

The structure of DinB from G. stearothermophilus is described and compared with a number of recently reported structures of this unusual fold. Structural similarities are revealed that unite several distant protein families.
Keywords: structural genomics, DinB, four-helix bundles, metal-binding proteins, Pfam families
Abstract
The crystal structure of the dinB gene product from Geobacillus stearothermophilus (GsDinB) is reported at 2.5 Å resolution. The dinB gene is one of the DNA-damage-induced genes and the corresponding protein, DinB, is the founding member of a Pfam family with no known function. The protein contains a four-helix up–down–down–up bundle that has previously been described in the literature in three disparate proteins: the enzyme MDMPI (mycothiol-dependent maleylpyruvate isomerase), YfiT and TTHA0303, a member of a small DUF (domain of unknown function). However, a search of the DALI structural database revealed similarities to a further 11 new unpublished structures contributed by structural genomics centers. The sequences of these proteins are quite divergent and represent several Pfam families, yet their structures are quite similar and most (but not all) seem to have the ability to coordinate a metal ion using a conserved histidine-triad motif. The structural similarities of these diverse proteins suggest that a new Pfam clan encompassing the families that share this fold should be created. The proteins that share this fold exhibit four different quaternary structures: monomeric and three different dimeric forms.
1. Introduction
The din (DNA-damage induced) genes in Bacillus subtilis and related Gram-positive bacteria are under the control of the SOS-like repair system (Cheo et al., 1991 ▶; Gillespie & Yasbin, 1987 ▶). They are regulated by LexA repression and are induced in response to environmental stressors such as radiation or chemical mutagens. The function of the protein encoded by B. subtilis dinB has not been elucidated, except for speculation based on sequence comparisons, that a conserved set of histidines might indicate metal-binding properties as often found in metal-dependent hydrolases (Makarova et al., 2000 ▶). The Pfam (Finn et al., 2008 ▶) DinB family (PF05163) groups together 238 proteins related to B. subtilis DinB.
Here, we describe the crystal structure of the Geobacillus stearothermophilus (formerly Bacillus stearothermophilus) DinB protein (GsDinB) determined at 2.5 Å resolution (PDB code 3gor). This protein shares 52.9% sequence identity with B. subtilis DinB. The protein shows a tertiary fold consisting of an up–down–down–up four-helix bundle with long connecting loops. This fold was originally described for the unrelated YfiT protein (Rajan et al., 2004 ▶) and has since been found in the mycothiol-dependent isomerase MDMPI (Wang et al., 2007 ▶) and a protein of unknown function TTHA0303 (Nagata et al., 2008 ▶). However, a search of the Protein Data Bank using DALI (Holm et al., 2006 ▶, 2008 ▶) revealed a number of related structures recently deposited by structural genomics centers. Of these, the only protein with a biochemically demonstrated function is MDMPI. The annotations of the rest of the identified homologues are either speculative or absent. Although at this point it is not possible to infer a single biological function for this fold, we describe similarities and differences that should be helpful when further functional data become available. We show that DUF families 1993, 664 and 1569 group together proteins that are structurally related to GsDinB. Furthermore, GsDinB represents the third dimerization architecture to be reported for this fold.
