

Abstract
Cognitive deficits are core features of psychiatric disorders and contribute substantially to functional outcome. It is still unclear, however, how cognitive deficits are related to underlying genetic liability and overt clinical symptoms. Fortunately, animal models of susceptibility genes can illuminate how the products of disease-associated genetic variants affect brain function and ultimately alter behavior. Using as a reference findings from the Cognitive Neuroscience Treatment Research to Improve Cognition in Schizophrenia program and the SchizophreniaGene database, we review cognitive data from mutant models of rare and common genetic variants associated with schizophrenia.
Keywords: CNTRICS, MATRICS, executive control, working memory, 22q11, DISC1
Introduction
Psychiatric disorders are heterogeneous behavioral syndromes marked by significant cognitive impairments. These deficits are particularly prominent in psychotic disorders and are especially severe in schizophrenia. Given that cognitive capacity largely influences functional outcome, improving cognition is currently a major focus of schizophrenia research. It is clear that model systems will be instrumental in developing targeted treatments toward this end. As such, defining the proper role for and utilizing the full potential of animal models is of utmost importance. In this review, we discuss insights from mutant animal models and what they reveal about the cognitive architecture related to genetic risk for schizophrenia.
As our guide, we use suggestions from the Cognitive Neuroscience Treatment Research to Improve Cognition in Schizophrenia (CNTRICS) program.1 Recent comprehensive reviews on animal models have focused on the various behavioral paradigms available, their ability to measure specific cognitive constructs, and their potential use in drug development.2,3 These reviews are largely based on suggestions from the Measurement and Treatment Research to Improve Cognition in Schizophrenia (MATRICS) initiative.4 In order to achieve rapid utility, the cognitive test battery that emerged from this collaboration is based on well-characterized, classical neuropsychological instruments. CNTRICS emerged in recognition that these tests are rather imprecise and now dated and identified 7 cognitive domains for more refined characterization in schizophrenia.5 We therefore discuss findings from mutant animal models in light of these cognitive domains.
Carving up and Comparing Cognition
It is still not clear whether cognitive dysfunction in schizophrenia reflects discrete independent deficits or a generalized functional impairment. This ambiguity is due to the current statistical methods used for identifying different cognitive domains, the rather blunt neuropsychological instruments used for cognitive profiling, and/or the limitations of classifying cognitive processes based on folk psychology.6,7 For example, MATRICS identified independent cognitive domains based on analytic methods that a priori orthogonalize different factors.8 Performances on tests measuring these cognitive processes, however, are significantly correlated with each other, and these processes are not necessarily independent at the level of common genetic influence.9,10 While schizophrenia patients show cognitive deficits across a range of neuropsychological tests, when modern cognitive neuroscience tasks are used, deficits within a given task can be highly specific.11 For now, resolving the specificity vs generality of cognitive deficits awaits profiling the cognitive architecture of both healthy and clinical populations using more refined cognitive neuroscience tools.
Regardless of the exact architecture of cognitive deficits, measuring relevant cognitive processes in model animals usually relies on the neuronal and psychological homology of these processes. Thus, tasks that are dependent on the same neural systems as those in humans and thought to measure the same psychological constructs are usually the most relevant. Using these criteria alone, however, may be problematic. For example, although prefrontal cortical (PFC) dysfunction contributes to cognitive deficits in schizophrenia, not all PFC-dependent processes are affected in patients,11 and PFC dysfunction is implicated in most, if not all, psychiatric disorders. Moreover, this poor specificity also applies to the psychological constructs themselves, such as working memory (WM), which is impaired across a variety of disorders.12–14 As such, a given PFC-dependent, WM task used in animal models may be relevant to autism or attention deficit/hyperactivity disorder in addition to schizophrenia. This implies that, because of the poor specificity of any particular, or even set of, cognitive deficits, behavioral tests alone cannot be used to validate an animal model. Additional support, like a solid genetic foundation, is needed for animal models to provide reliable insight into a disease process.
Varieties of Mutant Animal Models
Model systems can be used for various means including mimicking an etiological factor, inducing a pathogenetic cascade, or recapitulating a final pathophysiological state. Here, we focus on mutant models of schizophrenia susceptibility genes and thus do not discuss all animal models that may be relevant for understanding schizophrenia neurobiology and cognition. Importantly, mutant animal models themselves are only as reliable as the genetic and clinical data upon which they are based, and choosing wisely among genetic findings is the first step in creating etiologically valid models. Although the genetic architecture of psychiatric disorders, as for other complex disorders, is still unknown, it likely consists of both highly penetrant rare alleles and common alleles of small effect. The potential, however, to translate genetic findings into etiologically valid animal models varies greatly for rare vs common risk alleles.
It is unclear to what extent or in what manner common genetic variants contribute to the etiology of psychiatric disorders, whether it is by incrementally increasing disease risk, through strong epistatic interactions, and/or by modifying the penetrance of rare variants. Even so, for most candidate genes with putative common risk variants, there is inconsistent statistical support, little consensus on the exact risk alleles, or scarce information on the alleles’ functional effects.15–19 This makes modeling efforts problematic, and, although models based on these genes may provide important insight into the biological functions of a particular gene, they do not identify which of these functions are relevant to disease pathogenesis.20,21 In light of the large number of potential schizophrenia susceptibility genes with common risk alleles,15 none of which have been unequivocally identified, we only discuss models available from the “top 30” candidates genes of the SchizophreniaGene database (http://www.szgene.org). This is an impartially compiled database of risk genes based on meta-analyses of genetic association studies (table 1). The list is by no means definitive and is constantly evolving, and we use it only to avoid a biased representation of genes. Critically, because most mutant models based on these genes do not recapitulate specific risk alleles, their relevance to disease pathogenesis remains unknown.
Table 1.
