

Abstract
6-[3-(1-adamantyl)-4-hydroxyphenyl]-2-naphthalenecarboxylic acid (CD437/AHPN) and 4-[3-(1-adamantyl)-4-hydroxyphenyl]-3-chlorocinnamic acid (3-Cl-AHPC/MM002) are inducers of apoptosis of malignant cells both in vitro and in vivo. Numerous mechanisms have been proposed for how these compounds exert this effect. This report shows that AHPN/3-Cl-AHPC binds specifically to the orphan nuclear receptor small heterodimer partner (SHP; NR0B2), and this binding promotes interaction of the receptor with a corepressor complex that minimally contains Sin3A, N-CoR, histone deacetylase 4, and HSP90. Formation of the SHP-Sin3A complex is essential for the ability of AHPN and 3-Cl-AHPC to induce apoptosis, as both knockout SHP and knockdown of Sin3A compromise the proapoptotic activity of these compounds but not other apoptosis inducers. These results suggest that AHPN/3-Cl-AHPC and their analogues are SHP ligands and their induction of apoptosis is mediated by their binding to the SHP receptor.
Introduction
The ability of the novel retinoid 6-[3-(1-adamantyl)-4-hydrox-yphenyl]-2-naphthalenecarboxylic acid (CD437/AHPN) to induce apoptosis in several different malignant cells has been well described (1–4). This observation led to the synthesis and development of a unique class of compounds, which are potent inducers of apoptosis and have been called retinoid-related molecules. The essential pharmacophoric elements in the AHPN scaffold necessary for apoptosis induction are the hydrophobic 1-adamantyl group, its adjacent hydrogen-donating hydroxyl group, and a distant carboxylic acid group (5). These compounds have also been shown to induce cell death in several animal models (1–7). Although some members of this class may bind and activate the nuclear retinoic acid receptors (RAR), recent data suggested that these compounds induce cell death through mechanisms that do not involve the RARs or the retinoid X receptors (RXR; refs. 8, 9). Further support for this premise was derived from the observation that the AHPN analogue 4-[3-(1-adamantyl)-4-hydroxyphenyl]-3-chlorocinnamic acid (3-Cl-AHPC/MM002), which binds to but does not activate the RARs, is a potent inducer of apoptosis (10). The mechanism by which AHPN and its analogues induce apoptosis remains unclear, although numerous potential pathways have been suggested (11–13). Our binding studies using labeled AHPN and nuclear extracts suggested the presence of a novel AHPN receptor (14). To further characterize this potential AHPN receptor, nuclear proteins, which specifically bound to AHPN linked to a Sepharose affinity support, were identified; one such protein was SAP18, a member of the mammalian Sin3A complex (15).
Mammalian Sin3A is a 145-kDa protein, which, through its interaction with selective proteins and histone deacetylases (HDAC), represses the expression of a large number of genes. Three classes of Sin3A-associated proteins have been identified. The first class of proteins consists of histone/chromatin-modifying enzymes, such as HDAC1, HDAC2, Swi/Snf, O-linked N-acetylglucosamine methyltransferase, and histone methyltransferases (16). The second class consists of a large number of DNA-binding transcription factors, which, when bound to their appropriate consensus sequences in association with Sin3A, results in gene repression. Finally, several docking proteins for specific repressor components are found in the Sin3A complex; these include SAP45 and SAP30, which mediate the association of Sin3A with HDAC1 and HDAC2 and other proteins (16, 17). Recently, the binding of the orphan nuclear receptor small heterodimer partner (SHP; NR0B2) to the Sin3A complex has been described (18, 19). SHP is a unique nuclear receptor, which lacks a DNA-binding domain but contains a putative ligand-binding domain. SHP has been proposed to interact with and inhibit the transcriptional activities of other nuclear proteins (20, 21).
We have now found that AHPN and 3-Cl-AHPC and their analogues specifically bind to SHP, resulting in the recruitment of a complex that minimally contains Sin3A, N-CoR, HDAC4, and HSP90 to the liganded receptor. Moreover, we found that AHPN and 3-Cl-AHPC enhanced binding of this SHP-Sin3A complex to the c-Myc promoter, resulting in transcriptional repression. Finally, both SHP and Sin3A were found to be necessary for AHPN and 3-Cl-AHPC–induced apoptosis, suggesting that this complex is central to the proapoptotic action of these compounds.
Materials and Methods
Materials/antibodies
3-Cl-AHPC, (E)-4-[3′-(1-adamantyl)-4′-hydroxyphenyl]-3-(3′-acetamidopropyloxy)cinnamic acid (3-A-AHPC) and [5,5′-3H2] AHPN, were synthesized as described previously (14, 22, 23). [11,12-3H2]tRA was purchased from Perkin-Elmer Life Sciences (Boston, MA). DMEM-F12, fetal bovine serum, and Trizol reagent were purchased from Invitrogen, Inc. (Grand Island, NY). Anti-c-Myc antibody was obtained from NeoMarkers (Fremont, CA) or Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Anti-mSin3A, anti-N-CoR, anti-GADD45, anti-HDAC1, anti-HDAC2, anti-HDAC4, anti-HSP90, and anti-SHP antibodies were from Santa Cruz Biotechnology. α-Tubulin antibody was from Oncogene Research Products (Boston, MA). HDAC colorimetric activity assay kit was purchased from Biomol Research Laboratories, Inc. (Plymouth Meeting, PA), and geldanamycin was obtained from the NIH (Bethesda, MD).
Cell culture and apoptosis
The human leukemia cell lines HL-60R and KG-1 and the breast carcinoma cell line MDA-MB-468 were maintained in RPMI 1640 and DMEM-F12, respectively, as described (10, 23). We have previously described our generation of the wild-type and SHP knockout cells (24). Apoptosis was assessed either using acridine orange staining or using ELISA apoptosis kits obtained from Roche Diagnostic Laboratory (Indianapolis, IN; ref. 22). The vehicle was 0.1% DMSO and 3-Cl-AHPC and AHPN were added to a final concentration of 1 μmol/L and [5,5′-3H2]AHPN was added to a final concentration of 5 nmol/L unless otherwise stated.