2. Materials and methods
2.1. Protein expression and purification
The gene encoding the DinB protein from G. stearothermophilus was cloned into the pMCSG7 vector by the Midwest Center for Structural Genomics using ligation-independent cloning as described previously (Stols et al., 2002 ▶). This clone, referred to as APC36150, encodes an N-terminal hexahistidine tag followed by a tobacco etch virus (TEV) protease cleavage site (Stols et al., 2002 ▶). The protein was expressed in Escherichia coli BL21 (DE3) cells using modified Terrific Broth (TB) medium (Research Products International Corp). Cell cultures were grown at 310 K to an OD600 of ∼1, the temperature was lowered to 293 K and protein expression was induced by the addition of 0.1 mg ml−1 IPTG. The His-tagged protein was purified using nickel-affinity chromatography (Ni–NTA agarose column, Qiagen). Pure fractions of protein were pooled together, dialyzed into rTEV cleavage buffer (50 mM Tris pH 8.0, 300 mM NaCl, 0.5 mM EDTA, 1 mM DTT) and incubated with recombinant TEV protease at 277 K. Cleavage efficiency was monitored using SDS–PAGE gels. Cleaved protein was then dialyzed to remove EDTA and DTT and again passed through an Ni–NTA column. Three residues of the TEV protease cleavage site (Ser-Asn-Ala) remained attached to the N-terminus after cleavage. The flowthrough of this column was collected and further purified by size-exclusion chromatography using a Superdex 75 (GE Healthcare) equilibrated with 20 mM Tris–HCl pH 8.0, 100 mM NaCl, 10 mM β-mercaptoethanol (β-ME). Protein samples were concentrated to 15 mg ml−1, divided into 35 µl aliquots in thin-walled PCR tubes, frozen in liquid nitrogen and stored at 193 K. Selenomethionyl protein was expressed in the methionine auxotroph B834 (LeMaster & Richards, 1985 ▶).
2.2. Crystallization, structure determination and refinement
The protein was screened for crystallization with a custom sparse-matrix screen (Cooper et al., 2007 ▶) using sitting-drop vapour diffusion. Drops containing 200 nl crystallization solution and 200 nl protein solution were set up at room temperature (∼295 K) and equilibrated against 60 µl of either crystallization solution or an alternate reservoir solution (1.5 M NaCl; Newman, 2005 ▶). Crystals readily formed in several conditions in which 1.5 M NaCl was present in the reservoir, but did not form in the traditional screen. Selenomethionyl protein behaved similarly and a single crystal was harvested directly from the sparse-matrix screen (1.0 M sodium acetate, 0.1 M imidazole pH 8.0) suspended over a reservoir containing 1.5 M NaCl. The rod-shaped crystals were approximately 10 × 10 × 75 nm in size and grew in 2–3 d. The crystals were briefly (10 s) transferred into a solution containing 20% glycerol (1.0 M sodium acetate, 0.1 M imidazole pH 8.0, 20% glycerol) before being plunged into liquid nitrogen for storage and transport.
Data were collected to 2.5 Å resolution at 100 K at SER-CAT (Southeast Regional Collaborative Access Team 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory) using 12 760 eV X-rays. The crystals displayed P212121 symmetry, with unit-cell parameters a = 68.5, b = 71.5, c = 123.3 Å and four copies in the asymmetric unit. This corresponds to a Matthews coefficient of 2.2 Å3 Da−1 and a solvent content of 39%. All data were processed using HKL-2000 (Otwinowski & Minor, 1997 ▶). The protein contains eight methionines, including the N-terminal methionine. The structure was solved by single-wavelength anomalous dispersion (SAD) phasing using SHELXC and SHELXD (Sheldrick, 2008 ▶) to locate heavy-atom sites and was phased and partially built with SOLVE/RESOLVE (Terwilliger, 2004 ▶) using f′ and f′′ values of −7.88 e and 4.82 e, respectively. SHELXD and SOLVE both found 32 heavy atoms, 28 of which corresponded to selenomethionines. The additional four sites corresponded to Ni2+ ions bound to the protein, which have an f′′ value of ∼1.8 e at 0.92 Å (Fig. 1 ▶ b). The initial solution had an overall figure of merit of 0.36. The model was extended using iterative cycles of RESOLVE and REFMAC5 (Vagin et al., 2004 ▶). This process dramatically improved the maps and the missing fragments were identified in intermediate models. A combination of ‘cut-and-paste’ model building, loop building within Coot (Emsley & Cowtan, 2004 ▶) and manual refinement resulted in a complete structure. The final refinements were performed in PHENIX (Adams et al., 2002 ▶) and included TLS refinement, NCS constraints and solvent modification. MolProbity (Davis et al., 2007 ▶) was used as a structure-validation tool. The structure and structure factors were deposited in the Protein Data Bank under PDB code 3gor. Analysis of the intermolecular contacts was performed using PISA (Krissinel & Henrick, 2007 ▶). All figures were generated using PyMOL (http://pymol.sourceforge.net/).
