

Preliminary crystallographic studies of recombinant cellobiose phosphorylase from Cellulomonas uda in complex with reaction substrates and products have led to high-quality diffraction data at high resolution, thus enabling structural studies to dissect the structural and mechanistic determinants of disaccharide phosphorylase activity.
Keywords: cellobiose phosphorylases, GH94, disaccharide phosphorylases, inverting hydrolases
Abstract
Disaccharide phosphorylases are able to catalyze both the synthesis and the breakdown of disaccharides and have thus emerged as attractive platforms for tailor-made sugar synthesis. Cellobiose phosphorylase from Cellulomonas uda (CPCuda) is an enzyme that belongs to glycoside hydrolase family 94 and catalyzes the reversible breakdown of cellobiose [β-d-glucopyranosyl-(1,4)-d-glucopyranose] to α-d-glucose-1-phosphate and d-glucose. Crystals of ligand-free recombinant CPCuda and of its complexes with substrates and reaction products yielded complete X-ray diffraction data sets to high resolution using synchrotron radiation but suffered from significant variability in diffraction quality. In at least one case an intriguing space-group transition from a primitive monoclinic to a primitive orthorhombic lattice was observed during data collection. The structure of CPCuda was determined by maximum-likelihood molecular replacement, thus establishing a starting point for an investigation of the structural and mechanistic determinants of disaccharide phosphorylase activity.
1. Introduction
Disaccharide phosphorylases (DSPs) catalyze the synthesis and degradation of disaccharides using phosphate-activated sugar donors (Kitaoka & Hayashi, 2002 ▶). DSPs can be found in both prokaryotic and eukaryotic organisms and can be either inverting or retaining, acting on α-glycosidic or β-glycosidic bonds. Their enzymatic versatility is reflected in their classification, as DSPs are found in both glycosyl hydrolase (GH) and glycosyl transferase (GT) families, e.g. chitobiose phosphorylase (GlcNAc-β1,4-GlcNAc, inverting, GH94; Honda et al., 2004 ▶), maltose phosphorylase (Glc-α1,4-Glc, inverting, GH65; Qian et al., 1994 ▶) and trehalose phosphorylase (Glc-α1,1α-Glc, retaining; Nidetzky & Eis, 2001 ▶). The ability of DSPs to catalyze reversible reactions arises from the fact that the energy of a glycosyl-phosphate bond is lower than those of other glycosyl adducts such as glycosyl nucleotides, which are the usual substrates for glycosyl transferases. As a result, the catalyzed reaction is reversible. This, together with their high substrate specificity, has led to the emergence of DSPs as attractive enzymes for on-demand synthesis or breakdown of sugar chains. However, only a handful of DSPs are known to date and their high substrate specificity has proven to be a limiting factor in the development of a generic sugar-synthesis platform. Thus, the determination of the three-dimensional structures of these enzymes, especially in complex with their respective substrates, would greatly contribute to our understanding of their reaction mechanism and the determinants for substrate specificity and promiscuity and would thus provide important insights towards the development of enzymes with altered specificities.
Cellobiose phosphorylase (CP; EC 2.4.1.20) is present as an intracellular enzyme in bacteria that utilize cellulose (Ayers, 1958 ▶; Hulcher & King, 1958 ▶; Sato & Takahashi, 1967 ▶; Yernool et al., 2000 ▶). The enzyme is thought to be involved in the utilization of extracellular cellobiose formed through the action of cellulase. CP is a member of GH family 94, together with chitobiose phosphorylase and cellodextrin phosphorylase. All three enzymes catalyze the reversible phosphorolysis of a β-1,4-glycosidic bond with inversion of the anomeric configuration. The structures of two representatives of this family, cellobiose phosphorylase from Cellvibrio gilvus and chitobiose phosphorylase from Vibrio proteolyticus, have previously been determined (Hidaka, Honda et al., 2004 ▶; Hidaka et al., 2006 ▶). Cellobiose phosphorylase from Cellulomonas uda (CPCuda) shares 88% and 34% sequence identity, respectively, with these enzymes. The CPCuda phosphorolytic reaction was found to proceed via an ordered Bi-Bi mechanism (Nidetzky et al., 2000 ▶). According to this scenario, cellobiose binds first followed by phosphate; glucose is then released and finally α-d-glucose-1-phosphate (αGP) leaves the active site, thereby completing the reaction. It has further been shown that CPCuda shows a strong preference for the β-anomer of cellobiose and that the phosphorolysis reaction yields β-d-glucose. Therefore, the reaction catalyzed by CPCuda can be summarized as
In this paper, we report the production of recombinant CPCuda for crystallographic and enzymological studies and the X-ray diffraction analysis of crystals of CPCuda at high resolution.
