological pathways leading to alpha-synuclein deposition in LB diseases.
Acknowledgements
R. W.-M. is a Wellcome Trust Research Career Development Fellow. We would like to thank Carolyn Sloan for technical assistance and we are grateful to the patients and their families who support our brain bank collections.
We would like to thank C. Gloeckner (Institute of Human Genetics, Munich-Neuherberg, Germany) for kindly providing us with LRRK2 expression constructs.
References
- 1.Braak H, Tredici KD, Rub U, de Vos RAI, Jansen Steur ENH, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiology of Aging. 2003;24(2):197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 2.Dale GE, Probst A, Luthert P, Martin J, Anderton BH, Leigh PN. Relationships between Lewy bodies and pale bodies in Parkinson's disease. Acta Neuropathologica. 1992;83(5):525–9. doi: 10.1007/BF00310030. [DOI] [PubMed] [Google Scholar]
- 3.Kuusisto E, Parkkinen L, Alafuzoff I. Morphogenesis of Lewy bodies: dissimilar incorporation of alpha-synuclein, ubiquitin, and p62. J Neuropathol Exp Neurol. 2003;62(12):1241–53. doi: 10.1093/jnen/62.12.1241. [DOI] [PubMed] [Google Scholar]
- 4.Gibb WRG, Scott T, Lees AJ. Neuronal inclusions of Parkinson's disease. Movement Disorders. 1991;6(1):2–11. doi: 10.1002/mds.870060103. [DOI] [PubMed] [Google Scholar]
- 5.Braak H, Sandmann-Keil D, Gai W, Braak E. Extensive axonal Lewy neurites in Parkinson's disease: a novel pathological feature revealed by [alpha]-synuclein immunocytochemistry. Neuroscience Letters. 1999;265(1):67–9. doi: 10.1016/s0304-3940(99)00208-6. [DOI] [PubMed] [Google Scholar]
- 6.Galvin JE, Uryu K, Lee VM, Trojanowski JQ. Axon pathology in Parkinson's disease and Lewy body dementia hippocampus contains alpha-, beta-, and gamma-synuclein. Proc Natl Acad Sci U S A. 1999;96(23):13450–5. doi: 10.1073/pnas.96.23.13450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spillantini MG, Schmidt ML, Lee VM-Y, Trojanowski JQ, Jakes R, Goedert M. [alpha]-Synuclein in Lewy bodies. Nature. 1997;388(6645):839–40. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 8.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276(5321):2045–7. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 9.Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet. 1998;18(2):106–8. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 10.Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, Llorens V, Gomez Tortosa E, del Ser T, Munoz DG, de Yebenes JG. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55(2):164–73. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- 11.Gomez-Tortosa E, Newell K, Irizarry MC, Sanders JL, Hyman BT. a-Synuclein immunoreactivity in dementia with Lewy bodies: morphological staging and comparison with ubiquitin immunostaining. Acta Neuropathologica. 2000;99(4):352–7. doi: 10.1007/s004010051135. [DOI] [PubMed] [Google Scholar]
- 12.Braak E, Sandmann-Keil D, Rab U, Gai WP, Vos RAId, Steur ENHJ, Arai K, Braak H. a-Synuclein immunopositive Parkinson's disease-related inclusion bodies in lower brain stem nuclei. Acta Neuropathologica. 2001;101(3):195–201. doi: 10.1007/s004010000247. [DOI] [PubMed] [Google Scholar]
- 13.Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, Lopez de Munain A, Aparicio S, Gil AM, Khan N, Johnson J, Martinez JR, Nicholl D, Carrera IM, Pena AS, de Silva R, Lees A, Marti-Masso JF, Perez-Tur J, Wood NW, Singleton AB. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. 2004;44(4):595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 14.Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Muller-Myhsok B, Dickson DW, Meitinger T, Strom TM, Wszolek ZK, Gasser T. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44(4):601–7. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 15.Tolosa E. Movement disorders: advances on many fronts. The Lancet Neurology. 2007;6(1):7–8. doi: 10.1016/S1474-4422(06)70661-5. [DOI] [PubMed] [Google Scholar]
