

The catalytic subunits of the human pyruvate dehydrogenase phosphatase isoforms 1 and 2 have been purified and crystallized. The crystals of PDP1c and PDP2c diffracted to 2.45 and 2.0 Å resolution, respectively.
Keywords: glucose metabolism, pyruvate dehydrogenase complex, pyruvate dehydrogenase phosphatase
Abstract
Pyruvate dehydrogenase phosphatase (PDP) is a mitochondrial serine phosphatase that activates phosphorylated pyruvate dehydrogenase complex by dephosphorylation. In humans, two PDP isoforms (1 and 2) have been identified. PDP1 is composed of a catalytic subunit (PDP1c) and a regulatory subunit (PDP1r), whereas PDP2 consists of only a catalytic subunit (PDP2c). Both PDP1c and PDP2c have been crystallized individually and complete X-ray diffraction data sets have been collected to 2.45 and 2.0 Å resolution, respectively. The PDP1c crystals belonged to space group P41212 or P43212, with unit-cell parameters a = b = 65.1, c = 216.1 Å. The asymmetric unit is expected to contain one molecule, with a Matthews coefficient V M of 2.56 Å3 Da−1. The PDP2c crystals belonged to space group P212121, with unit-cell parameters a = 53.6, b = 69.1, c = 109.7 Å. The asymmetric unit is expected to contain one molecule, with a Matthews coefficient V M of 1.91 Å3 Da−1.
1. Introduction
The mitochondrial pyruvate dehydrogenase complex (PDC) is a macromolecular complex that catalyzes the oxidative decarboxylation of pyruvate to produce acetyl-CoA and NADH, linking glycolysis to the Krebs cycle and lipogenic pathways (Harris et al., 2002 ▶). Since the decarboxylation reaction is irreversible, PDC plays the role of a rate-limiting enzyme in glucose metabolism. PDC activity is tightly regulated by reversible phosphorylation in mammalian cells (Harris et al., 2002 ▶; Reed et al., 1985 ▶; Holness & Sugden, 2003 ▶). Pyruvate dehydrogenase kinase (PDK) inactivates PDC by phosphorylation of the E1p component of PDC, whereas pyruvate dehydrogenase phosphatase (PDP) restores PDC activity by dephosphorylation of phospho-E1p. Starvation and diabetes cause a decrease in PDP expression with a reciprocal increase in PDK expression to inactivate PDC for glucose conservation (Wu et al., 2000 ▶; Huang et al., 2003 ▶), which is an unfavourable condition for type 2 diabetes. In humans, many disease-causing mutations in PDC components have been reported to be associated with lactic acidosis (Robinson, 2001 ▶; Hengeveld & de Kok, 2002 ▶). Recently, two PDP mutations associated with diminished or abolished PDP activity in cells have been shown to result in PDC deficiency and death in one case (Maj et al., 2005 ▶; Cameron et al., 2009 ▶).
To date, two PDP (1 and 2) isoforms have been identified in mammalian mitochondria (Huang et al., 1998 ▶). PDP1 is a heterodimer consisting of subunits with molecular weights of 97 and 50 kDa. The smaller subunit (PDP1c) has phosphatase activity, whereas the larger subunit is an FAD-containing regulatory protein (PDP1r; Teague et al., 1982 ▶). Since native PDP2 has not been isolated to date, it is not known whether the PDP2 catalytic subunit (PDP2c) requires a regulatory subunit. PDP1c and PDP2c share a sequence identity of 53% and both belong to the Mg2+-dependent protein phosphatase M1A (PPM1A; formally PP2C) family (Huang et al., 1998 ▶). PDP1c, but not PDP2c, binds to a lipoyl-bearing domain of the E2p component of PDC in a Ca2+-dependent manner, resulting in a tenfold enhancement of PDP1c activity (Pettit et al., 1982 ▶; Maj et al., 2006 ▶; Turkan et al., 2002 ▶). Co-localization with the E1p substrate is apparently the primary factor in the Ca2+-dependent stimulation of PDP1c activity. A crystal structure of rat PDP1c alone has recently been determined (Vassylyev & Symersky, 2007 ▶). The structure revealed that PDP1c has a very similar structure to that of PPM1A. However, the locations of the Ca2+-binding and lipoyl domain-binding sites are still unclear. On the other hand, PDP2c binds to neither Ca2+ ion nor the lipoyl domain, despite its high sequence identity to PDP1c (Huang et al., 1998 ▶; Karpova et al., 2003 ▶). Since PDP2c has not been crystallized, the molecular basis for its inability to interact with the lipoyl domain is not known.