2. Materials and methods
2.1. Expression and purification
The full-length sequence of C. uda cellobiose phosphorylase (GenBank AAQ20920.1) was cloned into a pTrc99a expression vector (pXCP; De Groeve et al., 2009 ▶). No tags were introduced during the cloning process. The pXCP vector was used to transform Escherichia coli BL21 (DE3) cells. The transformed cells were plated on agar plates containing 100 µg ml−1 carbenicillin (Cb) for antibiotic selection and were grown overnight at 310 K. A single colony was selected and grown at 310 K in a rotary shaker in a small volume (50 ml) of LB–Cb broth medium. The overnight culture was used to inoculate 0.8 l LB–Cb broth medium at a ratio of 1:200; growth followed at 310 K until an OD600 of 0.6–1.0 was reached. Protein expression was induced by adding isopropyl β-d-1-thiogalactopyranoside (IPTG) to a final concentration of 1 mM. After induction, cells were grown overnight in a rotary shaker and harvested by centrifugation at 6000g for 30 min at 277 K.
Large amounts of recombinant CPCuda were purified in three steps. Cell pellets were suspended in lysis buffer (50 mM Tris–HCl pH 7.8, 50 mM NaCl) and lysed by ultrasonication on ice (2 × 90 s, 30% amplitude). Cell debris was removed by centrifugation (14 000 rev min−1, 277 K, 30 min). To remove residual debris and to avoid nonspecific binding, the supernatant was loaded onto a 2 ml Q-Sepharose FF pre-column (GE Healthcare) equilibrated with 50 mM Tris–HCl pH 7.8, 50 mM NaCl and eluted with 1 M NaCl. The sample was diluted to reduce the final NaCl concentration to 50 mM and was then loaded onto a 10 ml pre-equilibrated Q-Sepharose FF column (50 mM Tris–HCl pH 7.8, 50 mM NaCl). The protein was eluted with a stepwise NaCl gradient and eluted from the column at 500 mM NaCl. The fractions containing the protein of interest were pooled; excess salt was removed by three or four consecutive washes using a 30 kDa molecular-weight cutoff Centricon (Sartorius Stedim Biotech) and the sample was concentrated to 2 ml. In a last polishing step, the concentrated protein sample was loaded onto a Superdex 200 16/60 column (GE Healthcare) equilibrated with 50 mM Tris–HCl pH 7.8, 300 mM NaCl. The fractions containing CPCuda were pooled, concentrated in 20 mM Tris–HCl pH 7.8, 5 mM NaCl using a centrifugal filter device (30 kDa molecular-weight cutoff) and stored at 277 K. All purification steps were assessed on Coomassie-stained SDS–PAGE gels.
2.2. Dynamic light scattering
The suitability of the storage and precrystallization buffers for supporting a monodisperse protein sample was screened by dynamic light-scattering studies (DLS). DLS measurements on recombinant CPCuda were performed using a Zetasizer Nano dynamic light-scattering instrument (Malvern) equipped with a 633 nm He–Ne laser and a temperature-controlled measuring chamber. Prior to all measurements, samples of purified CPCuda (12 mg ml−1) were clarified by centrifugation at 13 200 rev min−1.