Binding assay
Binding of [5,5′-3H2]AHPN to glutathione S -transferase (GST)-SHP and fractionation over G25 Sephadex were done as we have previously published (14). In the immunoprecipitation binding assays, specific primary antibodies (1 μg) were added in 200 to 500 μg nuclear extracts in binding buffer containing labeled and nonlabeled ligands and incubated at 4°C for 2 h. Nuclear extracts were prepared using the method of Ausubel et al. (25). Protein G-Sepharose Cl-4B (20 μL) was then added and washed thrice with binding buffer. Washed beads were added directly to the scintillation liquid or the [5,5′-3H2]AHPN (CD437) was eluted using elution buffer (50 mmol/L NaHCO3 and 1% SDS), and the eluate was added to the scintillation fluid to assess bound radioactivity. In the GST-SHP binding assays, glutathione-Sepharose bead-bound GST-SHP (0.5–0.7 μg) was used in the GST-SHP binding assays.
Radioligand binding data were analyzed using GraphPad Prism 4.0 software (GraphPad Software, San Diego, CA). Saturation radioligand binding data were analyzed by fitting an equation for one site binding hyperbola to the untransformed data: Y = Bmax × X / (KD + X).
This describes the binding of a ligand to a receptor that follows the law of mass action. B max is the maximal binding of [5,5′-3H2]AHPN and KD is the concentration of ligand required to reach half-maximal binding. Linear regression was used to analyze transformed data in Scatchard plots.
Correlation of binding to GST-SHP protein and level of apoptosis induction were examined using 3-Cl-AHPC and several analogues: (E)-2-4-(4-[3-(1-adamantyl)-4-hydroxyphenyl]-3-chlorophenyl)ethylenyl-5-tetrazole (BI-2002), 5-(4-[3-(1-adamantyl)-4-hydroxyphenyl]-3-chlorophenyl)methylene-thiazoleidene-2,4-dione (BI-2003), (E)-3-2-[3-(1-adamantyl)-4hydroxyphenyl] pyrimidinyl)propenoic acid (BI-2005), (E)-4-[3-(1-adamantyl)-4-hydroxyphenyl]-3-chlorocinnamyl hydroxamic acid (BI-2007), (E)-2-(4-[3-(1-adamantyl)-4-hydroxyphenyl]-3-chlorophenyl)ethyleneboronic acid (BI-2009), and 3-A-AHPC. [5,5′-3H2]AHPN was added to a concentration of 5 nmol/L, whereas competing nonlabeled 3-Cl-AHPC analogues were added to a final concentration of 50 μmol/L. Each point represents the mean value for each analogue determined from three independent experiments. The correlation coefficient and P were determined using Pearson correlation analysis.
Western blots
Western blots, RNA preparation, and Northern blots were done as we have previously described (26).
HDAC assay
Histone deacetylation activation assays were done using the HDAC colorimetric assay kit and the manufacturer’s instructions. Sin3A was immunoprecipitated from 500 μg nuclear extracts and added to 1 mmol/L substrate for each HDAC activation assay in 96-well microtiter plates; the HDAC activity was measured at 405 nm in a microtiter plate reader.
Small interfering RNA transfection
Cells were transfected with small interfering RNA (siRNA) using Oligofectamine and Oligofectamine plus reagent according to Elbashir et al. (27) and the manufacturer’s instructions. The cells were incubated with two primers, 21-nucleotide RNA with 3′-dAA overhangs synthesized by Dharmacon RNA Technology (Dharmacon Research, Lafayette, CO). The primer sequences were 5′-GGUUGCUCGUCUCUUUAAAAA-3′(mS3-1) and 5′-UUAUCGUUGUGAAGAUGAAAA-3′(mS3-2) corresponding to base pair start from 1,342 to 2,086 of the coding sequence of the Sin3A gene (PubMed accession no. NM_015477). The siRNA sequences for human SHP were 5′-GCAGUGGCUUCAAUGCUGUUAA-3′(si-SHP1) and 5′-CUAUGUGCACCUCAUCGCA-3′(si-SHP2). Control cells were treated with nontargeting siRNA control duplex sequence from Dharmacon Research. Cells were harvested 24, 48, and 72 h following the transfection to assess protein levels by Western blot. Cells were treated with 1 μmol/L 3-Cl-AHPC after 24 h of transfection and harvested 24 and 48 h after incubation with 1 μmol/L 3-Cl-AHPC for protein and apoptosis assays.
Chromatin immunoprecipitation assay
KG-1 and MDA-MB-468 cells were treated with 1 μmol/L 3-Cl-AHPC for 24 h and proteins were cross-linked to DNA by adding formaldehyde (final concentration of 1%) to the culture medium and fixing at room temperature for 10 min followed by washing twice with cold PBS and then chromatin immunoprecipitation (ChIP) assays were done using a modified procedures of Weinmann et al. (28). The antibody/protein/DNA complex was eluted with elution buffer (50 mmol/L NaHCO3, 1% SDS), and the cross-link was reversed by addition of NaCl to a final concentration of 250 mmol/L, 5 μL proteinase K (15 mg/mL) and 10 μg RNaseA and incubated at 65°C overnight. The samples were extracted with phenol-chloroform-isoamylalcohol (25:24:1) and then chloroform-isoamylalcohol (24:1). The DNA was precipitated with 3 mol/L sodium acetate (pH 5.3; 1:10 by volume) and 5 μg glycogen, and 1 mL of ethanol precipitate was resuspended with Tris-EDTA buffer and analyzed by PCR. c-Myc primers were designed from the promoter region exon 1 (forward, 5′-ATAATGCGAGGGTCTGGA-3′; reverse, 5′-ATACTCAGCHCGATCCCT-3′) and from the primer set specific for SHP (forward, 5′-CCCAAGATGCTGTGACCTTT-3′; reverse, 5′-CCAGAAGGACTCCAGACAGC-3′). The primer sequences for β-actin were 5′-TCCTTCCTGGGCATGGAG-3′(forward) and 5′-AGGAGGAGCAATGATCTT-3′(reverse).