Figure 1.

(a) Ribbon diagram of a monomer of DinB shown in rainbow representation and colored with a spectrum from the N-terminus (blue) to the C-terminus (red). The Ni2+-coordinating histidine triad, as well as Gln134, are represented as sticks. The Ni2+ ion is shown as a green sphere. (b) The experimental electron-density map and an anomalous difference map after the initial round of RESOLVE are shown around the refined structure. The blue 2F o − F c map is shown at 1.5σ and the red anomalous difference map is shown at 2.5σ. Notice that the size of the Ni2+ ion is approximately half that of the peak of the selenium of selenomethionine.
3. Results and discussion
3.1. Crystal structure of G. stearothermophilus DinB
The final model of GsDinB was refined using data collected from a selenomethione-containing crystal to crystallographic R and R free values of 18.6% and 24.7%, respectively, with satisfactory geometry. Data-collection and refinement statistics are presented in Table 1 ▶. The asymmetric unit contained four monomers of the protein, each displaying a four-helix-bundle topology with an atypical up–down–down–up topology (Fig. 1 ▶). The N-terminal helix (α1) is a seven-turn helix with a bend at Leu12. This helix is followed by a flexible but ordered 11-residue-long loop that wraps over the top of the helical bundle before leading into α2, a six-turn helix. Thus, the α2 helix is across from the α1 helix. A long cross-over loop connects α2 to the parallel helix α3, which lies in the groove between α1 and α2. Finally, the α3 helix is followed by another long loop which wraps under the helical bundle, so that α3 and α4 are across from each other. The last loop contains one turn of a helix and a β-hairpin before connecting to the C-terminal α4 helix.
Table 1. Data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Macromolecule details | |
| Database codes | PDB code 3gor; PUMA2 accession RBSTP2382 |
| No. of residues per monomer | 154 [157 with cloning residues] |
| Molecular weight (Da) | 17954 [18227 with tag] |
| Molecular assembly | Dimer |
| Data-collection statistics | |
| Wavelength (Å) | 0.97928 |
| Resolution range (Å) | 40–2.5 (2.59–2.50) |
| Space group | P212121 |
| Unit-cell parameters (Å) | a = 68.5, b = 71.5, c = 123.3 |
| Total reflections | 134829 |
| Unique reflections | 20004 (1631) |
| Redundancy | 6.7 (5.4) |
| Completeness (%) | 94.2 (78.1) |
| Rmerge† (%) | 8.8 (28.9) |
| Average I/σ(I) | 31.8 (4.3) |
| Wilson B factor (Å2) | 50.6 |
| Refinement statistics | |
| Resolution (Å) | 35.6–2.51 (2.59–2.51) |
| Completeness (%) | 92.7 (78.1) |
| Working reflections‡ | 36943 (2036) |
| Test reflections‡ | 1887 (113) |
| Rwork§ (%) | 18.6 (26.5) |
| Rfree§ (%) | 24.7 (38.1) |
| No. of waters | 58 |
| R.m.s. deviations from ideal geometry | |
| Bonds (Å) | 0.007 |
| Angles (°) | 0.9 |
| Average B factors (Å2) | |
| Main chain | 62.0 |
| Side chain | 74.2 |
| Waters | 52.3 |
| MolProbity results | |
| All-atom clash score | 13.26 [91st percentile] |
| Ramachandran favored (%) | 98.0 |
| Ramachandran outliers (%) | 0.33 |
R
merge = 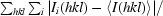
 , where I
i(hkl) is the intensity of the ith observation and 〈I(hkl)〉 is the mean intensity of the reflections. The values are for unmerged Friedel pairs.
, where I
i(hkl) is the intensity of the ith observation and 〈I(hkl)〉 is the mean intensity of the reflections. The values are for unmerged Friedel pairs.
Phenix.refine treats F + obs and F − obs as independent reflections.
The crystallographic R factor R = 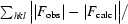
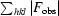 ; R
free =
where all reflections belong to a test set of randomly selected data.
; R
free =
where all reflections belong to a test set of randomly selected data.