2.3. Crystallization
In-house crystallization trials were set up based on the hanging-drop vapour-diffusion method. Droplets containing either 1 µl reservoir solution and 1 µl protein solution (12 mg ml−1 CPCuda in 20 mM Tris–HCl pH 7.8, 5 mM NaCl; the protein concentration was determined using the Bradford method; Bradford, 1976 ▶) or 1 µl reservoir solution, 1 µl protein solution and 1 µl 20 mM α-d-glucose-1-phosphate (αGP; final concentration) were mixed on 18 mm siliconized round cover slides, sealed and equilibrated against 400 µl reservoir solution at room temperature (293 K). Both types of droplet were screened against Crystal Screens 1 and 2 (Hampton Research). Additionally, crystallization screens were set up at the High Throughput Crystallization facilities at EMBL Hamburg (Mueller-Dieckmann, 2006 ▶) using the Classics and JCSG+ screens (Qiagen/Nextal), the Grid, Index, Natrix and SaltRX screens (Hampton Research) and the JBScreen Classic 1–10 (Jena Bioscience). In this case, 300 nl protein solution was mixed with 300 nl well solution in a sitting-drop vapour-diffusion setup at 293 K.
Promising leads were further optimized with the hanging-drop vapour-diffusion method via additional crystallization that probed a number of crystallization parameters.
2.4. Cryoprotection and soaking
Efficient cryoprotection of the crystals could be achieved by supplementing the crystals with 30%(v/v) glycerol. Alternatively, the presence of sodium formate in the crystallization condition allowed us to exploit the possibility of using high concentrations of sodium formate as a cryoprotectant (Lamour et al., 2002 ▶). The crystals were transferred from the crystallization drop to drops containing increasing concentrations of cryoprotectant using a nylon loop. Between transfer steps, crystals were left to equilibrate for 2–3 min. One to three rounds of transfer were sufficient to achieve efficient cryoprotection. The crystals were then mounted in a nylon loop and flash-cooled by submerging them in liquid nitrogen.
Protein–substrate complexes were obtained by incubating crystals of CPCuda in cryo-solutions containing appropriate amounts of substrate. Further handling and flash-cooling of the crystals were carried out as described above.
2.5. Data collection
Early batches of CPCuda crystals were tested for diffraction on the beamlines of EMBL Hamburg (DESY, Hamburg, Germany). A first complete data set (CP_SO4) was measured at SLS (Swiss Light Source, Villigen, Switzerland) on beamline X06SA operating at a wavelength of 1.0015 Å from a single crystal grown in 1.5 M ammonium sulfate, 0.1 M Tris–HCl pH 8.5 and cryoprotected with 30%(v/v) glycerol. 1440 images with an oscillation range of 0.25° were recorded on a Pilatus6M counting pixel detector system (Schlepütz et al., 2005 ▶).
Two additional data sets were recorded from single crystals on the ID23 beamline at the European Synchrotron Radiation Facility (ESRF; Grenoble, France) equipped with an ADSC Q315R detector. Both crystals grew from 4 M sodium formate, 20 mM αGP. A first crystal was cryoprotected by transfer to a solution containing 4 M sodium formate, 20 mM αGP, 30%(v/v) glycerol. A complete data set (CP_nat) was recorded at a wavelength of 0.9835 Å by rotating the crystal over 180° with an oscillation range of 1.0°. A second crystal (CP_cbi) was transferred to 5 M sodium formate, 20 mM cellobiose for cryoprotection. Owing to the crystal moving out of the beam centre, the data set was measured in two sweeps of 317 and 560 images, respectively, with an oscillation range of 0.25°.
A final data set (CP_glc) was collected at SLS at a wavelength of 1 Å over a range of 110° with an oscillation range of 0.5°. The crystal grew from 3.4 M sodium formate, 0.1 M sodium acetate trihydrate pH 4.6, 20 mM αGP and was transferred to a solution containing 5 M sodium formate, 0.1 M sodium acetate trihydrate pH 4.6, 50 mM αGP, 50 mM glucose for cryoprotection. Data-collection statistics can be found in Table 1 ▶.