In order to obtain structural insights into the different characteristics of human PDP1c and PDP2c, both proteins have been crystallized as the first step towards solving their structures.
2. Materials and methods
2.1. Expression and purification of PDP1c and PDP2c proteins
Expression plasmids for N-terminally His6-tagged mature forms of human PDP1c (residues 71–537) and PDP2c (residues 67–529) in pET28 vector (EMD Chemicals, Gibbstown, New Jersey, USA) were kindly supplied by Dr Brian Robinson, University of Toronto, Canada. The expressed proteins from these plasmids have 21 residues including the His6 tag (MGSSHHHHHHSSGLVPRGSHM) at the N-terminus of the mature sequences of PDP1c and PDP2c. For PDP1c preparation, the original plasmid was used without modifications. For PDP2c expression, the NheI–XhoI fragment coding the mature PDP2c sequence in the parent plasmid was subcloned into the pSUMO (Small Ubiquitin MOdifier) vector (Lifesensors, Malvern, Philadelphia, USA). In the resulting vector, the protein is linked to an N-terminally His6-tagged SUMO protein moiety. A tobacco etch virus (TEV) protease recognition sequence (ENLYFQ↓AS, with the arrow showing the cleavage site) was engineered into the linker region. The primer sequences used for PCR amplification were TCAAGCTAGCTCAACAGAGGAAGATGATTTTC (forward, NheI site in bold) and GGTGCTCGAGTAGGATGG (reverse, XhoI site in bold). The sequence of the resulting plasmids was confirmed by DNA sequencing.
The His6-tagged PDP1c or His6-tagged SUMO-PDP2c protein was expressed by co-transformation with pGroESL, which overexpresses the chaperonins GroEL and GroES in the Escherichia coli BL21 cell line. An initial culture was established by overnight incubation at 310 K in 50 ml LB medium. 1 l fresh LB medium was subsequently inoculated with 10 ml of the overnight culture and allowed to grow for 3.5 h. The incubation temperature was subsequently lowered to 301 K and maintained for 3 h, followed by 296 K for an additional 2 h. In the final incubation step, isopropyl β-d-1-thiogalactopyranoside was added to a final concentration of 0.5 mM and the cells were grown overnight. Harvested cells were ruptured on ice by sonication in a lysis buffer consisting of 100 mM potassium phosphate buffer pH 7.5, 500 mM NaCl, 0.1% Triton X-100, 0.1% Tween-20, 10% glycerol, 10 mM imidazole, 5 mM β-mercaptoethanol (BME), 0.5 mM benzamidine, 1 mM phenylmethylsulfonyl fluoride and 1 mg ml−1 lysozyme. The cell lysate was clarified by centrifugation and the supernatant was loaded onto an Ni–NTA column (Qiagen, Valencia, California, USA) at room temperature. After extensive washing with wash buffer (100 mM potassium phosphate pH 7.5, 500 mM NaCl, 10% glycerol, 20 mM imidazole and 5 mM BME), the proteins were eluted with an elution buffer consisting of the wash buffer and 250 mM imidazole. Further purification was conducted on a Superdex 200 column (GE Healthcare, Piscataway, New Jersey, USA) with a gel-filtration buffer consisting of 50 mM potassium phosphate pH 7.5, 100 mM KCl, 5% glycerol and 5 mM BME. Peak fractions were combined and the purified proteins in the gel-filtration buffer were concentrated by ultrafiltration for storage at 193 K (Fig. 1 ▶).
Figure 1.

