

The subtilisin-like proteases AprV2 and BprV from virulent strains and AprB2 and BprB from benign strains of D. nodosus have been crystallized and X-ray diffraction data have been collected to 2.0, 2.0, 1.7 and 1.8 Å resolution, respectively.
Keywords: Dichelobacter nodosus, footrot, subtilisin-like serine proteases
Abstract
Dichelobacter nodosus is the principal causative agent of ovine footrot, a disease of significant economic importance to the sheep industry. D. nodosus secretes a number of subtilisin-like serine proteases which mediate tissue damage and presumably contribute to the pathogenesis of footrot. Strains causing virulent footrot secrete the proteases AprV2, AprV5 and BprV and strains causing benign footrot secrete the closely related proteases AprB2, AprB5 and BprB. Here, the cloning, purification and crystallization of AprV2, AprB2, BprV and BprB are reported. Crystals of AprV2 and AprB2 diffracted to 2.0 and 1.7 Å resolution, respectively. The crystals of both proteases belonged to space group P1, with unit-cell parameters a = 43.1, b = 46.0, c = 47.2 Å, α = 97.8, β = 115.2, γ = 115.2° for AprV2 and a = 42.7, b = 45.8, c = 45.7 Å, α = 98.4, β = 114.0, γ = 114.6° for AprB2. Crystals of BprV and BprB diffracted to 2.0 and 1.8 Å resolution, respectively. The crystals of both proteases belonged to space group P21, with unit-cell parameters a = 38.5, b = 89.6, c = 47.7 Å, β = 113.6° for BprV and a = 38.5, b = 90.5, c = 44.1 Å, β = 109.9° for BprB. The crystals of all four proteases contained one molecule in the asymmetric unit, with a solvent content ranging from 36 to 40%.
1. Introduction
Ovine footrot is a devastating and highly contagious disease that affects the feet of sheep. The disease has been estimated to cost the British and Australian sheep industries £24 million and $A20 million, respectively, each year owing to the reduction in meat and wool production and the costs associated with prevention and treatment programs (Green & George, 2008 ▶; Wani & Samanta, 2006 ▶). Footrot is caused by a mixed bacterial infection, although the Gram-negative anaerobic bacterium Dichelobacter nodosus is the principal causative agent of the disease. Footrot varies in severity, ranging from a mild interdigital dermatitis (benign footrot) to an aggressive necrotic separation of the horn from the underlying tissue (virulent footrot) (Egerton & Parsonson, 1969 ▶; Green & George, 2008 ▶). D. nodosus isolates are classified as virulent or benign according to the severity of the disease that they cause.
Both virulent and benign strains of D. nodosus secrete a number of subtilisin-like serine proteases that have been suggested to be involved in the pathogenesis of footrot through their role in tissue destruction (Billington et al., 1996 ▶). Virulent strains secrete the acidic proteases AprV2 and AprV5 and the basic protease BprV (Lilley et al., 1992 ▶, 1995 ▶; Kortt et al., 1993 ▶, 1994 ▶; Riffkin et al., 1993 ▶). Benign strains secrete the proteases AprB2, AprB5 and BprB, which vary from their virulent counterparts by only a few amino acids (Lilley et al., 1992 ▶, 1995 ▶; Kortt et al., 1993 ▶, 1994 ▶; Riffkin et al., 1993 ▶). AprV2 and AprB2 differ in sequence by one amino acid (Y92R), whereas BprV and BprB differ in sequence by nine amino acids. These subtilases are initially expressed as inactive precursors. Subsequent proteolytic processing, which removes the N-terminal pre-domain and pro-domain and the C-terminal extension domain, yields mature active protease with a molecular mass of 36 kDa (Kortt et al., 1994 ▶). Subtilases isolated from the culture supernatant of virulent strains display a higher elastinolytic and caseinolytic activity than those secreted by benign strains (Liu & Yong, 1993 ▶; Stewart, 1979 ▶; Depiazzi & Richards, 1979 ▶). It has been suggested that these differences underpin the differences observed between the virulent and benign forms of the disease.