Table 1. Data-collection statistics.
Values in parentheses are for the highest resolution shell.
| CP_nat | CP_SO4 | CP_glc | CP_cbi | |
|---|---|---|---|---|
| X-ray source | ESRF/ID23 | SLS/X06SA | SLS/X06SA | ESRF/ID23 |
| Temperature (K) | 100 | 100 | 100 | 100 |
| Cryoprotectant | Glycerol | Glycerol | Sodium formate | Sodium formate |
| Ligand | αGP | — | Glucose | Cellobiose |
| Wavelength (Å) | 0.9835 | 1.0015 | 1.0000 | 0.9330 |
| Crystal-to-detector distance (mm) | 211 | 320 | 260 | 150 |
| Data range (°) | 110 | 150 | 110 | 140 |
| Frame oscillation (°) | 1.0 | 0.25 | 0.5 | 0.25 |
| Data-processing software | XDS/XSCALE | XDS/XSCALE | MOSFLM/SCALA | PROTEUM2 |
| Space group | P21 | P21 | P21 | P212121 |
| Resolution (Å) | 50–1.7 (1.74–1.70) | 50–1.8 (1.86–1.81) | 50–2.4 (2.53–2.40) | 98–2.0 (2.05–2.00) |
| Unit-cell parameters | ||||
| a (Å) | 85.69 | 86.10 | 85.41 | 85.69 |
| b (Å) | 103.80 | 103.78 | 102.74 | 103.64 |
| c (Å) | 98.50 | 99.15 | 98.31 | 195.56 |
| β (°) | 96.51 | 96.57 | 96.44 | 90.00 |
| No. of molecules in ASU | 2 | 2 | 2 | 2 |
| No. of observed reflections | 389149 | 425258 | 149337 | 227963 |
| No. of unique reflections | 181274 | 148898 | 54077 | 118758 |
| Redundancy | 2.15 | 2.86 | 2.76 | 1.92 |
| Completeness (%) | 96.5 (95.5) | 94.6 (87.1) | 81.6 (81.2) | 98.8 (90.8) |
| Rmeas†/Rint‡ | 12.0 (69.6) | 11.8 (60.6) | 20.5 (66.4) | 8.07 (23.7) |
| 〈I/σ(I)〉 | 8.66 (1.94) | 9.62 (2.41) | 5.7 (1.7) | 4.26 (1.95) |
R
meas is reported for CP_nat, CP_SO4 and CP_glc. It is the multiplicity-weighted R
merge, i.e.
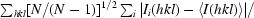
 , where I
i(hkl) is the intensity of the ith measurement of reflection hkl and 〈I(hkl)〉 is the average value over multiple measurements (Diederichs & Karplus, 1997 ▶).
, where I
i(hkl) is the intensity of the ith measurement of reflection hkl and 〈I(hkl)〉 is the average value over multiple measurements (Diederichs & Karplus, 1997 ▶).
R
int is reported for CP_cbi. It is 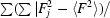
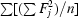 , where the inner sums are over the symmetry-equivalent reflections and the outer sums are over the unique hkl data. The term n is the number of equivalent data for a given hkl being merged.
, where the inner sums are over the symmetry-equivalent reflections and the outer sums are over the unique hkl data. The term n is the number of equivalent data for a given hkl being merged.
3. Results
CPCuda was recombinantly produced in E. coli in large amounts and was purified to homogeneity (10–15 mg protein per litre of culture). The purified enzyme migrated as a single band on SDS–PAGE corresponding to approximately 90 kDa, which is consistent with the calculated molecular weight of 90 814.72 Da for the monomer (Fig. 1 ▶). However, the elution profile of CPCuda on a calibrated gel-filtration column corresponded to a protein with a molecular weight of 180 kDa. This is consistent with the results from DLS, which revealed a hydrodynamic radius of 10.4 nm, corresponding to a molecular weight of 164 kDa for a globular protein. These results suggest that in contrast to previous reports (Nidetzky et al., 2000 ▶) CPCuda is a dimer in solution, as has been reported previously for closely related disaccharide phosphorylases (Hidaka, Kitaoaka et al., 2004 ▶; Hidaka et al., 2006 ▶).