The four monomers of GsDinB in the asymmetric unit are arranged into two dimers. Analysis of intermolecular contacts shows that the dimerization interfaces are virtually identical and result in ∼1000 Å2 being buried in each monomer. The complexation significance score (Krissinel & Henrick, 2007 ▶) is 0.52, suggesting that this interface is likely to have a functional role. The dimerization is primarily mediated by the two α4 helices and generates a twofold axis of symmetry that is roughly parallel to the bundle center and positioned between the two monomers. The hydrophobic core of the interface is formed by main-chain/side-chain interactions that result in the crossing of the two α4 helices near Gly133 at an ∼45° angle. The residues involved in this contact include Asp126, Ile129, His130, Gly133, Gln134, Phe136 and Val137. Three other regions of the protein also make intermolecular contacts with the α4 helix of the adjacent monomer: the residues flanking the kink of helix α1 (Tyr10 and His14), the loop that precedes α2 which posits residues Pro34 and Thr35 against Val137 and Gly141, and the C-terminal fragment downstream of α4 which brings the side chain of Phe150 to the center of the dimerization interface.
Based on the electron-density map, we identified a metal-binding site that is partly exposed to the solvent and is located in a groove between helices α2 and α4. A large positive peak is located between the imidazole rings of three histidines, His47, His127 and His131, so that the distances between the putative metal ion and the N∊2 atoms are 2.28, 2.31 and 2.28 Å, respectively, and are well within the expected range. Based on the purification protocol, which utilized Ni-affinity chromatography, we assume that the metal ion is Ni2+, although we did not verify this experimentally. The octahedral coordination appears to be completed by water molecules, although the limited resolution combined with the high mobility of the structure in this area precludes detailed interpretation of the electron density.
The metal-binding center lacks some features typically associated with catalytic centers. Histidines are preferentially protonated on Nδ1 (Reynolds et al., 1973 ▶), but they coordinate metal ions via N∊2, which is stereochemically more favorable (Chakrabarti, 1990 ▶). Consequently, within metal-binding sites it is common to see histidines interact via Nδ1 with acidic groups or backbone carbonyls, both of which act as hydrogen-bond acceptors, to stabilize the less favourable tautomeric form of the imidazole that leaves N∊2 unprotonated (Argos et al., 1978 ▶; Christianson & Alexander, 1989 ▶). This is evident in such examples as the FCD domains of GntR transcription factors (Zheng et al., 2009 ▶) and in homoserine lactone hydrolase (Kim et al., 2005 ▶) and related enzymes from the metallo-β-lactamase superfamily. In contrast, GsDinB shows none of this chemical sophistication. Neither His47 nor His127 is hydrogen bonded to any strong hydrogen-bond acceptors via Nδ1 and only His131 is hydrogen bonded in this way to Gln134. However, as Gln134 has no other hydrogen-bonding partners, it can function as both a hydrogen-bond donor via N∊2 as well as a hydrogen-bond acceptor via O∊1.
3.2. Comparison with related structures in the PDB
A comparison of GsDinB with structures in the PDB using the DALI server (Holm et al., 2008 ▶) identified 14 structures with a similar fold as defined by a Z score of >10.0 (Table 2 ▶). Only one of these proteins (PDB entry 3dka; Joint Center for Structural Genomics, unpublished work) shows a significant amino-acid sequence identity to GsDinB (54%). The remaining proteins show sequence-identity levels within the range 8–19%, which are consistent with the functional divergence of these proteins. 11 of these structures have been deposited recently by structural genomics groups, mostly by the Joint Center for Structural Genomics (Lesley & Wilson, 2005 ▶), but none of them have been published. Only one of these proteins has a conclusively identified function: mycothiol-dependent maleylpyruvate isomerase (MDMPI; Wang et al., 2007 ▶). It is also the only one in which the DinB-like moiety exists as a domain in a longer two-domain protein.