Owing to the proposed importance of the proteases secreted by D. nodosus in the pathogenesis of ovine footrot, the subtilases AprV2, AprB2, BprV and BprB have been expressed, purified and crystallized for comparative structural studies. The structures of these proteases will provide a valuable insight into the mechanism of protease-mediated footrot pathogenesis.
2. Experimental methods
2.1. Cloning
The region of the aprV2 gene encoding the pre-domain, pro-domain and mature domain was amplified from genomic DNA isolated from the virulent D. nodosus strain VCS1703A. The primers used for cloning are shown in Table 1 ▶. The amplified PCR product was digested with NdeI and XhoI and ligated into pET22b expression vector (Novagen) digested with the same enzymes. Clones were screened by colony PCR and their orientation and sequence were confirmed by DNA sequencing. The resultant recombinant expression vector pET22b-AprV2 encoded residues 1–474 of the AprV2 precursor (NCBI Accession No. ABQ13853) with a C-terminal His6 tag for affinity chromatography.
Table 1. PCR and site-directed mutagenesis (SDM) primers used in this study.
For the primers used for cloning, the restriction sites are shown in bold. For the primers used for SDM, the mutation is shown in bold. VCS1703A is a virulent stain of D. nodosus. C305 is a benign stain of D. nodosus.
| Use | Sequence | Template |
|---|---|---|
| AprV2 cloning (forward) | GACAGAATTCCCATATGAAACGATTCATTATGAACAAAATG | VCS1703A genomic DNA |
| AprV2 cloning (reverse) | GTATTGGGCAACGAAGGCAATCGTCTCGAGAAGCTTACA | VCS1703A genomic DNA |
| AprB2 SDM (forward) | CTGGGCATGCGGTGGTCGTCCTGATCCACGCAAAG | pET22b-AprV2 |
| AprB2 SDM (reverse) | CTTTGCGTGGATCAGGACGACCACCGCATGCCCAG | pET22b-AprV2 |
| BprV cloning (forward) | GACAGAATTCCCATATGAATCTATCGAACATTTCTGCGG | VCS1703A genomic DNA |
| BprV cloning (reverse) | CTGTAAGCTTCTCGAGACGATTTTGATCGCCTAAAACAGC | VCS1703A genomic DNA |
| BprB cloning (forward) | GACAGAATTCCCATATGAATCTATCGAACATTTCTGCGG | C305 genomic DNA |
| BprB cloning (reverse) | CTGTAAGCTTCTCGAGACGATTTTGATCGCCTAAAACAGC | C305 genomic DNA |
To generate pET22b-AprB2, site-directed mutagenesis was performed by polymerase chain reaction using the QuikChange Site-Directed Mutagenesis Kit (Stratagene) to change the Tyr92 codon to an Arg codon. pET22b-AprV2 was used as the template. The primers used are listed in Table 1 ▶. The amplified pET22-AprB2 expression vector was used for transformation into DH5α and the resultant plasmid was confirmed by DNA sequencing. The resultant recombinant expression vector pET22b-AprB2 encoded residues 1–474 of the AprB2 precursor (NCBI Accession No. ABQ13853) with a C-terminal His6 tag for affinity chromatography.
The regions of the bprV and bprB genes encoding the pre-domain, pro-domain and mature domain were amplified from genomic DNA isolated from the virulent D. nodosus strain VCS1703A and the benign strain C305, respectively. The primers used are listed in Table 1 ▶. The primers are identical for both proteases as they share identical sequences at the 5′ and 3′ end of the cloned regions. The amplified PCR products were digested with the restriction enzymes NdeI and XhoI and ligated into expression vector pET22b (Novagen) digested with the same enzymes. Clones were screened by colony PCR and confirmed by DNA sequencing. The recombinant expression vector pET22b-BprV encoded residues 1–476 of BprV (NCBI Accession No. ABQ13116) and pET22b-BprB encoded residues 1–476 of BprB (NCBI Accession No. AAA80562). Both contained a C-terminal His6 tag for affinity chromatography.