- 16.Gaig C, Marti MJ, Ezquerra M, Rey MJ, Cardozo A, Tolosa E. G2019S LRRK2 mutation causing Parkinson's disease without Lewy bodies. J Neurol Neurosurg Psychiatry. 2007;78(6):626–8. doi: 10.1136/jnnp.2006.107904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dachsel JC, Ross OA, Mata IF, Kachergus J, Toft M, Cannon A, Baker M, Adamson J, Hutton M, Dickson DW, Farrer MJ. Lrrk2 G2019S substitution in frontotemporal lobar degeneration with ubiquitin-immunoreactive neuronal inclusions. Acta Neuropathol (Berl) 2006;113(5):601–6. doi: 10.1007/s00401-006-0178-1. [DOI] [PubMed] [Google Scholar]
- 18.Mata IF, Wedemeyer WJ, Farrer MJ, Taylor JP, Gallo KA. LRRK2 in Parkinson's disease: protein domains and functional insights. Trends in Neurosciences. 2006;29(5):286–93. doi: 10.1016/j.tins.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 19.Bancroft JD, Gamble M. Theory and practice of Histological Techniques 5th Ed. Churchil Livingstone. 2002 [Google Scholar]
- 20.Giasson BI, Covy JP, Bonini NM, Hurtig HI, Farrer MJ, Trojanowski JQ, Van Deerlin VM. Biochemical and pathological characterization of Lrrk2. Annals of Neurology. 2006;59(2):315–22. doi: 10.1002/ana.20791. [DOI] [PubMed] [Google Scholar]
- 21.Hatano T, Kubo S, Imai S, Maeda M, Ishikawa K, Mizuno Y, Hattori N. Leucine-rich repeat kinase 2 associates with lipid rafts. Hum Mol Genet. 2007;16(6):678–90. doi: 10.1093/hmg/ddm013. [DOI] [PubMed] [Google Scholar]
- 22.Gloeckner CJ, Kinkl N, Schumacher A, Braun RJ, O'Neill E, Meitinger T, Kolch W, Prokisch H, Ueffing M. The Parkinson disease causing LRRK2 mutation I2020T is associated with increased kinase activity. Hum Mol Genet. 2006;15(2):223–32. doi: 10.1093/hmg/ddi439. [DOI] [PubMed] [Google Scholar]
- 23.Miklossy J, Arai T, Guo JP, Klegeris A, Yu S, McGeer EG, McGeer PL. LRRK2 expression in normal and pathologic human brain and in human cell lines. J Neuropathol Exp Neurol. 2006;65(10):953–63. doi: 10.1097/01.jnen.0000235121.98052.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Melrose H, Lincoln S, Tyndall G, Dickson D, Farrer M. Anatomical localization of leucine-rich repeat kinase 2 in mouse brain. Neuroscience. 2006;139(3):791–4. doi: 10.1016/j.neuroscience.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 25.Simon-Sanchez J, Herranz-Perez V, Olucha-Bordonau F, Perez-Tur J. LRRK2 is expressed in areas affected by Parkinson's disease in the adult mouse brain. European Journal of Neuroscience. 2006;23(3):659–66. doi: 10.1111/j.1460-9568.2006.04616.x. [DOI] [PubMed] [Google Scholar]
- 26.Galter D, Westerlund M, Carmine A, Lindqvist E, Sydow O, Olson L. LRRK2 expression linked to dopamine-innervated areas. Annals of Neurology. 2006;59(4):714–9. doi: 10.1002/ana.20808. [DOI] [PubMed] [Google Scholar]
- 27.Biskup S, Moore DJ, Celsi F, Higashi S, West AB, Andrabi SA, Kurkinen K, Yu SW, Savitt JM, Waldvogel HJ, Faull RL, Emson PC, Torp R, Ottersen OP, Dawson TM, Dawson VL. Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann Neurol. 2006;60(5):557–69. doi: 10.1002/ana.21019. [DOI] [PubMed] [Google Scholar]