Table 2. Structures related to GsDinB.
| PDB code | Source | DALI Z | R.m.s.d.† | Identity (%) | PFAM | Dimerization | Triad |
|---|---|---|---|---|---|---|---|
| 3gor (GsDinB) | Geobacillus stearothermophilus | DinB (1.1 × 10−25) | 2 noncrystallographic dimers (α4, α1 parallel) | His47, His127, His131, with Ni2+ | |||
| 3dka | Bacillus subtilis | 24.2 | 1.0 | 54 | DinB (3.6 × 10−54) | Noncrystallographic dimer(like GsDinB) | His47, His127, His131, with Ni2+ |
| 2hkv | Exiguobacterium sibiricum | 14.7 | 2.7 | 17 | DinB (0.015) | Crystallographic dimer (like GsDinB) | His48, His123, His127, with Ni2+ |
| 3di5 | B. cereus | 14.5 | 2.7 | 19 | DinB (2.9 × 10−39) | Crystallographic dimer (like GsDinB) | His48, His127, His131, with Ni2+ |
| 2p1a | B. cereus | 13.9 | 2.4 | 10 | DinB (0.27) | Noncrystallographic dimer (like GsDinB) | His42, His114, His118, no metal |
| 2qe9 | B. subtilis | 13.7 | 3.2 | 13 | DinB (1 × 10−25) | Noncrystallographic dimer (like GsDinB) | His46, His128, His132, with Ni2+ |
| 2f22 | B. halodurans | 13.5 | 2.5 | 18 | DinB (0.0032) | Noncrystallographic dimer (like GsDinB) | His44, His124, His128, with Ni2+ |
| 2oqm | Shewanella denitrificans | 13.2 | 3.3 | 8 | DUF1993 (1.2 × 10−87) | 2 noncrystallographic dimers (like GsDinB) | Ile56, Asn138, His142, no metal |
| 1rxq (YfiT) | B. subtilis | 12.8 | 3.2 | 15 | No match | 2 noncrystallographic dimers, unique (α2, α3 antiparallel) | His67, His160, His164, with Ni2+ |
| 3cex | Enterococcus faecalis | 12.4 | 2.7 | 14 | No match | 2 crystallographic dimers (like GsDinB) | His47, Asp151, His155, no metal |
| 2ou6 | Deinococcus radiodurans | 12.2 | 2.5 | 12 | DinB (0.16), DUF664 (0.0018) | Crystallographic dimer (like GsDinB) | His76, Asp168, His172, with Ni2+ |
| 2qnl | Cytophaga hutchinsonii | 11.9 | 3.2 | 11 | No match | Crystallographic dimer (like GsDinB) | His52, His148, His152, no metal |
| 2nsf (MDMPI) | Corynebacterium glutamicum | 11.7 | 3.5 | 13 | No match | Monomeric | His52, Glu144, His148, with Zn2+ |
| 2yqy (TTHA0303) | Thermus thermophilus | 11.3 | 2.6 | 15 | DUF664 (0.18), DUF1569 (0.012) | Monomeric | His53, His155, His159, no metal |
| 2rd9 | B. halodurans | 10.8 | 3.2 | 10 | DinB (0.27) | 2 noncrystallographic, dimers unique (α4, α1 antiparallel) | His48, His142, His131, with Ni2+ |
Root-mean-square deviation of the structure when aligned with GsDinB.
Table 2 ▶ also shows the results of amino-acid sequence analysis of all these proteins against Pfam families. Several, including the two YfiT-like proteins and MDMPI, are not readily identifiable as members of any Pfam family. Three structures are annotated in the PDB as DinB (PF05163) family members. These are PDB entries 3dka (B. subtilis), 2hkv (Exiguobacterium sp.), 3di5 (B. cereus) and 2oqm (Shewanella denitrificans). Pfam does not include the latter as a member of DinB and instead groups it with DUF1993 (PF09351, 167 sequences), but identifies the unannotated 2qe9 (B. subtilis) and 2f22 (B. halodurans) as members of the DinB family. Three other structures, 2p1a (B. cereus), 2ou6 (Deinococcus radiodurans) and 2rd9 (B. halodurans), show weak similarity to the DinB family. Of these, 2p1a (B. cereus) is annotated in the PDB as a DNA-binding protein and 2ou6 (D. radiodurans) as a member of DUF664 (PF04978, 83 sequences). Pfam indicates that 2yqy (Thermus thermophilus) has weak similarity to DUF664 as well as to DUF1569 (PF07606, 17 sequences). The remaining protein with a weak similarity to DinB is 2rd9 (B. halodurans), which is currently annotated as a YfiT-like putative hydrolase. It should also be noted that the hydrolase activity of YfiT has not been confirmed experimentally (Rajan et al., 2004 ▶; Nagata et al., 2008 ▶).