Figure 1.
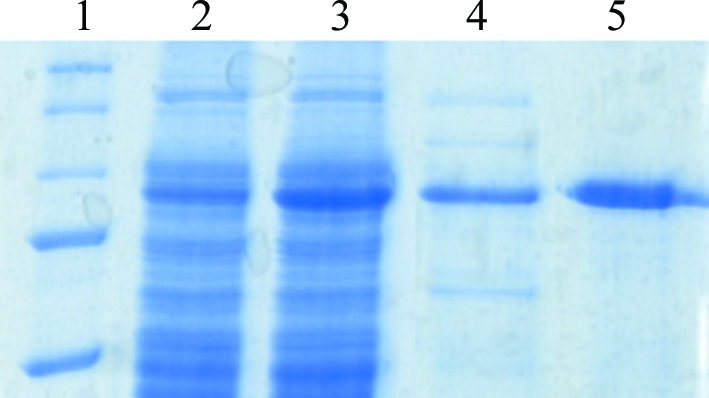
Coomassie-stained SDS–PAGE analysis of recombinant CPCuda. Lane 1, molecular-weight markers; lane 2, total cell lysate before induction; lane 3, total cell lysate after induction; lane 4, eluate from Q-Sepharose at 500 mM NaCl; lane 5, pooled fractions after gel filtration (Superdex 200).
Crystallization trials on pure and monodisperse CPCuda yielded a number of promising lead conditions based on ammonium and/or lithium sulfate as the precipitant. Optimization of the initial clusters of thin needles from these conditions led to thin rod-like crystals with approximate dimensions of 20 × 20 × 200 µm (Fig. 2 ▶ a). Further optimization yielded single diffracting crystals, which could be reproducibly grown overnight from 1–1.8 M ammonium sulfate, 0.1 M Tris–HCl pH 8.5. A complete data set (CP_SO4) to 1.8 Å resolution was recorded at at beamline X06SA of the SLS. However, the diffraction quality varied dramatically between crystals, so that a large number of crystals needed to be screened before crystals of good diffraction quality could be identified. We therefore initiated new crystallization trials in the presence of the substrates and products of the reaction catalyzed by CPCuda. This approach led to the identification of a new crystallization condition in the presence of 20 mM αGP (3–5 M sodium formate, unbuffered). The crystals grew reproducibly within 3–4 d as rectangular rods measuring 40 × 40 × 200 µm (Fig. 2 ▶ b). These crystals proved to be more consistent in terms of diffraction quality and offered the additional advantage that they could be cryoprotected in the absence of glycerol, a molecule that is known to occasionally occupy sugar-binding sites (Hidaka et al., 2006 ▶). Two data sets, CP_nat and CP_cbi, were recorded from these crystals to 1.7 and 2.0 Å resolution, respectively, at at beamline ID23 of the ESRF. The addition of 0.1 M sodium acetate trihydrate pH 4.6 to the original crystallization buffer allowed us to initiate soaking trials to obtain a ternary complex of the enzyme, as CPCuda is not catalytically active at this pH (unpublished results). One data set (CP_glc) from such a crystal soaked with glucose and αGP was recorded to a resolution of 2.4 Å at beamline X06SA of the SLS.
Figure 2.

CPCuda crystals grown from (a) 1.5 M ammonium sulfate, 0.1 M Tris–HCl pH 8.5 and (b) 4.0 M sodium formate.