Results
AHPN binds to a Sin3A complex(es)
We previously showed specific binding of [5,5′-3H2]AHPN to nuclear extracts of several malignant cells (14). AHPN binding to specific nuclear proteins was examined using an AHPN affinity column. One of the proteins isolated from this column was SAP18, a member of the Sin3A complex (15). [5,5′-3H2]AHPN binding to a Sin3A complex was therefore assessed using nuclear extracts from the HL-60 and MDA-MB-468 cell lines as described in Fig. 1. [5,5′-3H2]AHPN was found to bind to the Sin3A complex in a manner that was inhibited by unlabeled AHPN, 3-Cl-AHPC, and the AHPN analogue 3-A-AHPC (Fig. 1A). The specificity of [5,5′-3H2]AHPN binding to the Sin3A complex was further shown by the lack of [5,5′-3H2]AHPN binding to nuclear extracts depleted of Sin3A as well as by the lack of binding to other nuclear proteins, such as the deacetylase SIRT1 and GADD45 (Fig. 1B and C). [5,5′-3H2]AHPN coimmunoprecipitated with N-CoR in MDA-MB-468 where N-CoR was found associated with the Sin3A complex (see Figs. 1C and 2 below), but [5,5′-3H2]AHPN did not coimmunoprecipitate with N-CoR in KG-1 cells where N-CoR was not found in the Sin3A complex (Fig. 1D, inset). Thus, [5,5′-3H2]AHPN binding to N-CoR was dependent on N-CoR being part of the Sin3A complex N-CoR. [5,5′-3H2]AHPN binding to the Sin3A complex was only found when conducted in the presence of nuclear extracts; [5,5′-3H2]AHPN did not display specific binding to the Sin3A complex when it was added after immunoprecipitation of Sin3A, implying that [5,5′-3H2] AHPN binding to the Sin3A complex requires the association of other nuclear factors with the Sin3A complex (Fig. 1C, lane 5).
Figure 1.

[5,5′-3H2]AHPN binding to the Sin3A complex. [5,5′-3H2]AHPN was added in the presence or absence of 50 μmol/L unlabeled AHPN, 3-Cl-AHPC, or 3-A-AHPC to the nuclear extracts, and Sin3A, SIRT1, GADD45, and N-CoR were immunoprecipitated. Nuclear extracts were prepared as described in Materials and Methods. A, nonlabeled AHPN, 3-Cl-AHPC, and 3-A-AHPC inhibit [5,5′-3H2]AHPN binding to the Sin3A complex. B, [5,5′-3H2]AHPN binding in nuclear extracts depleted of Sin3A (ID-Sin3A). C, [5,5′-3H2]AHPN binding to Sin3A, SIRT1, GADD45, N-CoR, and immunoprecipitated Sin3A. D, [5,5′-3H2]AHPN binding to N-CoR in KG-1 cells. Inset, N-CoR in KG-1 cells. NS, nonspecific band. Bars, SD.
Figure 2.

3-Cl-AHPC enhances the association of N-CoR, HDAC4, HSP90, TBL1, and GPS2 with the Sin3A complex. Nuclear extracts were incubated with 3-Cl-AHPC (50 μmol/L) for 2 h, Sin3A was immunoprecipitated and fractionated on SDS-PAGE, and silver staining (A) and Western blots (B and C) were done as described in Materials and Methods. C, 3-Cl-AHPC enhances binding in cells of N-CoR, HDAC4, and HSP90 to Sin3A. Cells were incubated with 1 μmol/L 3-Cl-AHPC for varying times. D, 3-Cl-AHPC exposure increased the expression of Sin3A protein. Cells were exposed to 1 μmol/L 3-Cl-AHPC for varying times.
AHPN and 3-Cl-AHPC recruit proteins to the Sin3A complex and enhances Sin3A-associated HDAC activity
Several approaches were used to determine whether exposure of cells or cell extracts to AHPN or 3-Cl-AHPC resulted in the recruitment of proteins to a Sin3A complex. MDA-MB-468 nuclear extract exposure to 3-Cl-AHPC or AHPN resulted in the recruitment of several proteins to Sin3A, including HDAC4, HSP90, N-CoR, and the N-CoR components TBL1 and GPS2, as identified by silver staining and Western blot (Fig. 2A and B; Supplementary Fig. S2). When MDA-MB-468 cells were incubated with either vehicle or 3-Cl-AHPC for varying times following which the Sin3A complex was immunoprecipitated and Western blots were done, AHPN or 3-Cl-AHPC exposure also resulted in Sin3A recruitment of HDAC4, HSP90, and N-CoR (Fig. 2C). Thus, exposure of nuclear extracts or whole cells to AHPN or 3-Cl-AHPC resulted in Sin3A complex modification. AHPN and 3-Cl-AHPC exposure also resulted in a 1.5-and 2-fold increased Sin3A expression in KG-1 and MDA-MB-468 cells, respectively (Fig. 2D).
Sin3A complexes repress gene expression through HDAC1-and HDAC2-mediated histone deacetylation (29). We therefore assessed whether 3-Cl-AHPC–mediated modification of the Sin3A complex resulted in the modulation of the Sin3A-associated HDAC activities. Exposure of MDA-MB-468 cells to 3-Cl-AHPC resulted in a 2-fold increase of HDAC activity in Sin3A immunoprecipitates (Fig. 3A). This increase in Sin3A-associated HDAC activity may be secondary to the 3-Cl-AHPC increase in Sin3A expression.
Figure 3.
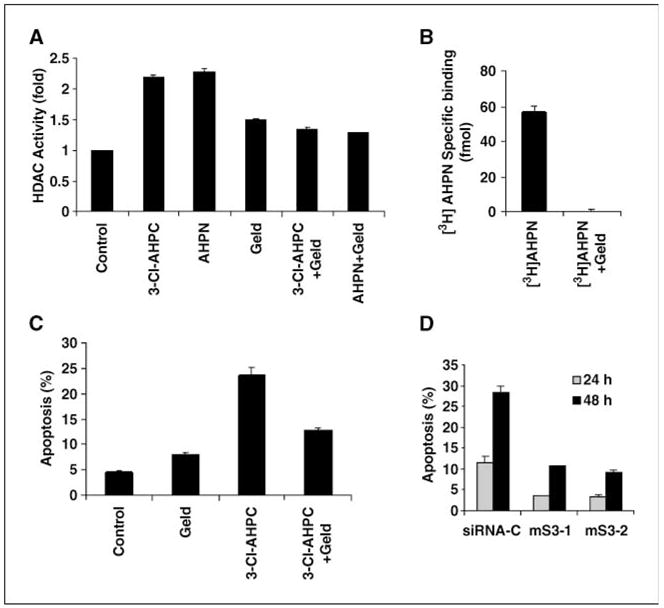
[5,5′-3H2]AHPN binding to Sin3A- and 3-Cl-AHPC–mediated apoptosis is dependent on HSP90 ATPase activity and Sin3A expression. MDA-MB-468 cells were incubated with either vehicle or 1 μg/mL geldanamycin (Geld) for 2 h and then exposed to AHPN and 3-Cl-AHPC or vehicle for 24 h. 3-Cl-AHPC induction of Sin3A-associated HDAC activity (A), [5,5′-3H2]AHPN Sin3A binding (B), and 3-Cl-AHPC–mediated apoptosis (C) in cells treated with and without geldanamycin. D, effect of Sin3A loss of expression in MDA-MB-468 cells on 3-Cl-AHPC–mediated apoptosis. Cells were exposed to vehicle or 1 μmol/L 3-Cl-AHPC for varying times.