Most of the DinB-related proteins listed in Table 2 ▶ form dimers very similar to that of GsDinB, with the dimerization interface being mediated by α4, α1 and associated loops (Fig. 2 ▶). These dimers contain an intermolecular dyad parallel to the axes of the helical bundles. A total surface area of between 950 Å2 and as much as 2150 Å2 is buried on dimerization. Among the crystal structures containing this fold we find both noncrystallographic dimers (3dka, 2p1a, 2qe9, 2f22 and 2oqm) and crystallographic dimers (2hkv, 3di5, 3cex, 2ou6 and 2qnl) that are similar to the GsDinB dimer. The extent of the involvement of α1 varies within these dimers and is related to whether or not the α1 helix contains a kink and to the length of the helix. For example, the S. denitrificans protein (2oqm) has a long straight α1 that interacts not only with α4 but also with α1 from its dimer partner. There is no clear conservation of residues within the dimerization interface.
Figure 2.

The three different dimerization architectures of GsDinB-related proteins. Each protein is shown from the side and from the top. (a, b) GsDinB, (c, d) 2oqm (DinB-like), (e, f) YfiT, (g, h) 2rd9. Each monomer is colored with a spectrum from the N-terminus (blue) to the C-terminus (red). The three residues located at the conserved metal-binding site are shown as sticks and metals are shown as spheres. Note that (a) and (b) share the same dimerization motif.
Only four of the structurally characterized proteins deviate from the dimerization paradigm seen in GsDinB, yet they include three different dimerization architectures. In YfiT an alternative interface is mediated by α2 and α3, creating a twofold perpendicular to these helices. YfiT has been reported to be dimeric in solution and the two nearly identical noncrystallographic dimers seen in the asymmetric unit are thought to be representative of the dimer in solution (Rajan et al., 2004 ▶). The dimerization mode seen in GsDinB is not possible for YfiT because an N-terminal extension in YfiT creates steric hindrance for the α4–α1-mediated interface. Conversely, the orientation of the cross-over helix between α2 and α3 in GsDinB precludes the dimerization mode seen in YfiT. Another dimerization architecture is seen in the B. halodurans protein (2rd9), in which the dimer is generated by an antiparallel rather than parallel arrangement of α4 and α1, thereby forming a twofold perpendicular to these helices.
The MDMPI protein, which contains an extra domain in addition to the four-helix bundle, is monomeric. The YfiT-like protein from T. thermophilus TTHA0303 (2yqy) also crystallized with two independent monomers in the asymmetric unit (Nagata et al., 2008 ▶).
The overarching question concerns the biological function of the proteins that belong to the various DinB-like families, i.e. DinB, YfiT, DUF664, DUF1569 and DUF1993. Analysis of amino-acid conservation reveals that the only highly conserved residues in the DinB family are associated with the putative metal-binding site, including the three key histidines and to a lesser extent Gln127 (which is often replaced by Asn). Both the YfiT and DUF1569 families have the same small sequence signature suggesting metal-binding capabilities, while both DUF644 and DUF1993 lack the relevant histidines. Interestingly, the presence of the three conserved histidines does not in itself guarantee the presence of a metal in the crystal structure. For example, 3dka, which is the closest homologue of GsDinB (54% identity), has no metal bound, but instead His47 and His127 are close enough for an unusual hydrogen bond between the two N∊2 atoms (2.8 Å). No metal is found in 2p1a, 2qnl and 2yqy, even though complete histidine triads are present in these proteins. Neither 2oqm nor 3cex have complete triads and they contain no metals. Of course, most of these proteins were expressed in a heterologous host with a hexahistidine affinity tag and were purified using an Ni2+-affinity step. Thus, metal ions may have been inadvertently introduced in the purification step. It is not clear whether all of the DinB-related proteins in fact bind metals in vivo or whether the metals that are found in the structures are the physiologically relevant metals. Zn2+ and Ni2+ can both assume an octahedral geometry (Auld, 2001 ▶). The only protein with well characterized metal-binding properties is MDMPI, which binds Zn2+ ions.