Rare and Common Alleles Associated With Schizophrenia and Related Mutant Modelsa
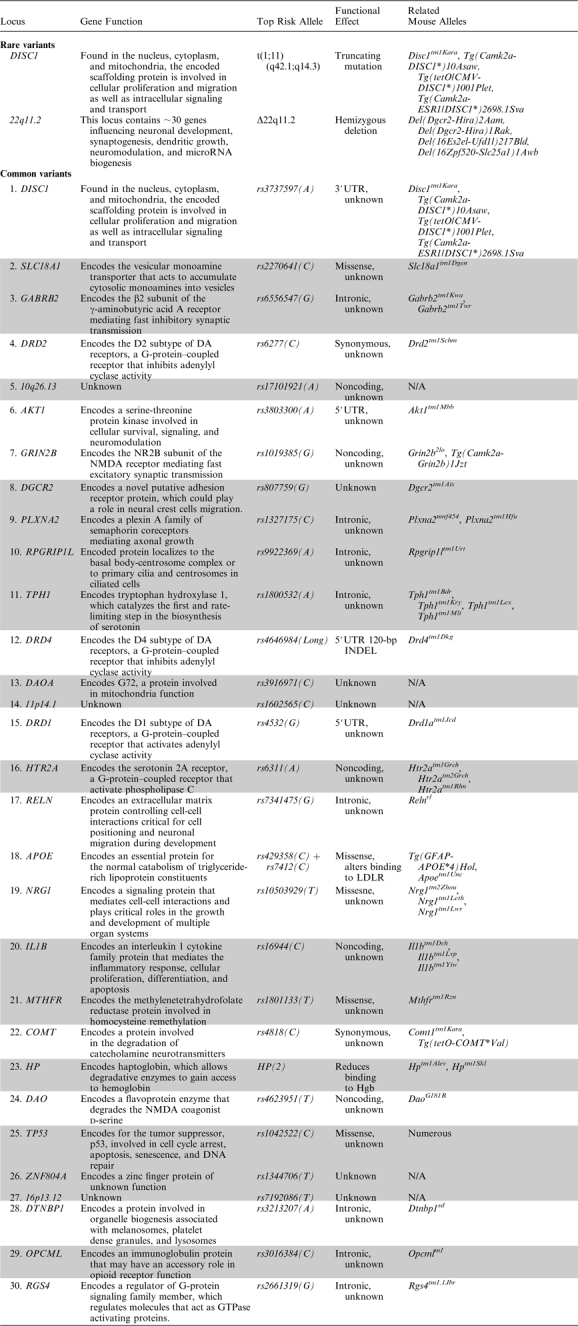 |
Although individual rare alleles (even recurrent ones) are so far associated with only a small fraction of cases,22,23 their statistical associations with disease status are robust and their functional effects clear. Due to the limited resolution of available genotyping platforms, most rare alleles identified thus far are large structural variants.24 These include a balanced chromosomal translocation (1;11)(q42.1;q14.3)25 and copy number variants (CNVs) at 22q11.2 as well as possibly at 1q21.1, 2p16.3, 15q13.3, 16p11.2, and 17p12.22,23,26–30 The ability to model these genetic lesions accurately makes models of rare alleles etiologically valid and more likely to identify disease-relevant biological pathways.31 As of yet, there are few animal models of CNVs, although some of them encompass or disrupt promising candidate genes. Two examples are the neurexin 1 gene (NRXN1) within the 2p16.3 locus and the α7 cholinergic nicotinic receptor gene (CHRNA7) within the 15q13.3 locus. Mutant NRXN1 mice have not been extensively characterized cognitively.32 Mice missing a single copy of CHRNA7 do not have robust cognitive deficits,33 and in the absence of a model of the entire 15q13.3 microdeletion it remains unclear how CHRNA7 deficiency may contribute to the diverse phenotypes associated with this CNV. We thus primarily focus on available mutant models from 2 of the few structural variants to be unequivocally associated with schizophrenia, t(1;11) and 22q11.2 microdeletion.
Different methodological approaches have been used to model the t(1;11) translocation that disrupts the DISC1 and DISC2 (Disrupted-In-Schizophrenia-1 and -2) genes and segregates with psychiatric disorders in a large Scottish family. Most models assume that the translocation produces a truncated DISC1 protein that interferes with the intact copy's function and thus overexpresses a truncated form of human DISC1 (figure 1). These include mice expressing an N-terminal fragment under the αCAMKII promoter (Tg(Camk2a-DISC1)10Asaw),34 tet-off double transgenic mice expressing human DISC1 under the CMV promoter with tetracycline under the αCAMKII promoter (Tg(tetO/CMV-DISC1*)1001Plet × Tg(Camk2a-tTA)1Mmay),35 and mice expressing a C-terminal fragment of DISC1 under the αCAMKII promoter using a single transgenic inducible and reversible system (Tg(Camk2a-ESR1/DISC1*)2698.1Sva).36 Another set of models was generated using an N-ethyl-N-nitrosourea-induced mutagenesis screen that uncovered 2 lines of mice carrying missense mutations in exon 2 of Disc1.37 Only one model (Disc1tm1Kara) directly targeted the endogenous murine Disc1 ortholog in a way that mimics the effect of the Scottish mutation (figure 1).38 These mice carry a truncating lesion in Disc1 that abolishes expression of the major Disc1 isoforms and, despite an artificial polyadenylation signal, causes low expression of the truncated protein indicating its potential instability under physiological conditions.39
Fig. 1.

Animal Models of the DISC1 Locus. Top: chromosomal location and genetic structure of the human DISC1 locus. The t(1;11) translocation break point occurs between exons 8 and 9 (arrow) with exons 9–13 (black) relocated to chromosome 11. Above it are the allele symbols and transgenic constructs based on the human DISC1 gene used to model the functional effects of the translocation. These models interfere with endogenous mouse Disc1 function in a dominant negative manner. Bottom: syntenic chromosomal location and genetic structure of the mouse Disc1 locus and the corresponding break point location (arrow). Below it are 5 of the Disc1 alleles so far described in mice. Disc1del arose from a spontaneous 25–base pair (bp) deletion within exon 6 that introduces a premature stop codon in exon 7 (TGA) of 129 and related strains of mice. Disc1tm1Kara was engineered to carry a premature stop codon at the end of exon 8 (TGA) followed by a polyadenylation signal in an attempt to recapitulate the translocation, but because this was created in a 129S6/SveV background the allele also carries the endogenous 25–bp deletion. Disc1Rgsc1390 and Disc1Rgsc1393 carry missense mutations in exon 2.
22q11.2 microdeletions were the first recurrent genetic lesion unequivocally associated with increased risk for schizophrenia.26 Carriers of these microdeletions, which occur predominantly de novo, are at inordinately high risk of developing schizophrenia.22,23,26,27 Fortunately, the human 22q11.2 locus is conserved within the syntenic region of mouse chromosome 16 and harbors nearly all orthologs of the human genes. There are now various single-gene and multigene deletions of this locus as well as mice hemizygous for all genes within the minimal 1.5-Mb region associated with schizophrenia (figure 2; Lgdel, Del(Dgcr2-Hira)1Rak, and Df(16)A, Del(Dgcr2-Hira)2Aam).40,41 Given the close similarity of the lesion in these mice to that occurring in humans, they afford an unprecedented opportunity for characterizing the cognitive consequences and underlying neural correlates associated with genetic risk for the disease.
Fig. 2.

Animal Models of the 22q11.2 Locus. Left: chromosomal location and genetic organization of the 22q11.2 locus. Each closed circle represents one gene. This 1.5-megabase critical region is flanked by low-copy repeat sequences (gray boxes) making it prone to nonhomologous recombination. PRODH-P and DGCR6-like are pseudogenes. Right: syntenic region of mouse chromosome 16 and the genetic organization of the corresponding orthologs. Single-gene deletion models that have been analyzed within the relevant CNTRICS cognitive domains are indicated with open circles. Various multigene deletion models that have been cognitively characterized and are discussed in the main text are also shown. The official allele symbols for the models are Lgdel, Del(Dgcr2-Hira)1Ra; Df(16)A, Del(Dgcr2-Hira)2Aam; Df1, Del(16Es2el-Ufd1l)217Bld; and Smdel, Del(16Zpf520-Slc25a1)1Awb.
Findings Across Cognitive Domains
Perception
Patients with schizophrenia show deficits even at the lowest levels of sensory function. This is evident across all sensory modalities and can detrimentally impact downstream cortical processing. Consequently, many deficits in higher order cognitive operations may result from primary sensory deficits.42 Although this area of research has a long history in animal models,43,44 few mutant models of putative risk genes have directly assessed perceptual disturbances.