2.2. Protein expression and purification
The expression and purification procedures for the AprV2, AprB2, BprV and BprB proteases were as follows. The appropriate expression construct was used to transform the Escherichia coli strain Rosetta-Gami(DE3)pLysS (Novagen). A single colony was inoculated into 100 ml 2YT medium containing 100 µg ml−1 ampicillin and 34 µg ml−1 chloramphenicol and the culture was grown overnight at 310 K with shaking. 10 ml overnight culture was used to inoculate 1 l 2YT medium containing 100 µg ml−1 ampicillin and 34 µg ml−1 chloramphenicol and the culture was grown at 310 K with shaking. When the cultures reached an OD600 of 0.6, protein expression was induced by the addition of 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG). Following the addition of IPTG the cultures were grown for ∼16 h at 289 K with shaking at 250 rev min−1. The cells were harvested by centrifugation (3500g, 277 K, 20 min) and resuspended in ice-cold lysis buffer (25 mM bis-tris pH 6.8, 300 mM NaCl, 2.5 mM CaCl2, 10 mM imidazole and 5% glycerol). The resuspended cells were homogenized and incubated with 1 mg ml−1 lysozyme for 30 min at 277 K. The cells were sonicated and insoluble material was pelleted by centrifugation (15 000g, 277 K, 30 min). The supernatant was filtered (0.2 µm) and applied onto a 1 ml His-Trap FF column (Amersham Biosciences) pre-equilibrated with lysis buffer. His-tagged protease was eluted from the column using a linear gradient of 25–350 mM imidazole in 25 mM bis-tris pH 6.8, 300 mM NaCl, 2.5 mM CaCl2 and 5% glycerol. The eluted fractions containing protease (determined by SDS–PAGE analysis) were pooled and further purified by size-exclusion chromatography using a Superdex S75 10/30 column (GE Healthcare Life Sciences) with 25 mM bis-tris pH 6.5, 150 mM NaCl, 5 mM CaCl2 and 5% glycerol as the elution buffer. The purity of the purified protease was assessed by SDS–PAGE. Typically, ∼2 mg protease was produced per litre of culture. The expressed protease precursors were processed during expression, yielding mature protease with a C-terminal RLEHHHHHH tag for purification (the residues RLE are a cloning artefact). N-terminal sequencing was used to determine the N-terminal sequence of the purified mature proteases. AprV2 and AprB2 contained residues 130–474 of the protease precursor. BprB and BprV contained residues 133–476 of the protease precursor. The predicted molecular mass of the mature proteases was ∼37 kDa.
Purified proteases were shown to be active by measuring the cleavage of the low-molecular-weight elastin-like peptide N-methoxysuccinyl-Ala-Ala-Pro-Val p-nitroanilide (AAPVn; Sigma). The activity assay was performed by incubating 1 µM purified protease with varying concentrations of AAPVn in 20 mM Tris pH 8, 5 mM CaCl2 at 310 K for 20 h. The release of the free nitroanilide over time was detected at 405 nm using a fluorescence microplate reader (FLUOstar Optima; Betsuyaku et al., 1996 ▶).
2.3. Crystallization
Purified AprV2 and AprB2 proteases were concentrated to ∼6 mg ml−1 by ultracentrifugation in a buffer containing 25 mM bis-tris pH 6.5, 150 mM NaCl, 5 mM CaCl2 and 5% glycerol. Crystallization experiments were carried out using the hanging-drop vapour-diffusion method in 24-well Linbro tissue-culture plates. The Crystallization Basic Kit (Sigma) was initially used to screen for conditions that promoted crystal formation. 1 µl protein solution was mixed with 1 µl reservoir buffer and allowed to equilibrate over 0.5 ml reservoir buffer at 293 K. Both proteases crystallized using 0.2 M sodium acetate, 0.1 M sodium cacodylate pH 6.5 and 30% PEG 8000 as the reservoir buffer. Crystals of AprV2 and AprB2 typically grew within 2 d (Figs. 1 ▶ a and 1 ▶ b). While fine screens were performed by varying both the pH and the PEG 8000 concentration, diffraction-quality crystals were obtained for both AprV2 and AprB2 in the original condition. Single crystals isolated from these drops were used to collect data sets for the AprV2 and AprB2 proteases.