- 28.West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM. Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. PNAS. 2005:16842–7. doi: 10.1073/pnas.0507360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith WW, Pei Z, Jiang H, Moore DJ, Liang Y, West AB, Dawson VL, Dawson TM, Ross CA. Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proc Natl Acad Sci U S A. 2005;102(51):18676–81. doi: 10.1073/pnas.0508052102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dachsel JC, Taylor JP, Mok SS, Ross OA, Hinkle KM, Bailey RM, Hines JH, Szutu J, Madden B, Petrucelli L, Farrer MJ. Identification of potential protein interactors of Lrrk2. Parkinsonism Relat Disord. 2007 doi: 10.1016/j.parkreldis.2007.01.008. Ahead of print, doi:10.1016/j.parkreldis.2007.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Greggio E, Jain S, Kingsbury A, Bandopadhyay R, Lewis P, Kaganovich A, van der Brug MP, Beilina A, Blackinton J, Thomas KJ. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiology of Disease. 2006;23(2):329–41. doi: 10.1016/j.nbd.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 32.Zhu X, Babar A, Siedlak SL, Yang Q, Ito G, Iwatsubo T, Smith MA, Perry G, Chen SG. LRRK2 in Parkinson's disease and dementia with Lewy bodies. Mol Neurodegener. 2006;1:17. doi: 10.1186/1750-1326-1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Covy JP, Van Deerlin VM, Giasson BI. Lack of evidence for Lrrk2 in alpha-synuclein pathological inclusions. Ann Neurol. 2006;60(5):618–9. [Google Scholar]
- 34.Greggio E, Lewis PA, van der Brug MP, Ahmad R, Kaganovich A, Ding J, Beilina A, Baker AK, Cookson MR. Mutations in LRRK2/dardarin associated with Parkinson disease are more toxic than equivalent mutations in the homologous kinase LRRK1. J Neurochem. 2007;102(1):93–102. doi: 10.1111/j.1471-4159.2007.04523.x. [DOI] [PubMed] [Google Scholar]
- 35.Schlossmacher MG, Frosch MP, Gai WP, Medina M, Sharma N, Forno L, Ochiishi T, Shimura H, Sharon R, Hattori N, Langston JW, Mizuno Y, Hyman BT, Selkoe DJ, Kosik KS. Parkin Localizes to the Lewy Bodies of Parkinson Disease and Dementia with Lewy Bodies. American Journal of Pathology. 2002;160(5):1655–67. doi: 10.1016/S0002-9440(10)61113-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lewy FH. Untersuchungen zur Klinik, Physiologie, Pathologie u. Pathogenese d. Paralysis agitans. Julius Springer; Berlin: 1923. Die Lehre vom Tonus und der Bewegung : Zugleich systemat. [Google Scholar]
- 37.Periquet M, Fulga T, Myllykangas L, Schlossmacher MG, Feany MB. Aggregated {alpha}-Synuclein Mediates Dopaminergic Neurotoxicity In Vivo. J Neurosci. 2007;27(12):3338–46. doi: 10.1523/JNEUROSCI.0285-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee H-J, Lee S-J. Characterization of Cytoplasmic alpha -Synuclein Aggregates. Fibril formation is tightly linked to the inclusion-forming process in cells. J Biol Chem. 2002;277(50):48976–83. doi: 10.1074/jbc.M208192200. [DOI] [PubMed] [Google Scholar]
- 39.Goldberg MS, Lansbury PT., Jr. Is there a cause-and-effect relationship between alpha-synuclein fibrillization and Parkinson's disease? Nat Cell Biol. 2000;2(7):E115–9. doi: 10.1038/35017124. [DOI] [PubMed] [Google Scholar]
- 40.Zach S, Bueler H, Hengerer B, Gillardon F. Predominant Neuritic Pathology Induced by Viral Overexpression of a-Synuclein in Cell Culture. Cellular and Molecular Neurobiology. 2007;27(4):505–15. doi: 10.1007/s10571-007-9141-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McLean PJ, Ribich S, Hyman BT. Subcellular localization of alpha-synuclein in primary neuronal cultures: effect of missense mutations. J Neural Transm Suppl. 2000;(58):53–63. doi: 10.1007/978-3-7091-6284-2_5. [DOI] [PubMed] [Google Scholar]
- 42.Lauwers E, Debyser Z, Dorpe J, Strooper B, Nuttin B, Baekelandt V. Neuropathology and Neurodegeneration in Rodent Brain Induced by Lentiviral Vectormediated Overexpression of alpha-Synuclein. Brain Pathology. 2003;13(3):364–72. doi: 10.1111/j.1750-3639.2003.tb00035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]