While recognizing the widespread perceptual deficits in schizophrenia,45 CNTRICS prioritized 2 constructs within visual perception for further clinical investigation: (1) visual gain control and (2) visual integration. The nominated tasks within these domains do not have clear rodent analogs, but CNTRICS recognized that prepulse inhibition (PPI) and mismatch negativity (MMN) might serve as a useful measures of gain control.46
PPI is a relatively robust preattentive assay with great translational potential that is well characterized at the neural circuit level. Its sensitivity and specificity, however, are rather low. Moreover, drugs that are not clinically effective cognitive enhancers nonetheless improve PPI suggesting that it has low discriminatory power for novel therapeutic agents. PPI in the context of mutant animal models has been recently reviewed,47 and thus, we do not review it further here.
MMN is another preattentive measure that is well characterized and reflects early stages of auditory processing. Similar to perceptual deficits in other domains, levels of MMN are directly related to overall functional ability in patients.48 Only one mutant strain of a “top 30” candidate gene has assessed MMN. These mice lack functional isoforms of Nrg1 due to a targeted mutation of the EGF domain (Nrg1tm1Cbm).49 When presented with novel auditory stimuli following a stream of familiar stimuli, Nrg1+/− mice do not show the characteristic negative deflection in the event-related potential indicating changes in sensory integration.50 In order to compare these findings with human studies, it will be important to know if and how the early perceptual changes in these mutants, and potentially other models, are related to higher order cognitive function.
Social and Affective Regulation
Directly relating social behavior in nonhuman animals to human social cognition is inherently problematic. Like for other domain-specific operations, it is uncertain what gives social cognition its uniqueness, whether it is specialization at the level of information processing or information selectivity.51 In fact, in humans, developmentally early changes in perceptual biases52 and faulty perceptual processing in the adult53,54 can be responsible for higher order social cognitive dysfunction making directly modeling social deficits in animals difficult. CNTRICS chose to focus on 2 constructs within social cognition: (1) emotional identification and (2) emotional responding.55 While there are rodent social interaction tasks that may, at face value, seem similar to these constructs, whether the relevant underlying mechanisms are the same is unknown. Nevertheless, social behavior in mutant animal models is commonly assessed and is discussed further in the accompanying review of negative symptoms.
Executive Control
Executive control encompasses an array of higher order cognitive processes. It is broadly defined as the ability to coordinate thoughts and actions in accordance with changing external demands or internal goals.56 Achieving this requires dynamically updating, manipulating, selecting, retrieving, and integrating information across sensory and motor modalities. Executive control is thus a domain-general process influencing various other cognitive operations. Deficits in executive control can potentially explain patients’ poor performance in a variety of cognitive tasks including those measuring attention,57 long-term memory (LTM),58 and WM.59 CNTRICS selected 2 constructs within executive control for further clinical development: (1) rule generation and selection and (2) dynamic adjustments in control.60
For rule generation and selection, CNTRICS nominated an intra/extradimensional set-shifting task that has been recently adapted for use in rodents and is based on the Wisconsin card sorting test. Mice lacking dopamine (DA) D2 receptors (Drd2tm1Schm) have selective deficits in this task. Under some conditions, these mice show deficits in early discrimination stages but not later rule abstraction or set-shifting stages.61 Using a different testing procedure that produces the expected performance profile in control animals, mutant mice exhibit selective deficits during the rule reversal phase.62 This later result is consistent with other work showing that D2 knockout (KO) mice have robust reversal learning deficits.63,64 In contrast, transgenic mice overexpressing the high-activity, human catechol-O-methyl transferase (COMT) Val allele (Tg(tetO –COMT-Val) × Tg(Eno2tTA)) seem to show selective impairments in the set-shifting phase of this task and perform normally at all other stages.65 Interestingly, while patient performance in this task varies according to clinical status, the most consistent impairments are during the reversal and set-shifting phases.66
Although there is a rodent analog to the task recommended by CNTRICS for dynamic adjustments in control, there is, as of yet, no data for mutant animal models. Other findings that may be relevant to executive control deficits in schizophrenia are from tasks looking at other forms of behavioral flexibility. In an operant-based reversal learning task and an inhibitory control task, mice carrying a spontaneous null allele of the reelin gene (Relnrl, reeler mice) learn and perform these tasks normally.67 For the related construct of impulsivity, as measured with a delayed discounting and a go/no-go task, mice lacking D4 receptors (Drd4tm1Dkg) also perform normally.68 Given the central role executive control deficits may play in schizophrenia in particular and in psychiatric disorders in general, the further development and use of tasks measuring these various cognitive processes are critically needed to behaviorally characterize mutant models.
Control of Attention
As for many psychological terms, the precise meaning of attention varies and spans basic bottom-up, exogenously driven, and top-down, endogenously driven, modulation of processing within other cognitive or perceptual domains. Attention is closely related to and often intimately involved in executive control and WM.57 CNTRICS focused on the single construct of attentional control because other basic forms of attention seem to be intact in patients. Although there are several rodent paradigms for measuring attentional control,69,70 there are no findings from models based on the “top 30” gene list or from models of the DISC1 or 22q11 rare alleles. Considering the central importance of attentional function to cognition and its impairment in schizophrenia, there is an obvious dearth of studies measuring attention in mutant models of candidate genes.
Long-term Memory
Learning and memory deficits are well known in schizophrenia and are one of the strongest predictors of functional outcome. As mentioned earlier, executive control deficits may account for many aspects of memory impairments in patients including LTM deficits.71 CNTRICS focused on 3 constructs within learning and memory for further development: (1) relational encoding and retrieval, (2) item encoding and retrieval, and (3) reinforcement learning.58 There are several animal behavioral paradigms for each of these constructs, although there are few direct analogs to the specific tests recommended by CNTRICS. We nonetheless summarize some findings in mutant animals that may be relevant to learning and memory deficits in schizophrenia.
There are many classical learning and memory paradigms that are widely used to characterize genetically modified mice. As such, there is a rich literature for mutant models with potential relevance to schizophrenia and other psychiatric disorders. Transgenic mice overexpressing the N-methyl-D-aspartic acid receptor NR2B subunit (Tg(Camk2a-Grin2b)1Jzt), encoded by the Grin2B gene, show enhancements in novel object recognition, conditioned fear and extinction, and spatial reference memory in the Morris water maze (MWM).72 Not surprisingly, postnatal deletion of NR2B from principle forebrain neurons (Grin2b2lox × Tg(Camk2a-cre)1Gsc) produces not only widespread deficits in learning and memory including impaired spatial reference memory in the MWM and elevated Y-maze but also deficits in spatial navigation and visual discrimination.73 Homozygous DA D1A receptor KO mice (Drd1atm1Jcd) are consistently impaired in the spatial reference memory version of the MWM, and, while they learn conditioned fear normally, they retain fear memory for longer and show delayed extinction.74–76 Reeler mice show normal acquisition and retention of spatial reference memory in the MWM but show less consistent retention of conditioned fear.67,77 Sdy mice (Dtnbp1sdy) carry a spontaneous null mutation of Dtnbp1, the dysbindin gene, and have impaired spatial reference memory and object recognition memory but enhanced conditioned fear.78–80 In contrast, mice carrying a spontaneous null mutation of DAO (DaoG181R) perform better in the spatial reference memory version of the MWM.81 Finally, heterozygous Nrg1 mutant animals (Nrg1tm1Cbm) have impaired conditioned fear but normal object recognition memory.50
Only a few of the mutant DISC1 mice so far described have been assessed in LTM tests. Transgenic DISC1 mice (Tg(Camk2a-DISC1)10Asaw) have normal spatial reference memory in the MWM,34 but double transgenic (Tg(tetO/CMV-DISC1*)1001Plet × Tg(Camk2a-tTA)1Mmay) female mice are impaired in this same task.35 Mice carrying the truncated Disc1 allele (Disc1tm1Kara) have intact spatial reference memory in the MWM and also express normal conditioned fear and object recognition memory.39 Thus, despite varying genetic approaches, DISC1 mutant mice seem to have essentially normal LTM using traditional learning and memory paradigms.