Figure 1.

(a) Crystals of AprB2. The crystals are 80 µm in the longest dimension. (b) Crystals of AprV2. The crystals are 60 µm in the longest dimension. (c) Crystals of BprB. The crystals are 80 µm in the longest dimension. (d) Crystals of BprV. The crystals are 60 µm in the longest dimension.
Purified BprV and BprB proteases were concentrated to ∼5 mg ml−1 by ultracentrifugation in a buffer containing 25 mM bis-tris pH 6.5, 150 mM NaCl, 5 mM CaCl2 and 5% glycerol immediately prior to performing crystallization experiments. Crystallization experiments were carried out using the sitting-drop vapour-diffusion method in 96-well Intelli-Plates set up using a Phoenix RE liquid-handling robot (Rigaku). 100 nl protein solution was mixed with 100 nl reservoir buffer and allowed to equilibrate over 120 µl reservoir buffer at 293 K. The Crystallization Basic Kit (Sigma), PEG/Ion kit (Hampton Research) and Index kit (Hampton Research) were used to screen for conditions that promoted crystal formation. BprV crystallized using 0.1 M bis-tris pH 6.5 and 25% PEG 3350 as the reservoir buffer, whereas BprB crystallized using 30% PEG 1500 as the reservoir buffer. Fine screens, altering the pH and the PEG concentration, were then performed using the hanging-drop vapour-diffusion method in 24-well Linbro tissue-culture plates at 293 K. Crystals of BprV and BprB typically grew within 2 d (Figs. 1 ▶ c and 1 ▶ d). Diffraction-quality crystals of BprV and BprB were obtained in 0.1 M sodium acetate pH 5.5, 22.5% PEG 3350 and 32.5% PEG 1500, respectively. Single crystals isolated from these drops were used to collect data sets for the BprV and BprB proteases.
2.4. Data collection and processing
A single crystal of AprV2, AprB2, BprV or BprB was mounted in a nylon cryoloop before being flash-frozen in a nitrogen stream at 100 K (Oxford Cryosystems). No additional cryoprotectants were added. X-ray diffraction data were measured in-house using a Rigaku RU-H3RHB rotating-anode X-ray generator equipped with Osmic mirrors and a Rigaku R-AXIS IV++ image-plate detector. Data were collected to resolutions of 2.0 Å for AprV2, 1.7 Å for AprB2, 2.0 Å for BprV and 1.8 Å for BprB (Fig. 2 ▶). For the AprV2 and AprB2 data sets 720 images were collected at an oscillation width of 0.5° with an exposure time of 20 min per image. The crystal-to-detector distances for the AprV2 and AprB2 data sets were 150 and 100 mm, respectively. For the BprV data set 440 images were collected at an oscillation width of 0.5° with an exposure time of 7 min per image and for BprB 504 images were collected at an oscillation width of 0.5° with an exposure time of 5 min per image. The crystal-to-detector distances for the BprV and BprB data sets were 150 and 130 mm, respectively. All data were indexed, integrated and scaled using MOSFLM and SCALA within CCP4 and CCP4i (Potterton et al., 2003 ▶; Leslie, 1992 ▶; Evans, 2006 ▶; Collaborative Computational Project, Number 4, 1994 ▶). A σ cutoff was not used during data processing. Data-collection and processing statistics are summarized in Table 2 ▶.
Figure 2.

Diffraction patterns from AprV2 (a), AprB2 (b), BprV (c) and BprB (d) crystals. The resolution limit at the edge of the plate is 2.0 Å for AprV2 and BprV, 1.6 Å for AprB2 and 1.8 Å for BprB.