4. Conclusion
The crystal structure of GsDinB revealed a unique four-helix-bundle fold shared by proteins that belong to Pfam families DinB, DUF664, DUF1569 and DUF1993 and also by proteins whose sequences have not yet been classified into any family, such as the YfiT proteins and the enzyme MDMPI. We suggest that owing to those structural similarities all these families should be included in a single Pfam clan.
It is important to note that there is confusion in the literature regarding the DinB nomenclature. A gene from E. coli, also denoted dinB, encodes an error-prone type IV DNA polymerase (Silvian et al., 2001 ▶; Wagner et al., 1999 ▶). Homologous polymerases are found in other prokaryotes. These proteins are also regulated by the SOS-like repair system in response to similar stimuli, but are only related to the protein described here by their position in the din operon. Two genes in B. anthracis (BAS2215 and BA2379) constitute examples of proteins from the DinB family that have been erroneously annotated as polymerases. The literature related to E. coli DinB is abundant compared with the modicum of papers that focus on the Bacillus DinB family. We suggest that the designation of the Pfam DinB family be altered, perhaps to DinBBs, in order to more clearly distinguish the Pfam DinB family from the DinB polymerases. The Pfam designation for E. coli DinB is IMS (PF00917), the ImpB/MucB/SamB family.
The function of the DinB proteins is elusive despite their structural characterization. While the majority of these proteins form distinct dimers, the dimerization mode is not conserved and neither is the dimerization interface. Finally, while a putative metal-binding site is found in a number of DinB and DinB-like proteins, the stereochemistry of this site is not consistent with a catalytic function.
Further studies are required in order to identify the biological role of DinB and its homologues. Hopefully, the structural characterization presented in this paper will be useful when such role is finally unveiled.
Supplementary Material
PDB reference: DinB, 3gor
Acknowledgments
Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. W-31-109-Eng-38. Supporting institutions of SER-CAT may be found at http://www.ser-cat.org/members.html. This study was supported by NIH–NIGMS Grant U54 GM074946-01US.
References
- Adams, P. D., Grosse-Kunstleve, R. W., Hung, L.-W., Ioerger, T. R., McCoy, A. J., Moriarty, N. W., Read, R. J., Sacchettini, J. C., Sauter, N. K. & Terwilliger, T. C. (2002). Acta Cryst. D58, 1948–1954. [DOI] [PubMed]
- Argos, P., Garavito, R. M., Eventoff, W., Rossmann, M. G. & Brändén, C. I. (1978). J. Mol. Biol.126, 141–158. [DOI] [PubMed]
- Auld, D. S. (2001). Biometals, 14, 271–313. [DOI] [PubMed]
- Chakrabarti, P. (1990). Protein Eng.4, 57–63. [DOI] [PubMed]
- Cheo, D. L., Bayles, K. W. & Yasbin, R. E. (1991). J. Bacteriol.173, 1696–1703. [DOI] [PMC free article] [PubMed]
- Christianson, D. W. & Alexander, R. S. (1989). J. Am. Chem. Soc.111, 6412–6419.