The CP_SO4 and CP_nat data sets were indexed and processed in the primitive monoclinic space group P21 using XDS (Kabsch, 1993 ▶; Table 1 ▶). The self-rotation function (SRF) as calculated using the program MOLREP (Vagin & Teplyakov, 1997 ▶) revealed three twofold-symmetry axes along x, y and z, but initial attempts to reindex the data in a primitive orthorhombic setting failed. The availability of the structure of the homologous cellobiose phosphorylase from Cellvibrio gilvus (CPCgil; PDB entry 2cqt; Hidaka et al., 2006 ▶) allowed us to approach the structure determination of CPCuda using maximum-likelihood molecular replacement as implemented in the program Phaser (McCoy et al., 2007 ▶). A semi-conservative monomeric search model was constructed with CHAINSAW (Stein, 2008 ▶) in which all nonconserved residues were truncated to the last common C atom. Analysis of crystal-packing densities suggested the presence of two monomers in the asymmetric unit for both crystals and for CP_SO4 Phaser could indeed convincingly place two monomers in the asymmetric unit, as demonstrated by the excellent scores (RFZ = 18.2, TFZ = 47.8, LLG = 3917; Fig. 3 ▶ a). Inspection of electron-density maps calculated with Fourier coefficients 2F o − F c,MR, αc,MR confirmed the solution and showed extra density for missing structural elements and side chains as well as unbiased F o − F c,MR, αc,MR density in the active site indicating the presence of sulfate and glycerol. Additionally, the monomers were found to form a dimer similar to that of the CPCgil enzyme. Consistent with the absence of peaks in the SRF away from the crystallographic axis, the twofold NCS was found to be nearly parallel to the unit-cell c axis. The combination of the crystallographic 21 screw axis along b with the NCS twofold nearly parallel to c then generates an extra 21 screw axis, yielding an apparent orthorhombic setting and explaining the observed self-rotation function.
Figure 3.

(a) Cα-atom traces and packing of CP_SO4 in the primitive monoclinic unit cell. Subunits in dimers making up the asymmetric unit of each lattice are coloured dark blue and cyan, respectively; symmetry-related molecules are depicted in greyscale. (b) Cα-atom traces and packing of CP_cbi in the primitive orthorhombic unit cell. Subunits in dimers making up the asymmetric unit of each lattice are coloured pink and magenta, respectively; symmetry-related molecules are depicted in greyscale. This figure was prepared with the program PyMOL (DeLano, 2002 ▶).
For the CP_nat data, a CP_SO4 monomer partially refined with REFMAC5 (Murshudov et al., 1997 ▶) was used as a search model. A rotation Z score of 21.1, a translation Z score of 48.0 and a log-likelihood gain of 4857 gave a dimer in the asymmetric unit, which was confirmed by the 2F o − F c,MR, αc,MR maps, although no additional density could be observed for bound αGP.
The CP_glc data set could not be processed by XDS, while MOSFLM (Leslie, 1992 ▶) was able to index and process the data in space group P21. Since indexing of the initial images prior to data collection suggested space group P212121, an efficient data-collection strategy based on a 110° sweep was applied, which resulted in only 81% completeness for the primitive monoclinic cell. However, despite the moderate completeness Phaser was able to place two monomers in the asymmetric unit (RFZ = 42.5, TFZ = 59.8, LLG = 8866) using a partially refined CP_nat monomer as the search model, corresponding to a V M of 2.41 Å3 Da−1 and a solvent content of 50.0%. The solution could be confirmed by 2F o − F c,MR, αc,MR electron-density maps and unbiased positive F o − F c,MR, αc,MR density could be observed in the +1 and −1 subsites of both active sites.
Owing to the crystal moving out of the beam centre, the CP_cbi data set was recorded in two sweeps of 80° and 140°. The first sweep could effectively be processed with XDS in space group P21; however, the overall completeness was only 60%. In contrast, the second part of the data set could not be processed with XDS or MOSFLM despite the apparent good quality of the diffraction images. We therefore employed the PROTEUM2 suite (Bruker AXS), which succeeded in indexing, processing and scaling the data in the primitive orthorhombic space group P212121 (Table 1 ▶). The switch in space group between the first and the second half of the data set raises some interesting possibilities. It is possible that the crystal moved not only out of the beam center but also, completely or partially, out of the cryostream. The associated warming of the crystal, followed by recooling when recentered, is comparable to the process of crystal annealing and might have caused structural changes inside the crystal. A space-group transition initiated by cryocooling has been reported previously (Campobasso et al., 1998 ▶) and it is possible that the annealing-like treatment of the crystal induced a comparable effect. The primitive orthorhombic cell shared the a and b axis with the primitive monoclinic cell, while its c axis was double that of the monoclinic cell.