LTM function in models of the 22q11.2 microdeletion are just beginning to be characterized despite the prominent learning and memory deficits in human deletion carriers. Mice hemizygous for 18 orthologs within the 1.5-Mb region associated with schizophrenia risk (figure 2, Df1) have impaired conditioned fear memory,82 but mice hemizygous for just 7 of the orthologs do not (figure 2, Smdel).83 Depending on the exact behavioral protocol, mice with a hemizygous deletion spanning all the orthologous genes within the 1.5-Mb region either exhibit (Df(16)A)41 or do not exhibit (Lgdel)40 impaired conditioned fear (figure 2).
Importantly, the tasks mentioned above do not directly assess the aspects of executive control that may contribute to LTM impairments in patients. Additionally, it is likely that the neural mechanisms underlying the executive control of LTM differ from those underlying the actual encoding and consolidation of memories. Although some models show clear LTM deficits, other models such as DISC1 mutants do not. It remains to be seen how these mutant mice and others perform in tasks more closely measuring the LTM constructs identified by CNTRICS.
Working Memory
WM is probably the best characterized cognitive domain in those with schizophrenia. It is also a constantly evolving construct in cognitive neuroscience making it extremely difficult to model in animals.84,85 Modern cognitive neuroscience definitions of WM emphasize executive control as a central feature and disagree mostly about the structure and localization of the memory stores or “slave systems.”86 Here, we distinguish WM from simple short-term memory (STM). While STM is a limited capacity system for transiently holding readily accessible information, WM is the manipulation and use of this information in accordance with internal goals. Thus, WM can be understood as the specific implementation of executive control over STM. In fact, CNTRICS has prioritized the translation of 2 executive components of WM: (1) goal maintenance and (2) interference control.87 Thus, in order to be clinically meaningful, it is critical that studies of mutant and other types of models examine the executive components of WM.
Unfortunately, many commonly used rodent tasks lack explicit executive demands and thus likely confound STM and WM. These demands for executive control recruit various processes including the manipulation and updating of information held in STM in addition to goal maintenance and interference resolution. Importantly, for a given task, animals, including humans, may use several cognitive processes in conjunction with different memory systems. This can make a behavioral test an imprecise measure of any particular psychological construct like WM. The exact load, however, placed on executive control, and thus WM, can be influenced by the number of trials per day, the intertrial interval, and the delay across which information is held active in memory. These parameters vary greatly across tasks, and while spontaneous alternation tasks likely measure simple STM, the extent that delayed alternation and delayed nonmatch to place (DNMTP) tasks measure WM depends on the exact task parameters influencing executive control as outlined above. It should be noted that the fact that these latter tasks may depend on the integrity of functionally homologous neural regions and are similarly neuromodulated as human WM tasks88 suggests that they may tap into related cognitive and physiological processes. Nevertheless, the need to model executive processes explicitly should not be overlooked because they largely determine WM capacity in general89 and disproportionately contribute to WM deficits in schizophrenia patients.90
Given that WM deficits have long played a central role in the neuropsychology of schizophrenia, many mutant models have been tested in putative WM tasks, although, for reasons mentioned above, the extent to which this is true depends on specific task parameters. Conditional NR2B KO mice (Grin2b2lox × Tg(Camk2a-cre)1Gsc) are severely impaired and show chance performance in a simple spontaneous alternation task73 as do mice lacking Rgs4 (Rgs4tm1.1Jbr).91 In contrast, mice heterozygous for a deletion of the transmembrane (Nrg1tm2Zhou) or immunoglobulin-like domain (Nrg1tm1Leth) of Nrg1 perform normally in this task.92,93 However, mice with decreased expression of the type III Nrg1 isoform (Nrg1tm1Lwr) are impaired in a delayed alternation task that included nonrandomly introduced delay periods and extensive training.94 Akt1-deficient mice (Akt1tm1Mbb) are impaired under various pharmacological challenges in a delayed alternation task with only a single delay period.95 Both D2 KO mice and COMT-Val transgenic mice show delay-dependent impairments during a delayed alternation and DNMTP task, respectively.65,96 Conditional NR2B KO mice are impaired in a DNMTP task with a single delay period and with relatively long intertrial intervals.73 Transgenic ApoE learn a delayed spatial win-shift task more slowly than wild-type controls97 and Sdy mice learn a DNMTP task more slowly than wild-type controls but then perform normally when the delay period is lengthened in an unpredictable manner.67 Finally, reeler mice perform normally in an operant-based delayed matching to place task with short intertrial intervals and parametrically varied delay periods.67
It is possible that some DISC1 mutant mice may have actual deficits in WM attributable to deficits in executive control in the context of normal STM. Transgenic DISC1 mice (Tg(Camk2a-DISC1)10Asaw) perform normally in a spontaneous alternation task,34 while mice transiently expressing the truncated C-terminal DISC1 protein (Tg(Camk2a-ESR1/DISC1*)2698.1Sva) have delay-dependent impairments in a DNMTP task.36 Mutant mice carrying missense mutations in Disc1 (Disc1Rgsc1390 and Disc1Rgsc1393) also show deficits in a DNMTP task, but they are impaired only at the briefest delay periods where STM load is the lowest suggesting that nonmnemonic processes may be at play. Interestingly, mice with the targeted disruption of Disc1 (Disc1tm1Kara) are not impaired in STM tasks but display deficits in 2 independent DNMTP tasks with pseudorandomly introduced delay periods.38,39 In these studies, one task is prone to proactive interference due to short intertrial intervals, and the other task requires the successful updating of information in order to concurrently remember 2 independent spatial locations. Thus, both implementations of these DNMTP tasks likely require some type of executive control. It is important to note, however, that these studies, like others, did not explicitly manipulate variables associated with executive control.