Table 2. Data-collection statistics for crystals of AprV2, AprB2, BprV and BprB.
Values in parentheses are for the highest resolution shell.
| AprV2 | AprB2 | BprV | BprB | |
|---|---|---|---|---|
| Wavelength (Å) | 1.54178 | 1.54178 | 1.54178 | 1.54178 |
| Space group | P1 | P1 | P21 | P21 |
| Unit-cell parameters (Å, °) | a = 43.06, b = 45.98, c = 47.24, α = 97.8, β = 115.2, γ = 113.9 | a = 42.68, b = 45.82, c = 45.75, α = 98.4, β = 114.0, γ = 114.6 | a = 38.5, b = 89.6, c = 47.7, β = 113.6 | a = 38.5, b = 90.5, c = 44.1, β = 109.9 |
| Resolution range (Å) | 30.5–2.0 (2.11–2.00) | 20.4–1.7 (1.79–1.70) | 29.9–2.0 (2.11–2.00) | 24.4–1.8 (1.90–1.80) |
| Observed/unique reflections | 40583/17666 | 114567/26960 | 90202/20040 | 126861/24844 |
| Multiplicity | 2.3 (2.3) | 4.2 (4.2) | 4.5 (4.4) | 5.1 (4.4) |
| 〈I/σ(I)〉 | 13.5 (3.2) | 22.6 (5.9) | 21.2 (6.4) | 23.8 (5.8) |
| Completeness (%) | 93.9 (89.3) | 92.0 (88.0) | 99.8 (99.0) | 94.7 (83.9) |
| Rmerge† | 0.060 (0.265) | 0.048 (0.223) | 0.060 (0.205) | 0.057 (0.216) |
R
merge = 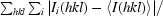
 , where I
i(hkl) and 〈I(hkl)〉 represent the diffraction-intensity values of the individual measurements and the corresponding mean values.
, where I
i(hkl) and 〈I(hkl)〉 represent the diffraction-intensity values of the individual measurements and the corresponding mean values.
3. Results and discussion
The crystals of both AprV2 and AprB2 belonged to the triclinic space group P1, with unit-cell parameters a = 43.1, b = 46.0, c = 47.2 Å, α = 97.8, β = 115.2, γ = 115.2° for AprV2 and a = 42.7, b = 45.8, c = 45.7 Å, α = 98.4, β = 114.0, γ = 114.6° for AprB2. Assuming the presence of one molecule in the asymmetric unit, the Matthews coefficient was calculated to be 1.94 Å3 Da−1 for AprV2 and 1.92 Å3 Da−1 for AprB2 (Matthews, 1968 ▶). These values correspond to an estimated solvent content of ∼36% for both proteases. The crystals of both BprV and BprB belonged to the monoclinic space group P21, with unit-cell parameters a = 38.5, b = 89.6, c = 47.7 Å, β = 113.6° for BprV and a = 38.5, b = 90.5, c = 44.1 Å, β = 109.9° for BprB. Assuming the presence of one molecule in the asymmetric unit, the Matthews coefficient was calculated to be 2.04 Å3 Da−1 for BprV and 1.94 Å3 Da−1 for BprB (Matthews, 1968 ▶). These values correspond to an estimated solvent content of 40 and 36% for BprV and BprB, respectively. An FFAS search (Jaroszewski et al., 2005 ▶) using the sequence of the mature domain of AprV2 (residues 130–474 of the protease precursor) as a query identified several close homologues to these proteases. These homologues included a thermitase from Thermoactinomyces vulgaris (36% identity over 277 residue pairs; PDB code 1tec; Gros et al., 1989 ▶), the Bacillus sp. Ak.1 serine protease (39% identity over 278 residue pairs; PDB code 1dbi; Smith et al., 1999 ▶) and the subtilisin BPN′ from B. amyloliquefaciens (38% identity over 272 residue pairs; PDB code 1sbt; Alden et al., 1971 ▶), suggesting that molecular replacement could be used to solve these structures.