These findings prompted us to reconsider the indexing of the previously collected data sets. CP_SO4 and CP_nat, but not CP_glc, could be successfully indexed in P212121 with the larger unit cell. However, the scaling statistics for P212121 indicated that P21 is indeed the true space group of these crystals [R meas (CP_SO4, P212121) = 22.5% (219.2%), R meas (CP_nat, P212121) = 17.8% (107.8%)]. On the other hand, the crystal packing in the primitive monoclinic versus the primitive orthorhombic cell is almost identical, indicating that the change in unit-cell parameters arises from very subtle differences between the two structures.
The asymmetric unit of CP_cbi was predicted to contain two monomers, corresponding to a solvent content of 49.6% and a Matthews coefficient of 2.44 Å3 Da−1. Consistent with the orthorhombic setting, the self-rotation function shows three perpendicular twofold axes, but no NCS peak away from the crystallographic axis could be observed. The native Patterson map calculated with the program phenix.xtriage (Adams et al., 2002 ▶) revealed a significant off-origin peak at (0.129, 0.5, 0) with 61% of the height of the origin peak, indicating strong pseudo-translational symmetry. This is caused by the presence of a twofold NCS axis parallel to the crystallographic c axis and might be the cause of the indexing problems experienced in XDS and MOSFLM. A refined monomeric CP_nat model was used for molecular replacement with Phaser, which placed two monomers in the asymmetric unit cell (Fig. 3 ▶ b) with unusually high statistics (RFZ = 50.1, TFZ = 87.7, LLG = 25413). These high values are probably partly a consequence of the presence of pseudo-translational symmetry. The pseudo-translation causes a systematic intensity modulation in the diffraction pattern which is mimicked by the molecular-replacement solution. This can result in a high correlation between F obs and F calc and thereby cause an artificial inflation of the Z scores and LLG values. Electron-density maps (2F o − F c,MR, αc,MR) confirmed the correctness of the solution and the presence of a dimer and showed clear unbiased F o − F c,MR, αc,MR density spanning the whole active site in both active sites, indicative of the presence of cellobiose.
The initial crystallization condition for the CPCuda crystals (1–1.8 M ammonium sulfate, 0.1 M Tris–HCl pH 8.5) is very similar to that for CPCgil (1.5 M ammonium sulfate, 0.1 M MES–NaOH pH 6.5, 5 mM glucose), as are the unit-cell parameters, despite the appreciable sequence difference in sequence (12%). Both proteins pack very similarly in the unit cell, although the CPCuda structure appears to adopt a more ‘open’ conformation. It is indeed remarkable that two proteins that differ by 12% (to a large extent with respect to their surface residues) crystallize in such similar fashion, especially since a single-residue substitution at the cell surface often results in a dramatically different crystal packing for a given protein sequence. We expect that the availability of the two closely homologous crystal structures at high resolution will allow a detailed comparison of crystal lattice contacts and might provide insights into the employment of well ordered surface epitopes to mediate lattice contacts, as has recently been proposed by Price et al. (2009 ▶).
Model building and refinement for CP_SO4, CP_nat, CP_glc and CP_cbi is under way and will be reported elsewhere.
Acknowledgments
We would like to thank the EMBL (Hamburg, Germany), the ESRF (Grenoble, France) and the SLS (Villigen, Switzerland) for beamtime allocation and technical support. This work was supported by grant IWT50191 from the ‘Instituut voor de Aanmoediging van Innovatie door Wetenschap en Technologie in Vlaanderen’ (IWT Flanders, Belgium).