HSP90 ATPase activity is essential for 3-Cl-AHPC binding to Sin3A- and 3-Cl-AHPC–induced apoptosis
3-Cl-AHPC exposure resulted in the recruitment of HSP90 to the Sin3A complex (Fig. 2). We used the HSP90 inhibitor geldanamycin to further delineate the role of HSP90 in AHPN and 3-Cl-AHPC binding to Sin3A and the AHPN- and 3-Cl-AHPC-mediated increase in Sin3A-associated HDAC activity and apoptosis. Geldanamycin pretreatment of cells resulted in inhibition of [5,5′-3H2]AHPN binding to the Sin3A complex as well as the 3-Cl-AHPC-mediated increase in HDAC activity and apoptosis (Fig. 3A–C). Geldanamycin did not inhibit HSP90 Sin3A binding (data not shown).
In view of AHPN and 3-Cl-AHPC binding to Sin3A, we examined the effect of loss of Sin3A expression on 3-Cl-AHPC–mediated apoptosis to determine the role of Sin3A in this process. Sin3A expression was markedly reduced in cells using siRNA and resulted in a significant inhibition of 3-Cl-AHPC–mediated apoptosis, suggesting a role for this complex in this process (Fig. 3D; Supplementary Fig. S3).
3-Cl-AHPC binds SHP, resulting in enhanced SHP-Sin3A association, which is essential for 3-Cl-AHPC binding to Sin3A complex and 3-Cl-AHPC–mediated apoptosis
The orphan receptor SHP has recently been found to be a member of the Sin3A complex, and it has been speculated that SHP-mediated repression of gene expression may be through its Sin3A association (18). Because we found that [5,5′-3H2]AHPN and 3-Cl-AHPC bound to a Sin3A complex but only in the presence of the nuclear extract, we speculated that [5,5′-3H2]AHPN/3-Cl-AHPC may be binding to SHP with subsequent recruitment of the Sin3A complex to the liganded receptor. We therefore assessed binding of [5,5′-3H2] AHPN to recombinant SHP. [5,5′-3H2]AHPN bound specifically and with high affinity (KD of 2.88 + 0.57 nmol/L; Fig. 4A) to recombinant SHP in a manner that was inhibited by 3-Cl-AHPC. Binding of [3H]trans-retinoic acid ([3H]tRA) to SHP was not observed (Fig. 4B). Preincubation of GST-SHP with SHP antibody totally inhibited [5,5′-3H2]AHPN binding to SHP (data not shown). In addition, specific [5,5′-3H2]AHPN binding to GST-SHP was observed when GST-SHP was incubated with [5,5′-3H2]AHPN, and then the GST-SHP was fractionated over a G25 column (Fig. 4C). [5,5′-3H2]AHPN binding coeluted with GST-SHP; this binding was inhibited by excess unlabeled 3-Cl-AHPC. No binding was noted if [5,5′-3H2]AHPN was incubated with only GST (data not shown). Thus, [5,5′-3H2]AHPN and 3-Cl-AHPC seem to be ligands for SHP, whereas the classic retinoid tRA is not.
Figure 4.

[5,5′-3H2]AHPN binding to SHP in MDA-MB-468 cells. A, Scatchard plot of [5,5′-3H2]AHPN binding to SHP. Scatchard plots and analysis were done as described in Materials and Methods. Results are representative of four independent experiments. B, [5,5′-3H2]AHPN and [3H]tRA binding to SHP. [5,5′-3H2]AHPN and [3H]tRA were added to a concentration of 5 nmol/L. Nonlabeled ligand was added to a final concentration of 50 μmol/L.C, [5,5′-3H2]AHPN binding to GST-SHP. D, correlation between [5,5′-3H2]AHPN binding to SHP and apoptosis induction in MDA-MB-468 cells.
The ability of several 3-Cl-AHPC analogues to inhibit [5,5′-3H2]AHPN binding to SHP was compared with their ability to induce apoptosis. A strong correlation of R = 0.96 (P = 0.002) was found between binding to SHP as assessed by the inhibition of [5,5′-3H2]AHPN binding to SHP and the induction of apoptosis (Fig. 4D). Furthermore, the 3-Cl-AHPC analogues 3-A-AHPC and BI-2002, which block AHPN- and 3-Cl-AHPC–mediated apoptosis, also inhibited [5,5′-3H2]AHPN binding to SHP, suggesting that AHPN and 3-Cl-AHPC binding to SHP plays an important role in AHPN/3-Cl-AHPC–mediated apoptosis (Fig. 4D; ref. 22).