Mice hemizygous for the orthologous 22q11.2 deletion also have deficits in delayed response tasks in addition to the learning and memory deficits described above. These mice (Del(Dgcr2-Hira)2Aam) are impaired in the acquisition of a delayed alternation T-maze task but perform normally once the task is acquired and delay periods are increased.41 Interestingly, this deficit may arise in part from impaired microRNA production due to deficiency of one gene within the deletion, Dgcr8, a microRNA processor; heterozygous deletion of Dgcr8 alone (Dgcr8Gt(xH157)Byg) similarly impairs acquisition of a DNMTP task.41 In contrast, haploinsufficiency of other examined genes within the deletion on their own do not cause robust deficits. Heterozygous and homozygous mice lacking the Nogo receptor (Rtn4rtm1Gogo) perform normally in a delayed alternation task,98 but homozygous animals of a different KO strain (Rtn4rtm1Stmr) are impaired in a delayed alternation task using a longer training schedule and with no manipulation of memory delay periods.99 This study, however, did not assess performance of heterozygous animals, and these are the most relevant to understanding how haploinsufficiency of individual genes contribute to the entire microdeletion syndrome. Heterozygous Comt KO mice show improvements in a DNMTP task, albeit at delays outside the range of immediate STM.65 Mice carrying a hypomorphic allele of Prodh (Prodhm1Kara) acquire a delayed alternation task as well as controls and perform normally when the memory delay is pseudorandomly lengthened.100 In Prodh mutant mice, however, there is an epistatic interaction between Prodh and Comt, such that Prodh deficiency causes a compensatory increase in Comt expression. Pharmacologically inhibiting this genetic feedback loop unmasks underlying DA dysfunction and reveals delayed alternation deficits in Prodh mutant mice using a single delay period.100 Taken together, this suggests that cognitive deficits associated with the 22q11.2 microdeletion result from the combined effects of genes acting individually and interactively.
Overall, it is clear that in order to better understand the neural and psychological constructs related to WM dysfunction and genetic risk for schizophrenia, novel rodent WM tasks are needed that parametrically vary the demand for executive control and thus isolate and specifically measure executive processes.
Cognition and Genetic Liability to Psychosis
Cognitive deficits are common to psychiatric disorders, but their exact relationship to the clinical syndromes is unclear. They are prominent in psychotic disorders, but psychosis is not a feature of all disorders with cognitive deficits, and within individuals, there is little relationship between cognition and psychotic symptoms. There is strong evidence that many cognitive deficits are mediated by the same genetic risk factors that lead to the overt disease. First-degree, unaffected relatives of those with schizophrenia, bipolar disorder, attention deficit/hyperactivity disorder, obsessive compulsive disorder, and autism, among others, show cognitive deficits, especially in executive control.12,101–103 This implies that genetic variants generally influencing cognition may also influence risk for psychiatric disorders.
The question remains, however, whether cognitive deficits lie along a disease pathway from genetic risk to clinical syndrome or arise independently due to genetic pleiotropy. Although cognitive deficits are common among psychiatric disorders, it is unclear if these are due to shared genetic liability across disorders. Given our limited understanding of the neural bases of cognitive processes and the exact nature of cognitive deficits across diagnostic groups, it is unknown whether deficits within a given domain, although superficially similar across different syndromes, result from the same abnormal physiological processes. The study of the mutant models reviewed above afford the opportunity to identify whether deficits within a cognitive domain converge merely at the behavioral level or whether there are common underlying neural correlates, and if so, how this may be related to diverse clinical phenotypes.
Conclusions
The focus on cognitive function in schizophrenia and other psychiatric disorders has grown tremendously. This is reflected by the recent establishment of both the MATRICS and CNTRICS programs and the subsequent basic science gap that animal models now need to fill. Although we have been critical of many current cognitive tasks used in animals, we do not suggest that all behavioral paradigms will have to necessarily mirror human tasks to be clinically relevant. A behavioral task may be extremely useful, regardless of its resemblance to any clinical test, if it is sensitive to and specific for some underlying disease process and predicts the clinical efficacy of therapeutic interventions. We speculate, however, that highly precise tools for probing cognition will be indispensable for identifying relevant disease processes. The fact that schizophrenia patients have executive control deficits across cognitive domains,104 are impaired even at the lowest levels of sensory perception,42 and have the most severe deficits in basic processing speed105 suggests a fundamental difference in some elementary and ubiquitous mechanism of cortical computation. Thus, carefully designed tasks for animal models are needed that dissect out and identify specific neural mechanisms underlying cognitive dysfunction. Ultimately, the integration of new cognitive neuroscience tools with those of new mutant animal models has the potential to clarify the relationships among genetic variation, cognition, and the psychopathology observed in those afflicted with mental illnesses.
Funding
National Institute of Mental Health (R01 MH077235, MH080234-01A1 to J.A.G.) National Alliance for Research on Schizophrenia and Depression (young investigator award); Simons Foundation.
Acknowledgments
We thank David Jentsch and Rebecca Levy for helpful comments and critical insights on an earlier draft of the manuscript. We apologize to our colleagues whose work we did not cite due to space limitations.
References
- 1.Carter CS, et al. Identifying cognitive mechanisms targeted for treatment development in schizophrenia: an overview of the first meeting of the Cognitive Neuroscience Treatment Research to Improve Cognition in Schizophrenia Initiative. Biol Psychiatry. 2008;64:4–10. doi: 10.1016/j.biopsych.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hagan JJ, Jones DN. Predicting drug efficacy for cognitive deficits in schizophrenia. Schizophr Bull. 2005;31:830–853. doi: 10.1093/schbul/sbi058. [DOI] [PubMed] [Google Scholar]
- 3.Young JW, Powell SB, Risbrough V, Marston HM, Geyer MA. Using the MATRICS to guide development of a preclinical cognitive test battery for research in schizophrenia. Pharmacol Ther. 2009;122:150–202. doi: 10.1016/j.pharmthera.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Green MF, et al. Approaching a consensus cognitive battery for clinical trials in schizophrenia: the NIMH-MATRICS conference to select cognitive domains and test criteria. Biol Psychiatry. 2004;56:301–307. doi: 10.1016/j.biopsych.2004.06.023. [DOI] [PubMed] [Google Scholar]
- 5.Cohen JD, Insel TR. Cognitive neuroscience and schizophrenia: translational research in need of a translator. Biol Psychiatry. 2008;64:2–3. doi: 10.1016/j.biopsych.2008.04.031. [DOI] [PubMed] [Google Scholar]