- Cooper, D. R., Boczek, T., Grelewska, K., Pinkowska, M., Sikorska, M., Zawadzki, M. & Derewenda, Z. (2007). Acta Cryst. D63, 636–645. [DOI] [PubMed]
- Davis, I. W., Leaver-Fay, A., Chen, V. B., Block, J. N., Kapral, G. J., Wang, X., Murray, L. W., Arendall, W. B. III, Snoeyink, J., Richardson, J. S. & Richardson, D. C. (2007). Nucleic Acids Res.35, W375–W383. [DOI] [PMC free article] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Finn, R. D., Tate, J., Mistry, J., Coggill, P. C., Sammut, S. J., Hotz, H. R., Ceric, G., Forslund, K., Eddy, S. R., Sonnhammer, E. L. & Bateman, A. (2008). Nucleic Acids Res.36, D281–D288. [DOI] [PMC free article] [PubMed]
- Gillespie, K. & Yasbin, R. E. (1987). J. Bacteriol.169, 3372–3374. [DOI] [PMC free article] [PubMed]
- Holm, L., Kaariainen, S., Rosenstrom, P. & Schenkel, A. (2008). Bioinformatics, 24, 2780–2781. [DOI] [PMC free article] [PubMed]
- Holm, L., Kaariainen, S., Wilton, C. & Plewczynski, D. (2006). Current Protocols in Bioinformatics, edited by A. D. Baxevanis, ch. 5, Unit 5.5. New York: Wiley. [DOI] [PubMed]
- Kim, M. H., Choi, W.-C., Kang, H. O., Lee, J. S., Kang, B. S., Kim, K.-J., Derewenda, Z. S., Oh, T.-K., Lee, C. H. & Lee, J.-K. (2005). Proc. Natl Acad. Sci. USA, 102, 17606–17611. [DOI] [PMC free article] [PubMed]
- Krissinel, E. & Henrick, K. (2007). J. Mol. Biol.372, 774–797. [DOI] [PubMed]
- LeMaster, D. M. & Richards, F. M. (1985). Biochemistry, 24, 7263–7268. [DOI] [PubMed]
- Lesley, S. A. & Wilson, I. A. (2005). J. Struct. Funct. Genomics, 6, 71–79. [DOI] [PubMed]
- Makarova, K. S., Aravind, L., Daly, M. J. & Koonin, E. V. (2000). Genetica, 108, 25–34. [DOI] [PubMed]
- Nagata, K., Ohtsuka, J., Takahashi, M., Asano, A., Iino, H., Ebihara, A. & Tanokura, M. (2008). Proteins, 70, 1103–1107. [DOI] [PubMed]
- Newman, J. (2005). Acta Cryst. D61, 490–493. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Rajan, S. S., Yang, X., Shuvalova, L., Collart, F. & Anderson, W. F. (2004). Biochemistry, 43, 15472–15479. [DOI] [PubMed]
- Reynolds, W. F., Peat, I. R., Freedman, M. H. & Lyerla, J. R. Jr (1973). J. Am. Chem. Soc.95, 328–331. [DOI] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Silvian, L. F., Toth, E. A., Pham, P., Goodman, M. F. & Ellenberger, T. (2001). Nature Struct. Biol.8, 984–989. [DOI] [PubMed]
- Stols, L., Gu, M., Dieckman, L., Raffen, R., Collart, F. R. & Donnelly, M. I. (2002). Protein Expr. Purif.25, 8–15. [DOI] [PubMed]
- Terwilliger, T. (2004). J. Synchrotron Rad.11, 49–52. [DOI] [PubMed]
- Vagin, A. A., Steiner, R. A., Lebedev, A. A., Potterton, L., McNicholas, S., Long, F. & Murshudov, G. N. (2004). Acta Cryst. D60, 2184–2195. [DOI] [PubMed]
- Wagner, J., Gruz, P., Kim, S.-R., Yamada, M., Matsui, K., Fuchs, R. P. & Nohmi, T. (1999). Mol. Cell, 4, 281–286. [DOI] [PubMed]
- Wang, R., Yin, Y.-J., Wang, F., Li, M., Feng, J., Zhang, H.-M., Zhang, J.-P., Liu, S.-J. & Chang, W.-R. (2007). J. Biol. Chem.282, 16288–16294. [DOI] [PubMed]
- Zheng, M., Cooper, D. R., Grossoehme, N. E., Yu, M., Hung, L.-W., Cieslik, M., Derewenda, U., Lesley, S. A., Wilson, I. A., Giedroc, D. P. & Derewenda, Z. S. (2009). Acta Cryst. D65, 356–365. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: DinB, 3gor
PDB reference: DinB, 3gor