SHP and Sin3A play essential roles in 3-Cl-AHPC–mediated apoptosis and inhibition of c-Myc expression
Exposure of cells to 3-Cl-AHPC resulted in the increased association of SHP as well as N-CoR, HDAC4, and HSP90 with Sin3A (Figs. 2 and 5A). The addition to cells of 3-A-AHPC, which blocks [5,5′-3H2]AHPN binding to SHP (Fig. 4D), also inhibited 3-Cl-AHPC–enhanced SHP association with the Sin3A complex (Fig. 5B) and blocked 3-Cl-AHPC–mediated apoptosis as we have previously shown (22). We speculated that if 3-Cl-AHPC binding to SHP plays an important role in 3-Cl-AHPC–mediated apoptosis, loss of SHP expression should inhibit 3-Cl-AHPC–mediated apoptosis. We therefore examined 3-Cl-AHPC–mediated apoptosis in MDA-MB-468 cells with loss of SHP expression as well as in SHP-null mouse embryonic fibroblasts (MEF; ref. 24). SHP expression was reduced 50% to 60% in MDA-MB-468 cells using siRNA (Supplementary Fig. S5). Knockdown of SHP in MDA-MB-468 cells and knockout of SHP in the MEF cells attenuated [5,5′-3H2]AHPN binding to Sin3A and resulted in resistance of the corresponding cells to 3-Cl-AHPC–mediated apoptosis (Fig. 5C and D). However, these manipulations of SHP did not alter apoptosis induction by the proteasome inhibitor N-acetyl-L-leucinyl-L-leucinyl-norleucinal (LLnL), Adriamycin, or the inhibitor of nuclear factor-κB (NF-κB) activation kamebakaurin (Fig. 5D). Although decreased, a low level of 3-Cl-AHPC–mediated apoptosis was still noted in the SHP knockout MEF cells. The mechanism for 3-Cl-AHPC–mediated apoptosis independent of SHP expression remains to be delineated.
Figure 5.

3-Cl-AHPC modulates SHP binding to Sin3A, and the loss of SHP expression results in inhibition of 3-Cl-AHPC (AHPN) binding to Sin3A- and 3-Cl-AHPC–mediated apoptosis. A, 3-Cl-AHPC exposure enhances SHP binding to Sin3A. Cells were exposed to 1 μmol/L 3-Cl-AHPC for varying times. B, the antagonist 3-A-AHPC blocks 3-Cl-AHPC–mediated SHP binding to Sin3A. Cells were exposed to the antagonist 3-A-AHPC (2 μmol/L) in the presence and absence of 3-Cl-AHPC (0.5 μmol/L) for 24 h. C, inhibition of SHP expression inhibits [5,5′-3H2]AHPN binding to Sin3A. D, loss of SHP expression specifically inhibits 3-Cl-AHPC–mediated (1 μmol/L) apoptosis in MDA-MB-468 knockdown (si-SHP1 and si-SHP2) and SHP knockout MEF cells but not apoptosis in cells exposed to 5 μmol/L kamebakaurin (KA), 50 μmol/L LLnL, or 10μmol/L Adriamycin for 48 h.
Sin3A inhibits gene expression at the transcriptional level through its increased binding to specific promoters (16, 30, 31). Gene array studies in several cell types showed a rapid inhibition of c-Myc expression following 3-Cl-AHPC exposure (data not shown). Exposure of MDA-MB-468 and KG-1 cells to 3-Cl-AHPC resulted in inhibition of c-Myc protein and mRNA expression within 6 h (Fig. 6A and B). 3-Cl-AHPC inhibition of c-Myc transcription in both cell types is associated with 3-Cl-AHPC–mediated enhanced binding of Sin3A and SHP to the c-Myc promoter as shown by ChIP assay (Fig. 6C and D). That this 3-Cl-AHPC–mediated SHP/Sin3A binding to the c-Myc promoter is specific is shown by a lack of 3-Cl-AHPC–increased Sin3A binding to the actin promoter (Fig. 6C).
Figure 6.

3-Cl-AHPC (1 μmol/L) exposure results in decreased c-Myc protein levels (A) and mRNA levels (B) and enhanced Sin3A (C) and SHP (D) binding to the c-Myc promoter. Cells were exposed to 1 μmol/L 3-Cl-AHPC for varying times. Northern blot, Western blot, and ChIP assays were done as described in Materials and Methods.
Discussion
The Sin3A protein serves as the scaffold to which several proteins bind with the subsequent formation of the Sin3A complex (16). Binding to Sin3A occurs through its amphipathic [paired amphipathic helix (PAH)] domains. Sin3A possesses four PAH domains (PAH1, PAH2, PAH3, and PAH4), and evidence suggests that they are not redundant but that each domain binds a unique protein(s) (30, 31); whereas PAH2 is responsible for the binding of the c-Myc family member Mad, PAH3 binds the corepressor N-CoR (30, 31). The Sin3A complex inhibits the transcription of a large number of genes through its unique binding of selective nuclear transcription factors, corepressors, as well as HDACs and methylases (30). Although Sin3A does not directly bind to DNA, it is recruited to promoter templates through its binding to other transcription factors harboring sequence-specific DNA-binding activity (32). We have found that 3-Cl-AHPC exposure resulted in enhanced binding of several proteins to the Sin3A complex that is involved in transcriptional repression as well as the nuclear orphan receptor SHP. We hypothesize that the initial event in this process is the binding of 3-Cl-AHPC or 3-Cl-AHPC to SHP. We have shown that SHP tightly binds AHPN with a KD of 2.8 nmol/L, which is similar to that noted for the binding of AHPN by nuclear extracts (14). Moreover, we found that both SHP and Sin3A play key roles in the proapoptotic activities of AHPN and 3-Cl-AHPC. Loss of Sin3A expression in MDA-MB-468 cells and SHP expression in MDA-MB-468 and MEF cells inhibits 3-Cl-AHPC–mediated apoptosis. Interestingly, MEF cells display increased sensitivity to 3-Cl-AHPC-mediated apoptosis. Furthermore, 3-Cl-AHPC analogues, which inhibit 3-Cl-AHPC-mediated apoptosis, also block AHPN binding to SHP and SHP binding to the Sin3A complex. Considered together, these results strongly suggest that SHP binding by 3-Cl-AHPC and the subsequent recruitment of a Sin3A-containing complex to the ligand-bound receptor play an important role in 3-Cl-AHPC–mediated apoptosis.
Interaction of SHP with a Sin3A complex has been previously documented in hepatocytes and has been shown to occur through the repression domain of the orphan receptor (18). More importantly, this association of SHP with Sin3A in these cells results in the recruitment of several proteins to the complex, association of the modified complex to the CYP7A1 gene promoter, and subsequent transcriptional repression (18).