SDS–PAGE gel of purified PDP1c, PDP2c and dissolved PDP2c crystals. The gel was stained with Coomassie Brilliant Blue. Lane M, molecular-weight markers (kDa).
For crystallization of PDP1c, the N-terminally His6-tagged PDP1c was used without removing the tag. For crystallization of PDP2c, the His6-tagged SUMO moiety was removed as follows. His6-SUMO-PDP2c and TEV protease were mixed in a mass ratio of 25:1 in digestion buffer (50 mM potassium phosphate pH 7.5, 250 mM KCl, 5% glycerol and 20 mM BME). The mixture was incubated at 277 K overnight with shaking. The completeness of the digestion was determined by SDS–PAGE. The digestion mixture was purified on an Ni–NTA column equilibrated with digestion buffer. The eluted fractions were concentrated and further purified on a Superdex 200 column as described above. The SUMO-free PDP2c contained two residual residues (AS) at the N-terminus of the mature sequence. The purified protein was concentrated to 15–20 mg ml−1 and stored in small aliquots at 193 K (Fig. 1 ▶).
2.2. Crystallization of PDP1c and PDP2c
For crystallization of PDP1c and PDP2c, the final concentrated protein solutions after the Superdex 200 gel filtration were used without a buffer change. Initial crystals were obtained by screening for crystallization conditions using a Phoenix RE crystallization robot (Rigaku, The Woodlands, Texas, USA) and Qiagen crystallization kits (Qiagen). Crystallization drops were set up on 96-well Intelli-Plates (Art Robbins, Sunnyvale, California, USA) by mixing 0.2 µl protein solution and 0.2 µl screening solution. The volume of the reservoir solutions was 50 µl. The plates were sealed with plastic film and stored at 293 K. The crystallization plates were observed with a Minstrel HP automated crystal-imaging system (Rigaku). Once initial crystallization conditions for PDP1c and PDP2c had been determined, optimization was carried out by the hanging-drop method with 24-well VDX plates (Hampton Research, Aliso Viejo, California, USA) at 293 K. The volumes of the crystallization drops and reservoir solution were 2 µl (1 µl protein solution plus 1 µl reservoir solution) and 1 ml, respectively.
To check the constitution of the PDP2c crystals, several PDP2c crystals of roughly 0.3 mm in size were removed from the crystallization drop and serially transferred into 5 µl reservoir solution ten times to wash the crystals. During washing, precipitate was removed from the crystal surface as much as possible. Subsequently, the crystals were dissolved in SDS sample buffer, which was followed by electrophoresis.
2.3. X-ray diffraction data collection and processing
PDP1c and PDP2c crystals were serially transferred to cryoprotectant solutions containing the respective well solutions and increasing concentrations of glycerol (5–25% in 5% increments) for 1 h. These crystals were flash-frozen in liquid propane and stored in liquid nitrogen. Diffraction data were collected at 100 K using an ADSC Quantum 315 imaging detector on beamline 19ID in the Structural Biology Center at the Advanced Photon Source (Argonne, Illinois, USA). The beam size and wavelength were 0.1 × 0.1 mm and 0.9787 Å, respectively. For data collection from PDP1c crystals, the exposure time was 5 s and the oscillation angle was 0.5° for each image and a total of 200 images were collected to obtain a complete data set. For PDP2c crystals, the exposure time was 5 s and the oscillation angle was 0.3° for each image and a total of 400 images were collected. The data were processed using HKL-2000 (Otwinowski & Minor, 1997 ▶). Data-collection statistics are summarized in Table 1 ▶.
Table 1. Data-collection statistics.
Values in parentheses are for the highest resolution shell.
| PDP1c | PDP2c | |
|---|---|---|
| Wavelength (Å) | 0.978 | 0.978 |
| Resolution (Å) | 2.45 (2.49–2.45) | 2.0 (2.03–2.00) |
| No. of reflections | 132449 | 125090 |
| No. of unique reflections | 17911 | 27044 |
| Multiplicity | 7.4 (5.5) | 4.6 (4.3) |
| Rmerge† (%) | 9.0 (48.6) | 5.8 (41.9) |
| Completeness (%) | 99.3 (88.8) | 95.0 (97.9) |
| Mean I/σ(I) | 20.7 (2.3) | 20.6 (3.3) |
| Space group | P41212 or P43212 | P212121 |
R
merge = 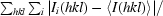
 , where I
i(hkl) is the ith observation of reflection hkl and 〈I(hkl)〉 is the mean intensity for all observations i of reflection hkl.