Acknowledgments
This research was supported by the Australian Research Council through funding to the Australian Research Council (ARC) Centre of Excellence in Structural and Functional Microbial Genomics. CJP is a National Health and Medical Research Council of Australia (NHMRC) Training Fellow (Australia). JCW is an ARC Federation Fellow and an NHMRC Honorary Principal Research Fellow. WW is supported by a Faculty scholarship provided by the Department of Biochemistry and Molecular Biology at Monash University and was previously supported by a scholarship provided by the Australian Research Council Centre of Excellence in Structural and Functional Microbial Genomics. We would also like to acknowledge the Protein Production Unit at Monash University for parallel purification of the proteases and the Monash Biomedical Proteomics facility for N-terminal sequencing.
References
- Alden, R. A., Birktoft, J. J., Kraut, J., Robertus, J. D. & Wright, C. S. (1971). Biochem. Biophys. Res. Commun.45, 337–344. [DOI] [PubMed]
- Betsuyaku, T., Nishimura, M., Yoshioka, A., Takeyabu, K., Miyamoto, K. & Kawakami, Y. (1996). Am. J. Respir. Crit. Care Med.154, 720–724. [DOI] [PubMed]
- Billington, S. J., Johnston, J. L. & Rood, J. I. (1996). FEMS Microbiol. Lett.145, 147–156. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Depiazzi, L. J. & Richards, R. B. (1979). Aust. Vet. J.55, 25–28. [DOI] [PubMed]
- Egerton, J. R. & Parsonson, I. M. (1969). Aust. Vet. J.45, 345–349. [DOI] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Green, L. E. & George, T. R. (2008). Vet. J.175, 173–180. [DOI] [PubMed]
- Gros, P., Fujinaga, M., Dijkstra, B. W., Kalk, K. H. & Hol, W. G. J. (1989). Acta Cryst. B45, 488–499. [DOI] [PubMed]
- Jaroszewski, L., Rychlewski, L., Li, Z., Li, W. & Godzik, A. (2005). Nucleic Acids Res.33, W284–W288. [DOI] [PMC free article] [PubMed]
- Kortt, A. A., Caldwell, J. B., Lilley, G. G., Edwards, R., Vaughan, J. & Stewart, D. J. (1994). Biochem. J.299, 521–525. [DOI] [PMC free article] [PubMed]
- Kortt, A. A., Riffkin, M. C., Focareta, A. & Stewart, D. J. (1993). Biochem. Mol. Biol. Int.29, 989–998. [PubMed]
- Leslie, A. G. W. (1992). Jnt CCP4/ESF–EACBM Newsl. Protein Crystallogr.26
- Lilley, G. G., Riffkin, M. C., Stewart, D. J. & Kortt, A. A. (1995). Biochem. Mol. Biol. Int.36, 101–111. [PubMed]
- Lilley, G. G., Stewart, D. J. & Kortt, A. A. (1992). Eur. J. Biochem.210, 13–21. [DOI] [PubMed]
- Liu, D. & Yong, W. K. (1993). Res. Vet. Sci.55, 124–129. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Potterton, E., Briggs, P., Turkenburg, M. & Dodson, E. (2003). Acta Cryst. D59, 1131–1137. [DOI] [PubMed]
- Riffkin, M. C., Focareta, A., Edwards, R. D., Stewart, D. J. & Kortt, A. A. (1993). Gene, 137, 259–264. [DOI] [PubMed]
- Smith, C. A., Toogood, H. S., Baker, H. M., Daniel, R. M. & Baker, E. N. (1999). J. Mol. Biol.294, 1027–1040. [DOI] [PubMed]
- Stewart, D. J. (1979). Res. Vet. Sci.27, 99–105. [PubMed]
- Wani, S. A. & Samanta, I. (2006). Vet. J.171, 421–428. [DOI] [PubMed]