SHP has been shown to interact with several nuclear receptors, including the thyroid hormone receptor, RAR, RXR, estrogen receptor, glucocorticoid receptor, and hepatocyte nuclear receptors FXR and LRH-1 (27). Interaction between SHP and these receptors is enhanced by the presence of their cognate ligand and results in SHP-mediated down-regulation of their respective signaling pathways (33). In addition, SHP is able to interact with Nur77 and inhibit transcriptional activation mediated by this orphan receptor (34). In contrast, SHP binding to the peroxisome-activated receptors α and γ as well as the transcriptional factor NF-κB results in their activation (35, 36). Although no ligand for SHP has been identified, recent SHP modeling studies by Macchiarulo et al. (37) suggest the presence of a phospholipid-binding site. SHP mRNA expression has been found in several tissues and cell lines. SHP protein is expressed in liver, heart, kidney, small intestine, stomach, spleen, adrenal, colon, breast adipose tissue, brain, epididymis, prostate, uterus, bladder, lung, and pancreas (20, 38–40). SHP expression is abundant in murine macrophage cell line (36) and also increases during monocytic differentiation with exposure of HL-60R leukemia cells to a 12-O-tetradecanoylphorbol-13-acetate response element (41).
In addition to SHP, 3-Cl-AHPC exposure results in the increased association of HSP90 with Sin3A. Several investigators have now shown that HSP90 is associated with and modulates the activity of a variety of steroid, hormone receptors (42). The role of HSP90 in the ability of these receptors to bind their respective ligands and activate gene transcription is controversial. Several groups have shown that the ligand-binding domain of the glucocorticoid receptor must be bound to HSP90 for receptor high-affinity ligand binding (43). Incubation of cells with the specific HSP90 inhibitor geldanamycin blocked glucocorticoid receptor ligand binding as well as its binding to its consensus sequence in the mouse mammary tumor virus promoter (44). We have found that the addition of geldanamycin to 3-Cl-AHPC–exposed cells inhibited 3-Cl-AHPC binding to the Sin3A complex as well as 3-Cl-AHPC–mediated induction of Sin3A-associated HDAC activity and apoptosis. Gene repression can be mediated through Sin3A-associated HDAC1- and HDAC2-mediated deacetylation of histones. The fact that HSP90 seems to be involved with 3-Cl-AHPC binding to Sin3A and the subsequent enhancement of Sin3A-associated HDAC activity suggests that HSP90 plays an important role in 3-Cl-AHPC-induced apoptosis. Studies to assess whether geldanamycin exposure inhibits 3-Cl-AHPC/Sin3A–mediated gene expression are under way.
AHPN and its analogues have been shown to have a wide variety of effects on malignant cells before the induction of apoptosis (45, 46). Diverse mechanisms have been proposed to more clearly define the pathway(s) through which AHPN and its analogues exert their effects. These include the induction of specific proteins, inhibition of the mitogen-activated protein kinase-1 phosphatase resulting in c-Jun NH2-terminal kinase kinase activation, induction of DNA adducts, translocation of the nuclear transcription factor TR3 to mitochondria, and TR3 binding to Bcl-2 as well other proposed mechanisms (13, 47, 48). How AHPN/3-Cl-AHPC exerts these diverse effects in malignant cells is not clear. Although AHPN activates RARγ and 3-Cl-AHPC does not, they seem to exhibit similar mechanisms of action in all aspects we have examined. In the present study, we have shown that AHPN, 3-Cl-AHPC, and their analogues bind to SHP and holo-SHP binds subsequently to a Sin3A complex that is likely involved in transcriptional repression. Whether AHPN/3-Cl-AHPC binding to SHP and the Sin3A complex is responsible for all of the documented effects of AHPN/3-Cl-AHPC on cellular function remains to be clarified. However, our results strongly suggest that SHP and Sin3A play an important role in AHPN/3-Cl-AHPC–mediated apoptosis. Studies to determine the role of SHP, if any, in the pathways detailed above by which AHPN, 3-Cl-AHPC, and their analogues induce apoptosis are being conducted. Crystalization of the 3-Cl-AHPC/SHP complex should allow us to delineate the 3-Cl-AHPC binding pocket and design more active SHP ligands.
Acknowledgments
Grant support: National Cancer Institute (M.I. Dawson and J.A. Fontana) and Veterans Affairs Merit Review (J.A. Fontana and M.I. Dawson).
We thank Dr. Robert G. Roeder (Rockefeller University, New York, NY) for providing anti-GPS2 and anti-TBL1 antibodies, Dr. Roy Frey (University of Pittsburgh, Pittsburgh, PA) for providing purified recombinant full-length SIRT with a 6-His tag and anti-SIRT1 antibody, and Dr. Anton Krumm (University of Washington School of Medicine, Seattle, WA) for providing the c-Myc gene containing the pHEBO-c-Myc plasmid.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
References
- 1.Shao ZM, Dawson MI, Li X-S, et al. p53 independent G0/G1 arrest and apoptosis induced by a novel retinoid in human breast cancer cells. Oncogene. 1995;11:493–504. [PubMed] [Google Scholar]
- 2.Schadendorf D, Kern MA, Artuc M, et al. Treatment of melanoma cells with the synthetic retinoid CD437 induces apoptosis via activation of AP-1 in vitro, and causes growth inhibition in xenografts in vivo. J Cell Biol. 1996;135:1889–98. doi: 10.1083/jcb.135.6.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sun SY, Yue P, Mao L, et al. Identification of receptor-selective retinoids that are potent inhibitors of the growth of human head and neck squamous cell carcinoma cells. Clin Cancer Res. 2000;6:1563–73. [PubMed] [Google Scholar]