- 6.Dickinson D, Ragland JD, Gold JM, Gur RC. General and specific cognitive deficits in schizophrenia: Goliath defeats David? Biol Psychiatry. 2008;64:823–827. doi: 10.1016/j.biopsych.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bilder RM, et al. Phenomics: the systematic study of phenotypes on a genome-wide scale. Neuroscience. 2009;164:30–42. doi: 10.1016/j.neuroscience.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nuechterlein KH, Robbins TW, Einat H. Distinguishing separable domains of cognition in human and animal studies: what separations are optimal for targeting interventions? A summary of recommendations from breakout group 2 at the measurement and treatment research to improve cognition in schizophrenia new approaches conference. Schizophr Bull. 2005;31:870–874. doi: 10.1093/schbul/sbi047. [DOI] [PubMed] [Google Scholar]
- 9.Dickinson D, Gold JM. Less unique variance than meets the eye: overlap among traditional neuropsychological dimensions in schizophrenia. Schizophr Bull. 2008;34:423–434. doi: 10.1093/schbul/sbm092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Butcher LM, Kennedy JK, Plomin R. Generalist genes and cognitive neuroscience. Curr Opin Neurobiol. 2006;16:145–151. doi: 10.1016/j.conb.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 11.MacDonald AW, III, et al. Specificity of prefrontal dysfunction and context processing deficits to schizophrenia in never-medicated patients with first-episode psychosis. Am J Psychiatry. 2005;162:475–484. doi: 10.1176/appi.ajp.162.3.475. [DOI] [PubMed] [Google Scholar]
- 12.Arts B, Jabben N, Krabbendam L, van Os J. Meta-analyses of cognitive functioning in euthymic bipolar patients and their first-degree relatives. Psychol Med. 2008;38:771–785. doi: 10.1017/S0033291707001675. [DOI] [PubMed] [Google Scholar]
- 13.Martinussen R, Hayden J, Hogg-Johnson S, Tannock R. A meta-analysis of working memory impairments in children with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2005;44:377–384. doi: 10.1097/01.chi.0000153228.72591.73. [DOI] [PubMed] [Google Scholar]
- 14.Nakao T, et al. Working memory dysfunction in obsessive-compulsive disorder: a neuropsychological and functional MRI study. J Psychiatr Res. 2009;43:784–791. doi: 10.1016/j.jpsychires.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 15.Purcell SM, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanders AR, et al. No significant association of 14 candidate genes with schizophrenia in a large European ancestry sample: implications for psychiatric genetics. Am J Psychiatry. 2008;165:497–506. doi: 10.1176/appi.ajp.2007.07101573. [DOI] [PubMed] [Google Scholar]
- 17.Need AC, et al. A genome-wide investigation of SNPs and CNVs in schizophrenia. PLoS Genet. 2009;5:e1000373. doi: 10.1371/journal.pgen.1000373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stefansson H, et al. Common variants conferring risk of schizophrenia. Nature. 2009;460:744–747. doi: 10.1038/nature08186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi J, et al. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature. 2009;460:753–757. doi: 10.1038/nature08192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bodmer W, Bonilla C. Common and rare variants in multifactorial susceptibility to common diseases. Nat Genet. 2008;40:695–701. doi: 10.1038/ng.f.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Todd JA. Statistical false positive or true disease pathway? Nat Genet. 2006;38:731–733. doi: 10.1038/ng0706-731. [DOI] [PubMed] [Google Scholar]
- 22.ISC. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–241. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stefansson H, et al. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Altshuler D, Daly MJ, Lander ES. Genetic mapping in human disease. Science. 2008;322:881–888. doi: 10.1126/science.1156409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.St Clair D, et al. Association within a family of a balanced autosomal translocation with major mental illness. Lancet. 1990;336:13–16. doi: 10.1016/0140-6736(90)91520-k. [DOI] [PubMed] [Google Scholar]
- 26.Karayiorgou M, et al. Schizophrenia susceptibility associated with interstitial deletions of chromosome 22q11. Proc Natl Acad Sci U S A. 1995;92:7612–7616. doi: 10.1073/pnas.92.17.7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu B, et al. Strong association of de novo copy number mutations with sporadic schizophrenia. Nat Genet. 2008;40:880–885. doi: 10.1038/ng.162. [DOI] [PubMed] [Google Scholar]
- 28.McCarthy SE, et al. Microduplications of 16p11.2 are associated with schizophrenia. Nat Genet. 2009;41:1223–1227. doi: 10.1038/ng.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rujescu D, et al. Disruption of the neurexin 1 gene is associated with schizophrenia. Hum Mol Genet. 2009;18:988–996. doi: 10.1093/hmg/ddn351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kirov G, et al. Support for the involvement of large copy number variants in the pathogenesis of schizophrenia. Hum Mol Genet. 2009;18:1497–1503. doi: 10.1093/hmg/ddp043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arguello PA, Gogos JA. Modeling madness in mice: one piece at a time. Neuron. 2006;52:179–196. doi: 10.1016/j.neuron.2006.09.023. [DOI] [PubMed] [Google Scholar]
- 32.Etherton MR, Blaiss CA, Powell CM, Sudhof TC. Mouse neurexin-1alpha deletion causes correlated electrophysiological and behavioral changes consistent with cognitive impairments. Proc Natl Acad Sci U S A. 2009;106:17998–18003. doi: 10.1073/pnas.0910297106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Young JW, et al. Impaired attention is central to the cognitive deficits observed in alpha 7 deficient mice. Eur Neuropsychopharmacol. 2007;17:145–155. doi: 10.1016/j.euroneuro.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 34.Hikida T, et al. Dominant-negative DISC1 transgenic mice display schizophrenia-associated phenotypes detected by measures translatable to humans. Proc Natl Acad Sci U S A. 2007;104:14501–14506. doi: 10.1073/pnas.0704774104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pletnikov MV, et al. Inducible expression of mutant human DISC1 in mice is associated with brain and behavioral abnormalities reminiscent of schizophrenia. Mol Psychiatry. 2008;13:115, 173–186. doi: 10.1038/sj.mp.4002079. [DOI] [PubMed] [Google Scholar]
- 36.Li W, et al. Specific developmental disruption of disrupted-in-schizophrenia-1 function results in schizophrenia-related phenotypes in mice. Proc Natl Acad Sci U S A. 2007;104:18280–18285. doi: 10.1073/pnas.0706900104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clapcote SJ, et al. Behavioral phenotypes of Disc1 missense mutations in mice. Neuron. 2007;54:387–402. doi: 10.1016/j.neuron.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 38.Koike H, Arguello PA, Kvajo M, Karayiorgou M, Gogos JA. Disc1 is mutated in the 129S6/SvEv strain and modulates working memory in mice. Proc Natl Acad Sci U S A. 2006;103:3693–3697. doi: 10.1073/pnas.0511189103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kvajo M, et al. A mutation in mouse Disc1 that models a schizophrenia risk allele leads to specific alterations in neuronal architecture and cognition. Proc Natl Acad Sci U S A. 2008;105:7076–7081. doi: 10.1073/pnas.0802615105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Long JM, et al. Behavior of mice with mutations in the conserved region deleted in velocardiofacial/DiGeorge syndrome. Neurogenetics. 2006;7:247–257. doi: 10.1007/s10048-006-0054-0. [DOI] [PubMed] [Google Scholar]