, where I
i(hkl) is the ith observation of reflection hkl and 〈I(hkl)〉 is the mean intensity for all observations i of reflection hkl.
3. Results and discussion
His6-tagged human PDP1c in the mature form was expressed in E. coli and was purified by metal-affinity chromatography and gel-filtration column chromatography (Fig. 1 ▶). The estimated final purity of PDP1c was >95%. PDP1c was soluble and stable at a concentration of 15 mg ml−1. Crystallization screening produced small crystals from JCSG+ suite (Qiagen) condition No. 45 consisting of 100 mM HEPES pH 7.5, 200 mM MgCl2 and 30% PEG 400. Crystals appeared from protein precipitation in 2 d and grew to 0.2 mm in a week (Fig. 2 ▶). Optimization of the crystal conditions by changing the pH or the concentration of protein or PEG 400 failed, suggesting that the initial conditions were optimal.
Figure 2.

Crystals of (a) PDP1c and (b) PDP2c. The crystal dimensions are approximately 0.2 × 0.05 × 0.05 mm for PDP1c crystals and 0.3 × 0.1 × 0.1 mm for PDP2c crystals.
The expression level of His6-tagged human PDP2c from the original pET28 plasmid was very low. Therefore, we constructed a new expression plasmid in which PDP2c was fused to a His6-tagged SUMO protein at the N-terminus. This plasmid robustly improved the expression level. SUMO-PDP2c was expressed and purified using a procedure similar to that used for PDP1c. After purification of SUMO-PDP2c, the SUMO moiety was removed by TEV protease digestion, which was followed by further purification by gel filtration to remove the cleaved SUMO moiety and TEV protease. The final PDP2c sample contained a smaller contaminant protein (about 42 kDa) which accounted for about 20% of the total protein (Fig. 1 ▶). As the smaller band was always present from the beginning of purification (data not shown), this contaminant is probably a degradation product of PDP2c caused by residual protease contamination. This SUMO-free PDP2c sample was concentrated to 20 mg ml−1 and initially crystallized in JCSG+ suite condition No. 30 consisting of 100 mM phosphate–citrate pH 4.2 and 40% PEG 300. The maximum crystal size was 0.05 mm. We optimized the conditions to improve the crystal size. The optimized condition was 100 mM sodium citrate pH 4.4 and 30% PEG 400. In this condition, PDP2c crystals grew to up to 0.3 mm in two weeks (Fig. 2 ▶). To check whether the crystals were formed by nondegraded PDP2c, the crystals were dissolved and analyzed by SDS–PAGE. As shown in Fig. 1 ▶, the dissolved crystal sample showed both the larger and the smaller bands, indicating that the crystals at least contained nondegraded PDP2c. However, it will not be completely clear whether the crystals really consist of both nondegraded and the probable degraded PDP2c until the structure has been solved.
Native data sets for the PDP1c and PDP2c crystals were collected on beamline 19ID in the Structural Biology Center at the Advanced Photon Source (Argonne, Illinois, USA). The PDP1c crystals diffracted X-rays to 2.45 Å resolution. The crystals belonged to space group P41212 or P43212 (tetragonal system), with unit-cell parameters a = b = 65.1, c = 216.1 Å, α = β = γ = 90°. The asymmetric unit is expected to contain one PDP1c molecule based on the value of the Matthews coefficient (2.56 Å3 Da−1). The PDP2c crystals diffracted to 2.0 Å resolution. The crystals belonged to space group P212121 (orthorhombic system), with unit-cell parameters a = 53.6, b = 69.1, c = 109.7 Å, α = β = γ = 90°. The asymmetric unit is expected to contain one PDP2c molecule based on the value of the Matthews coefficient (1.91 Å3 Da−1). The structure of rat PDP1c has previously been reported (PDB code 2pnq; Vassylyev & Symersky, 2007 ▶). The sequence identities between human PDP1c and rat PDP1c and between human PDP2c and rat PDP1c are 98% and 53%, respectively. Molecular replacement is being carried out using the rat PDP1c structure as a search model.