- 4.Mologni L, Ponzanelli I, Bresciani F, et al. The novel synthetic retinoid 6-[3-adamantyl-4-hydroxyphenyl]-2-naphthalene carboxylic acid (CD437) causes apoptosis in acute promyelocytic leukemia cells through rapid activation of caspases. Blood. 1999;93:1045–61. [PubMed] [Google Scholar]
- 5.Chun KH, Pfahl M, Lotan R. Induction of apoptosis by the synthetic retinoid MX3350-1 through extrinsic and intrinsic pathways in head and neck squamous carcinoma cells. Oncogene. 2005;24:3669–77. doi: 10.1038/sj.onc.1208339. [DOI] [PubMed] [Google Scholar]
- 6.Lu XP, Fanjul A, Picard N, et al. Novel retinoid-related molecules as apoptosis inducers and effective inhibitors of human lung cancer cells in vivo. Nat Med. 1997;3:686–90. doi: 10.1038/nm0697-686. [DOI] [PubMed] [Google Scholar]
- 7.Garattini E, Parrella E, Diomede L, et al. ST1926, a novel and orally active retinoid-related molecule inducing apoptosis in myeloid leukemia cells: modulation of intracellular calcium homeostasis. Blood. 2004;103:194–207. doi: 10.1182/blood-2003-05-1577. [DOI] [PubMed] [Google Scholar]
- 8.Hsu CA, Rishi AK, Li X-S, et al. Retinoid induced apoptosis in leukemia cells through a retinoic acid nuclear receptor-independent pathway. Blood. 1997;89:4470–9. [PubMed] [Google Scholar]
- 9.Sun SY, Yue P, Chandraratna RA, Testfaigi Y, Hong WK, Lotan R. Dual mechanisms of action of the retinoid CD437: nuclear retinoic acid receptor-mediated suppression of squamous differentiation and receptor-independent induction of apoptosis in UMSCC22B human head and neck squamous cell carcinoma cells. Mol Pharmacol. 2000;58:508–14. doi: 10.1124/mol.58.3.508. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y, Dawson MI, Ning Y, et al. Induction of apoptosis in retinoid-refractory acute myelogenous leukemia by a novel AHPN analog. Blood. 2003;102:3743–52. doi: 10.1182/blood-2003-01-0108. [DOI] [PubMed] [Google Scholar]
- 11.Marchetti P, Zanzani N, Joseph B, et al. The novel retinoid 6-[3-(1-adamantyl)-4-hydroxyphenyl]-2-naphthalene carboxylic acid can trigger apoptosis through a mitochondrial pathway independent of the nucleus. Cancer Res. 1999;59:6257–66. [PubMed] [Google Scholar]
- 12.Zhao X, Denmark K, Wong L, et al. Retinoic acid receptor-independent mechanism of apoptosis of melanoma cells by the retinoid CD437 (AHPN) Cell Death Differ. 2001;8:878–86. doi: 10.1038/sj.cdd.4400894. [DOI] [PubMed] [Google Scholar]
- 13.Lin B, Kolluri SK, Lin F, et al. Conversion of Bcl-2 from protector to killer by interaction with nuclear orphan receptor Nur77/TR3. Cell. 2004;116:527–40. doi: 10.1016/s0092-8674(04)00162-x. [DOI] [PubMed] [Google Scholar]
- 14.Fontana JA, Dawson MI, Leid M, et al. Identification of a unique binding protein specific for a novel retinoid inducing cellular apoptosis. Int J Cancer. 2000;86:474–9. doi: 10.1002/(sici)1097-0215(20000515)86:4<474::aid-ijc5>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 15.Boehmelt G, Antonio L, Iscove NN. Cloning of the murine transcriptional corepressor component SAP18 and differential expression of its mRNA in the hematopoietic hierarchy. Gene. 1998;207:267–75. doi: 10.1016/s0378-1119(97)00648-3. [DOI] [PubMed] [Google Scholar]
- 16.Cowley SM, Kang RS, Frangioni JV, et al. Functional analysis of the Mad1-3A repressor-corepressor interaction reveals determinants of specificity, affinity, and transcriptional response. Mol Cell Biol. 2004;24:2698–709. doi: 10.1128/MCB.24.7.2698-2709.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y, Sun Z-W, Iratni I, et al. SAP30, a novel protein conserved between human and yeast, is a component of a histone deacetylase complex. Mol Cell. 1998;1:1021–31. doi: 10.1016/s1097-2765(00)80102-1. [DOI] [PubMed] [Google Scholar]
- 18.Kemper JK, Kim H, Miao J, Bhalla S, Bae Y. Role of an mSin3A-Swi/Snf chromatin remodeling complex in the feedback repression of bile acid biosynthesis by SHP. Mol Cell Biol. 2004;24:7707–19. doi: 10.1128/MCB.24.17.7707-7719.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bae Y, Kemper JK, Kemper B. Repression of CAR-mediated transactivation of CYP2B genes by the orphan receptor short heterodimer partner (SHP) DNA Cell Biol. 2004;23:81–91. doi: 10.1089/104454904322759894. [DOI] [PubMed] [Google Scholar]
- 20.Seol W, Choi HS, Moore DD. An orphan nuclear hormone receptor that lacks a DNA binding domain and heterodimerizes with other receptors. Science. 1996;272:1336–9. doi: 10.1126/science.272.5266.1336. [DOI] [PubMed] [Google Scholar]
- 21.Seol W, Chung M, Moore DD. Novel receptor interaction and repression domains in the orphan receptor SHP. Mol Cell Biol. 1997;17:7126–31. doi: 10.1128/mcb.17.12.7126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dawson MI, Harris DL, Liu G, et al. Antagonist analogue of 6-[3′-(1-adamantyl)-4′-hydroxyphenyl]-2-naphthalenecarboxylic acid (AHPN) family of apoptosis inducers that effectively blocks AHPN-induced apoptosis but not cell-cycle arrest. J Med Chem. 2004;47:3518–36. doi: 10.1021/jm030524k. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Y, Mohamad R, Rishi AK, et al. Induction of apoptosis of human B-CLL and ALL cells by a novel retinoid and its nonretinoidal analog. Blood. 2002;100:2917–25. doi: 10.1182/blood.V100.8.2917. [DOI] [PubMed] [Google Scholar]
- 24.Wang L, Lee Y-K, Bundman D, et al. Redundant pathways for negative feedback regulation of bile acid production. Dev Cell. 2002;2:721–31. doi: 10.1016/s1534-5807(02)00187-9. [DOI] [PubMed] [Google Scholar]