- 41.Stark KL, et al. Altered brain microRNA biogenesis contributes to phenotypic deficits in a 22q11-deletion mouse model. Nat Genet. 2008;40:751–760. doi: 10.1038/ng.138. [DOI] [PubMed] [Google Scholar]
- 42.Javitt DC. When doors of perception close: bottom-up models of disrupted cognition in schizophrenia. Annu Rev Clin Psychol. 2009;5:249–275. doi: 10.1146/annurev.clinpsy.032408.153502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Geyer MA, Braff DL. Startle habituation and sensorimotor gating in schizophrenia and related animal models. Schizophr Bull. 1987;13:643–668. doi: 10.1093/schbul/13.4.643. [DOI] [PubMed] [Google Scholar]
- 44.Javitt DC, Steinschneider M, Schroeder CE, Arezzo JC. Role of cortical N-methyl-D-aspartate receptors in auditory sensory memory and mismatch negativity generation: implications for schizophrenia. Proc Natl Acad Sci U S A. 1996;93:11962–11967. doi: 10.1073/pnas.93.21.11962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Butler PD, Silverstein SM, Dakin SC. Visual perception and its impairment in schizophrenia. Biol Psychiatry. 2008;64:40–47. doi: 10.1016/j.biopsych.2008.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Green MF, et al. Perception measurement in clinical trials of schizophrenia: promising paradigms from CNTRICS. Schizophr Bull. 2009;35:163–181. doi: 10.1093/schbul/sbn156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Powell SB, Zhou X, Geyer MA. Prepulse inhibition and genetic mouse models of schizophrenia. Behav Brain Res. 2009;204:282–294. doi: 10.1016/j.bbr.2009.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Light GA, Braff DL. Mismatch negativity deficits are associated with poor functioning in schizophrenia patients. Arch Gen Psychiatry. 2005;62:127–136. doi: 10.1001/archpsyc.62.2.127. [DOI] [PubMed] [Google Scholar]
- 49.Meyer D, Birchmeier C. Multiple essential functions of neuregulin in development. Nature. 1995;378:386–390. doi: 10.1038/378386a0. [DOI] [PubMed] [Google Scholar]
- 50.Ehrlichman RS, et al. Neuregulin 1 transgenic mice display reduced mismatch negativity, contextual fear conditioning and social interactions. Brain Res. 2009;1294:116–127. doi: 10.1016/j.brainres.2009.07.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Adolphs R. The social brain: neural basis of social knowledge. Annu Rev Psychol. 2009;60:693–716. doi: 10.1146/annurev.psych.60.110707.163514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Klin A, Lin DJ, Gorrindo P, Ramsay G, Jones W. Two-year-olds with autism orient to non-social contingencies rather than biological motion. Nature. 2009;459:257–261. doi: 10.1038/nature07868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sergi MJ, Rassovsky Y, Nuechterlein KH, Green MF. Social perception as a mediator of the influence of early visual processing on functional status in schizophrenia. Am J Psychiatry. 2006;163:448–454. doi: 10.1176/appi.ajp.163.3.448. [DOI] [PubMed] [Google Scholar]
- 54.Leitman DI, et al. The neural substrates of impaired prosodic detection in schizophrenia and its sensorial antecedents. Am J Psychiatry. 2007;164:474–482. doi: 10.1176/ajp.2007.164.3.474. [DOI] [PubMed] [Google Scholar]
- 55.Carter CS, Barch DM, Gur R, Pinkham A, Ochsner K. CNTRICS final task selection: social cognitive and affective neuroscience-based measures. Schizophr Bull. 2009;35:153–162. doi: 10.1093/schbul/sbn157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barch DM, Braver TS, Carter CS, Poldrack RA, Robbins TW. CNTRICS final task selection: executive control. Schizophr Bull. 2009;35:115–135. doi: 10.1093/schbul/sbn154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Luck SJ, Gold JM. The construct of attention in schizophrenia. Biol Psychiatry. 2008;64:34–39. doi: 10.1016/j.biopsych.2008.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ragland JD, et al. CNTRICS final task selection: long-term memory. Schizophr Bull. 2009;35:197–212. doi: 10.1093/schbul/sbn134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Barch DM, Smith E. The cognitive neuroscience of working memory: relevance to CNTRICS and schizophrenia. Biol Psychiatry. 2008;64:11–17. doi: 10.1016/j.biopsych.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kerns JG, Nuechterlein KH, Braver TS, Barch DM. Executive functioning component mechanisms and schizophrenia. Biol Psychiatry. 2008;64:26–33. doi: 10.1016/j.biopsych.2008.04.027. [DOI] [PubMed] [Google Scholar]
- 61.Glickstein SB, Desteno DA, Hof PR, Schmauss C. Mice lacking dopamine D2 and D3 receptors exhibit differential activation of prefrontal cortical neurons during tasks requiring attention. Cereb Cortex. 2005;15:1016–1024. doi: 10.1093/cercor/bhh202. [DOI] [PubMed] [Google Scholar]
- 62.De Steno DA, Schmauss C. A role for dopamine D2 receptors in reversal learning. Neuroscience. 2009;162:118–127. doi: 10.1016/j.neuroscience.2009.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kruzich PJ, Mitchell SH, Younkin A, Grandy DK. Dopamine D2 receptors mediate reversal learning in male C57BL/6J mice. Cogn Affect Behav Neurosci. 2006;6:86–90. doi: 10.3758/cabn.6.1.86. [DOI] [PubMed] [Google Scholar]
- 64.Kruzich PJ, Grandy DK. Dopamine D2 receptors mediate two-odor discrimination and reversal learning in C57BL/6 mice. BMC Neurosci. 2004;5:12. doi: 10.1186/1471-2202-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Papaleo F, et al. Genetic dissection of the role of catechol-O-methyltransferase in cognition and stress reactivity in mice. J Neurosci. 2008;28:8709–8723. doi: 10.1523/JNEUROSCI.2077-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Leeson VC, et al. Discrimination learning, reversal, and set-shifting in first-episode schizophrenia: stability over six years and specific associations with medication type and disorganization syndrome. Biol Psychiatry. 2009;66:586–593. doi: 10.1016/j.biopsych.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Krueger DD, et al. Assessment of cognitive function in the heterozygous reeler mouse. Psychopharmacology. 2006;189:95–104. doi: 10.1007/s00213-006-0530-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Helms CM, Gubner NR, Wilhelm CJ, Mitchell SH, Grandy DK. D4 receptor deficiency in mice has limited effects on impulsivity and novelty seeking. Pharmacol Biochem Behav. 2008;90:387–393. doi: 10.1016/j.pbb.2008.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sarter M, Martinez V, Kozak R. A neurocognitive animal model dissociating between acute illness and remission periods of schizophrenia. Psychopharmacology. 2009;202:237–258. doi: 10.1007/s00213-008-1216-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jentsch JD, Arguello PA, Anzivino LA. Null mutation of the arginine-vasopressin gene in rats slows attentional engagement and facilitates response accuracy in a lateralized reaction time task. Neuropsychopharmacology. 2003;28:1597–1605. doi: 10.1038/sj.npp.1300194. [DOI] [PubMed] [Google Scholar]
- 71.Ranganath C, Minzenberg MJ, Ragland JD. The cognitive neuroscience of memory function and dysfunction in schizophrenia. Biol Psychiatry. 2008;64:18–25. doi: 10.1016/j.biopsych.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tang YP, et al. Genetic enhancement of learning and memory in mice. Nature. 1999;401:63–69. doi: 10.1038/43432. [DOI] [PubMed] [Google Scholar]