References
- Adams, P. D., Grosse-Kunstleve, R. W., Hung, L.-W., Ioerger, T. R., McCoy, A. J., Moriarty, N. W., Read, R. J., Sacchettini, J. C., Sauter, N. K. & Terwilliger, T. C. (2002). Acta Cryst. D58, 1948–1954. [DOI] [PubMed]
- Ayers, W. A. (1958). J. Bacteriol.76, 515–517. [DOI] [PMC free article] [PubMed]
- Bradford, M. M. (1976). Anal. Biochem.72, 248–254. [DOI] [PubMed]
- Campobasso, N., Begun, J., Costello, C. A., Begley, T. P. & Ealick, S. E. (1998). Acta Cryst. D54, 448–450. [DOI] [PubMed]
- De Groeve, M. R. M., De Baere, M., Hoflack, L., Desmet, T., Vandamme, E. & Soetaert, W. (2009). Protein Eng. Des. Sel.22, 393–399. [DOI] [PubMed]
- DeLano, W. (2002). The PyMOL Molecular Viewer. DeLano Scientific, Palo Alto, California, USA.
- Diederichs, K. & Karplus, P. A. (1997). Nature Struct. Biol.4, 269–275. [DOI] [PubMed]
- Hidaka, M., Honda, Y., Kitaoka, M., Nirasawa, S., Hayashi, K., Wakagi, T., Shoun, H. & Fushinobu, S. (2004). Structure, 12, 937–947. [DOI] [PubMed]
- Hidaka, M., Kitaoka, M., Hayashi, K., Wakagi, T., Shoun, H. & Fushinobu, S. (2004). Acta Cryst. D60, 1877–1878. [DOI] [PubMed]
- Hidaka, M., Kitaoka, M., Hayashi, K., Wakagi, T., Shoun, H. & Fushinobu, S. (2006). Biochem. J.398, 37–43. [DOI] [PMC free article] [PubMed]
- Honda, Y., Kitaoka, M. & Hayashi, K. (2004). Biochem. J.377, 225–232. [DOI] [PMC free article] [PubMed]
- Hulcher, F. H. & King, K. W. (1958). J. Bacteriol.76, 571–577. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (1993). J. Appl. Cryst.26, 795–800.
- Kitaoka, M. & Hayashi, K. (2002). Trends Glycosci. Glycotechnol.14, 35–50.
- Lamour, V., Hoermann, L., Jeltsch, J.-M., Oudet, P. & Moras, D. (2002). Acta Cryst. D58, 1376–1378. [DOI] [PubMed]
- Leslie, A. G. W. (1992). Jnt CCP4/ESF–EACBM Newsl. Protein Crystallogr.26
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst.40, 658–674. [DOI] [PMC free article] [PubMed]
- Mueller-Dieckmann, J. (2006). Acta Cryst. D62, 1446–1452. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Nidetzky, B. & Eis, C. (2001). Biochem. J.360, 727–736. [DOI] [PMC free article] [PubMed]
- Nidetzky, B., Eis, C. & Albert, M. (2000). Biochem. J.351, 649–659. [PMC free article] [PubMed]
- Price, W. N. et al. (2009). Nature Biotechnol.27, 51–57. [DOI] [PMC free article] [PubMed]
- Qian, N., Stanley, G. A., Hahn-Hägerdal, B. & Radström, P. (1994). J. Bacteriol.176, 5304–5311. [DOI] [PMC free article] [PubMed]
- Sato, M. & Takahashi, H. (1967). Agric. Biol. Chem.31, 470–474.
- Schlepütz, C. M., Herger, R., Willmott, P. R., Patterson, B. D., Bunk, O., Brönnimann, C., Henrich, B., Hülsen, G. & Eikenberry, E. F. (2005). Acta Cryst. A61, 418–425. [DOI] [PubMed]
- Stein, N. (2008). J. Appl. Cryst.41, 641–643.
- Vagin, A. & Teplyakov, A. (1997). J. Appl. Cryst.30, 1022–1025.
- Yernool, D. A., McCarthy, J. K., Eveleigh, D. E. & Bok, J. D. (2000). J. Bacteriol.182, 5172–5179. [DOI] [PMC free article] [PubMed]