Acknowledgments
We thank Dr Brian Robinson of University of Toronto, Canada for providing the expression plasmids of human PDP1c and PDP2c. We also thank Drs Diana Tomchick and Chad Brautigam in the UT Southwestern Structural Biology Laboratory for help in synchrotron data collection. The use of the Argonne National Laboratory Structural Biology Center beamlines at the Advanced Photon Source was supported by the US Department of Energy, Office of Energy Research under Contract No. W-31-109-ENG-38. JK and MK were supported by Human Frontier Science Program, Young Investigators’ Grant (RGY82/2008).
References
- Cameron, J. M., Maj, M., Levandovskiy, V., Barnett, C. P., Blaser, S., Mackay, N., Raiman, J., Feigenbaum, A., Schulze, A. & Robinson, B. H. (2009). Hum. Genet.125, 319–326. [DOI] [PubMed]
- Harris, R. A., Bowker-Kinley, M. M., Huang, B. & Wu, P. (2002). Adv. Enzyme Regul.42, 249–259. [DOI] [PubMed]
- Hengeveld, A. F. & de Kok, A. (2002). Curr. Med. Chem.9, 499–520. [DOI] [PubMed]
- Holness, M. J. & Sugden, M. C. (2003). Biochem. Soc. Trans.31, 1143–1151. [DOI] [PubMed]
- Huang, B., Gudi, R., Wu, P., Harris, R. A., Hamilton, J. & Popov, K. M. (1998). J. Biol. Chem.273, 17680–17688. [DOI] [PubMed]
- Huang, B., Wu, P., Popov, K. M. & Harris, R. A. (2003). Diabetes, 52, 1371–1376. [DOI] [PMC free article] [PubMed]
- Karpova, T., Danchuk, S., Kolobova, E. & Popov, K. M. (2003). Biochim. Biophys. Acta, 1652, 126–135. [DOI] [PubMed]
- Maj, M. C., Cameron, J. M. & Robinson, B. H. (2006). Mol. Cell. Endocrinol.249, 1–9. [DOI] [PubMed]
- Maj, M. C., MacKay, N., Levandovskiy, V., Addis, J., Baumgartner, E. R., Baumgartner, M. R., Robinson, B. H. & Cameron, J. M. (2005). J. Clin. Endocrinol. Metab.90, 4101–4107. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Pettit, F. H., Teague, W. M. & Reed, L. J. (1982). Methods Enzymol.90, 402–407. [DOI] [PubMed]
- Reed, L. J., Damuni, Z. & Merryfield, M. L. (1985). Curr. Top. Cell. Regul.27, 41–49. [DOI] [PubMed]
- Robinson, B. H. (2001). The Metabolic and Molecular Bases of Inherited Disease, edited by C. R. Scriver, A. L. Beaudet, D. Valle, W. S. Sly, B. Childs, B. Vogelstein & K. W. Kinzler, Vol. 2, pp. 2275–2295. New York: McGraw–Hill.
- Teague, W. M., Pettit, F. H., Wu, T. L., Silberman, S. R. & Reed, L. J. (1982). Biochemistry, 21, 5585–5592. [DOI] [PubMed]
- Turkan, A., Gong, X., Peng, T. & Roche, T. E. (2002). J. Biol. Chem.277, 14976–14985. [DOI] [PubMed]
- Vassylyev, D. G. & Symersky, J. (2007). J. Mol. Biol.370, 417–426. [DOI] [PMC free article] [PubMed]
- Wu, P., Blair, P. V., Sato, J., Jaskiewicz, J., Popov, K. M. & Harris, R. A. (2000). Arch. Biochem. Biophys.381, 1–7. [DOI] [PubMed]