- 73.von Engelhardt J, et al. Contribution of hippocampal and extra-hippocampal NR2B-containing NMDA receptors to performance on spatial learning tasks. Neuron. 2008;60:846–860. doi: 10.1016/j.neuron.2008.09.039. [DOI] [PubMed] [Google Scholar]
- 74.El-Ghundi M, et al. Spatial learning deficit in dopamine D(1) receptor knockout mice. Eur J Pharmacol. 1999;383:95–106. doi: 10.1016/s0014-2999(99)00573-7. [DOI] [PubMed] [Google Scholar]
- 75.El-Ghundi M, O'Dowd BF, George SR. Prolonged fear responses in mice lacking dopamine D1 receptor. Brain Res. 2001;892:86–93. doi: 10.1016/s0006-8993(00)03234-0. [DOI] [PubMed] [Google Scholar]
- 76.Smith DR, et al. Behavioural assessment of mice lacking D1A dopamine receptors. Neuroscience. 1998;86:135–146. doi: 10.1016/s0306-4522(97)00608-8. [DOI] [PubMed] [Google Scholar]
- 77.Qiu S, et al. Cognitive disruption and altered hippocampus synaptic function in Reelin haploinsufficient mice. Neurobiol Learn Mem. 2006;85:228–242. doi: 10.1016/j.nlm.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 78.Takao K, et al. Impaired long-term memory retention and working memory in sdy mutant mice with a deletion in Dtnbp1, a susceptibility gene for schizophrenia. Mol Brain. 2008;1:11. doi: 10.1186/1756-6606-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bhardwaj SK, et al. Behavioral characterization of dysbindin-1 deficient sandy mice. Behav Brain Res. 2009;197:435–441. doi: 10.1016/j.bbr.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 80.Feng YQ, et al. Dysbindin deficiency in sandy mice causes reduction of snapin and displays behaviors related to schizophrenia. Schizophr Res. 2008;106:218–228. doi: 10.1016/j.schres.2008.07.018. [DOI] [PubMed] [Google Scholar]
- 81.Maekawa M, Watanabe M, Yamaguchi S, Konno R, Hori Y. Spatial learning and long-term potentiation of mutant mice lacking D-amino-acid oxidase. Neurosci Res. 2005;53:34–38. doi: 10.1016/j.neures.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 82.Paylor R, et al. Mice deleted for the DiGeorge/velocardiofacial syndrome region show abnormal sensorimotor gating and learning and memory impairments. Hum Mol Genet. 2001;10:2645–2650. doi: 10.1093/hmg/10.23.2645. [DOI] [PubMed] [Google Scholar]
- 83.Kimber WL, et al. Deletion of 150 kb in the minimal DiGeorge/velocardiofacial syndrome critical region in mouse. Hum Mol Genet. 1999;8:2229–2237. doi: 10.1093/hmg/8.12.2229. [DOI] [PubMed] [Google Scholar]
- 84.Jonides J, et al. The mind and brain of short-term memory. Annu Rev Psychol. 2008;59:193–224. doi: 10.1146/annurev.psych.59.103006.093615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Baddeley A. Working memory: looking back and looking forward. Nat Rev Neurosci. 2003;4:829–839. doi: 10.1038/nrn1201. [DOI] [PubMed] [Google Scholar]
- 86.Postle BR. Working memory as an emergent property of the mind and brain. Neuroscience. 2006;139:23–38. doi: 10.1016/j.neuroscience.2005.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Barch DM, et al. CNTRICS final task selection: working memory. Schizophr Bull. 2009;35:136–152. doi: 10.1093/schbul/sbn153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Aultman JM, Moghaddam B. Distinct contributions of glutamate and dopamine receptors to temporal aspects of rodent working memory using a clinically relevant task. Psychopharmacology. 2001;153:353–364. doi: 10.1007/s002130000590. [DOI] [PubMed] [Google Scholar]
- 89.McNab F, Klingberg T. Prefrontal cortex and basal ganglia control access to working memory. Nat Neurosci. 2008;11:103–107. doi: 10.1038/nn2024. [DOI] [PubMed] [Google Scholar]
- 90.Cannon TD, et al. Dorsolateral prefrontal cortex activity during maintenance and manipulation of information in working memory in patients with schizophrenia. Arch Gen Psychiatry. 2005;62:1071–1080. doi: 10.1001/archpsyc.62.10.1071. [DOI] [PubMed] [Google Scholar]
- 91.Grillet N, et al. Generation and characterization of Rgs4 mutant mice. Mol Cell Biol. 2005;25:4221–4228. doi: 10.1128/MCB.25.10.4221-4228.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.O'Tuathaigh CM, et al. Phenotypic characterization of spatial cognition and social behavior in mice with ‘knockout’ of the schizophrenia risk gene neuregulin 1. Neuroscience. 2007;147:18–27. doi: 10.1016/j.neuroscience.2007.03.051. [DOI] [PubMed] [Google Scholar]
- 93.Rimer M, Barrett DW, Maldonado MA, Vock VM, Gonzalez-Lima F. Neuregulin-1 immunoglobulin-like domain mutant mice: clozapine sensitivity and impaired latent inhibition. Neuroreport. 2005;16:271–275. doi: 10.1097/00001756-200502280-00014. [DOI] [PubMed] [Google Scholar]
- 94.Chen YJ, et al. Type III neuregulin-1 is required for normal sensorimotor gating, memory-related behaviors, and corticostriatal circuit components. J Neurosci. 2008;28:6872–6883. doi: 10.1523/JNEUROSCI.1815-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lai WS, et al. Akt1 deficiency affects neuronal morphology and predisposes to abnormalities in prefrontal cortex functioning. Proc Natl Acad Sci U S A. 2006;103:16906–16911. doi: 10.1073/pnas.0604994103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Glickstein SB, Hof PR, Schmauss C. Mice lacking dopamine D2 and D3 receptors have spatial working memory deficits. J Neurosci. 2002;22:5619–5629. doi: 10.1523/JNEUROSCI.22-13-05619.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hartman RE, et al. Behavioral phenotyping of GFAP-apoE3 and -apoE4 transgenic mice: apoE4 mice show profound working memory impairments in the absence of Alzheimer's-like neuropathology. Exp Neurol. 2001;170:326–344. doi: 10.1006/exnr.2001.7715. [DOI] [PubMed] [Google Scholar]
- 98.Hsu R, et al. Nogo Receptor 1 (RTN4R) as a candidate gene for schizophrenia: analysis using human and mouse genetic approaches. PLoS One. 2007;2:e1234. doi: 10.1371/journal.pone.0001234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Budel S, et al. Genetic variants of Nogo-66 receptor with possible association to schizophrenia block myelin inhibition of axon growth. J Neurosci. 2008;28:13161–13172. doi: 10.1523/JNEUROSCI.3828-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Paterlini M, et al. Transcriptional and behavioral interaction between 22q11.2 orthologs modulates schizophrenia-related phenotypes in mice. Nat Neurosci. 2005;8:1586–1594. doi: 10.1038/nn1562. [DOI] [PubMed] [Google Scholar]
- 101.Snitz BE, Macdonald AW, III, Carter CS. Cognitive deficits in unaffected first-degree relatives of schizophrenia patients: a meta-analytic review of putative endophenotypes. Schizophr Bull. 2006;32:179–194. doi: 10.1093/schbul/sbi048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Goos LM, Crosbie J, Payne S, Schachar R. Validation and extension of the endophenotype model in ADHD patterns of inheritance in a family study of inhibitory control. Am J Psychiatry. 2009;166:711–717. doi: 10.1176/appi.ajp.2009.08040621. [DOI] [PubMed] [Google Scholar]
- 103.Delorme R, et al. Shared executive dysfunctions in unaffected relatives of patients with autism and obsessive-compulsive disorder. Eur Psychiatry. 2007;22:32–38. doi: 10.1016/j.eurpsy.2006.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Barch DM. The cognitive neuroscience of schizophrenia. Annu Rev Clin Psychol. 2005;1:321–353. doi: 10.1146/annurev.clinpsy.1.102803.143959. [DOI] [PubMed] [Google Scholar]
- 105.Dickinson D, Ramsey ME, Gold JM. Overlooking the obvious: a meta-analytic comparison of digit symbol coding tasks and other cognitive measures in schizophrenia. Arch Gen Psychiatry. 2007;64:532–542. doi: 10.1001/archpsyc.64.5.532. [DOI] [PubMed] [Google Scholar]