- 25.Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman J, Smith JA, Struhl K, editors. Current protocols in molecular biology. New York: John Wiley and Sons; 1990. p. 12.1.1. [Google Scholar]
- 26.Li X-S, Rishi AK, Shao Z-M, et al. Posttranscriptional regulation of p21WAF1/CIP1 expression in human breast carcinoma cells. Cancer Res. 1996;56:5055–62. [PubMed] [Google Scholar]
- 27.Elbashir SM, Lendeckel W, Tuschl T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 2001;15:188–200. doi: 10.1101/gad.862301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weinmann AS, Bartley SM, Zhang T, Zhang MQ, Farnham PJ. Use of chromatin immunoprecipitation to clone novel E2F target promoters. Mol Cell Biol. 2001;21:6820–32. doi: 10.1128/MCB.21.20.6820-6832.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alland L, Muhle R, Hou H, Jr, et al. Role for N-CoR and histone deacetylase in Sin3-mediated transcriptional repression. Nature. 1997;387:49–55. doi: 10.1038/387049a0. [DOI] [PubMed] [Google Scholar]
- 30.Ayer DE. Histone deacetylases: transcriptional repression with SINers and NuRDs. Trends Cell Biol. 1999;9:193–8. doi: 10.1016/s0962-8924(99)01536-6. [DOI] [PubMed] [Google Scholar]
- 31.Moehren U, Dresssel U, Reeb CA, et al. The highly conserved region of the co-repressor Sin3A functionally interacts with the co-repressor Alien. Nucleic Acids Res. 2004;32:2995–3004. doi: 10.1093/nar/gkh621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pile LA, Schlag EM, Wassarman DA. The SIN3/RPD3 deacetylase complex is essential for G(2) phase cell cycle progression and regulation of SMRTER corepressor levels. Mol Cell Biol. 2002;22:4965–76. doi: 10.1128/MCB.22.14.4965-4976.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Y, Dufau ML. Gene silencing by nuclear orphan receptors. Vitam Horm. 2004;68:1–48. doi: 10.1016/S0083-6729(04)68001-0. [DOI] [PubMed] [Google Scholar]
- 34.Yeo MG, Yoo YG, Choi HS, Pak YK, Lee MO. Negative cross-talk between Nur77 and small heterodimer partner and its role in apoptotic cell death of hepatoma cells. Mol Endocrinol. 2005;19:950–63. doi: 10.1210/me.2004-0209. [DOI] [PubMed] [Google Scholar]
- 35.Nishizawa H, Yamagata K, Shimomura, et al. Small heterodimer partner, an orphan nuclear receptor, augments peroxisome proliferator-activated receptor γ transactivation. J Biol Chem. 2002;277:1586–92. doi: 10.1074/jbc.M104301200. [DOI] [PubMed] [Google Scholar]
- 36.Kim YS, Han C-Y, Kim S-W, et al. The orphan nuclear receptor small heterodimer partner as a novel coregulator of nuclear factor-κB in oxidized low density lipoprotein-treated macrophage cell line RAW 264.7. J Biol Chem. 2001;276:33736–40. doi: 10.1074/jbc.M101977200. [DOI] [PubMed] [Google Scholar]
- 37.Macchiarulo A, Rizzo G, Costantino G, Fiorucci S, Pellicciari R. Unveiling hidden features of orphan nuclear receptors: the case of the small heterodimer partner (SHP) J Mol Graph Model. 2006;24:362–72. doi: 10.1016/j.jmgm.2005.09.016. [DOI] [PubMed] [Google Scholar]
- 38.Johansson L, Thomsen JS, Damdimopolos AE, et al. The orphan nuclear receptor SHP inhibits agonist-dependent transcriptional activity of estrogen receptors ERα and ERβ. J Biol Chem. 1999;274:345–53. doi: 10.1074/jbc.274.1.345. [DOI] [PubMed] [Google Scholar]
- 39.Saynal S, Kim JY, Kim HJ, et al. Differential regulation of the orphan nuclear receptor small heterodimer partner (SHP) gene promoter by orphan nuclear receptor ERR isoforms. J Biol Chem. 2002;277:1739–48. doi: 10.1074/jbc.M106140200. [DOI] [PubMed] [Google Scholar]
- 40.Kovacic A, Speed CJ, Simpson ER, Clyne CD. Inhibition of aromatase transcription via promoter II by short heterodimer partner in human preadipocyte. Mol Endocrinol. 2004;18:252–9. doi: 10.1210/me.2003-0211. [DOI] [PubMed] [Google Scholar]
- 41.Choi YH, Park MJ, Kim KW, et al. The orphan nuclear receptor SHP involved in monocytic differentiation, and its expression is increased by c-Jun. J Leukoc Biol. 2007;76:1082–8. doi: 10.1189/jlb.1203658. [DOI] [PubMed] [Google Scholar]
- 42.Segnitz B, Gehring U. The function of steroid hormone receptors is inhibited by the hsp90-specific compound geldanamycin. J Biol Chem. 1997;272:18694–701. doi: 10.1074/jbc.272.30.18694. [DOI] [PubMed] [Google Scholar]
- 43.Kanelakis KC, Shewach DS, Pratt WB. Nucleotide binding states of hsp70 and hsp90 during sequential steps in the process of glucocorticoid receptor. hsp90 heterocomplex assembly J Biol Chem. 2002;277:33698–703. doi: 10.1074/jbc.M204164200. [DOI] [PubMed] [Google Scholar]
- 44.Stavreva DA, Muller WG, Hager GL, Smith CL, McNally JG. Rapid glucocorticoid receptor exchange at a promoter is coupled to transcription and regulated by chaperones and proteasomes. Mol Cell Biol. 2004;24:2682–97. doi: 10.1128/MCB.24.7.2682-2697.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun SY, Yue P, Lotan R. Implication of multiple mechanisms in apoptosis induced by the synthetic retinoid CD437 in human prostate carcinoma cells. Oncogene. 2000;19:4513–22. doi: 10.1038/sj.onc.1203810. [DOI] [PubMed] [Google Scholar]
- 46.Zhao X, Spanjaard RA. The apoptotic action of the retinoid CD437/AHPN: diverse effects, common basis. J Biomed Sci. 2003;10:44–9. doi: 10.1007/BF02255996. [DOI] [PubMed] [Google Scholar]
- 47.Pfahl M, Piedrafita FJ. Retinoid targets for apoptosis induction. Oncogene. 2003;22:9058–62. doi: 10.1038/sj.onc.1207109. [DOI] [PubMed] [Google Scholar]
- 48.Li H, Kolluri SK, Gu J, et al. Cytochrome c release and apoptosis induced by mitochondrial targeting of nuclear orphan receptor TR3. Science. 2000;289:1159–64. doi: 10.1126/science.289.5482.1159. [DOI] [PubMed] [Google Scholar]