

Abstract
The ribonucleotide reductases from three ancient eubacteria, the hyperthermophilic Thermotoga maritima (TM), the radioresistant Deinococcus radiodurans (DR), and the thermophilic photosynthetic Chloroflexus aurantiacus, were found to be coenzyme-B12 (class II) enzymes, similar to the earlier described reductases from the archaebacteria Thermoplasma acidophila and Pyrococcus furiosus. Reduction of CDP by the purified TM and DR enzymes requires adenosylcobalamin and DTT. dATP is a positive allosteric effector, but stimulation of the TM enzyme only occurs close to the temperature optimum of 80–90°C. The TM and DR genes were cloned by PCR from peptide sequence information. The TM gene was sequenced completely and expressed in Escherichia coli. The deduced amino acid sequences of the two eubacterial enzymes are homologous to those of the archaebacteria. They can also be aligned to the sequence of the large protein of the aerobic E. coli ribonucleotide reductase that belongs to a different class (class I), which is not dependent on B12. Structure determinations of the E. coli reductase complexed with substrate and allosteric effectors earlier demonstrated a 10-stranded β/α-barrel in the active site. From the conservation of substrate- and effector-binding residues we propose that the B12-dependent class II enzymes contain a similar barrel.
Keywords: deoxyribonucleotide synthesis, allosteric regulation, evolution, thermophilic
All living cells produce the four deoxyribonucleotides required for DNA replication and repair by reduction of ribonucleotides. Three classes of ribonucleotide reductases exist (1, 2). All use free radical chemistry for catalysis but differ in the way in which they produce a protein radical required for the activation of the substrate. Class I enzymes, with the aerobic Escherichia coli enzyme as prototype, are α2β2 proteins. The large α protein (R1) harbors catalytic and allosteric sites, whereas β contains a diferric center and a stable tyrosyl radical. They occur in eukaryotes and some aerobic eubacteria. Class II enzymes, with the Lactobacillus leichmannii enzyme as prototype, have an α or α2 structure and use adenosylcobalamin as radical generator. They occur in aerobic and anaerobic bacteria. Class III enzymes finally, with the anaerobic E. coli enzyme as prototype, have an α2β2 structure and use S-adenosylmethionine to generate a stable glycyl radical. They occur in anaerobic bacteria.
The prototypes for the three classes were not homologous and it seemed possible that they had arisen separately during evolution (3). However, functional aspects are in favor of a common root (1, 2). Members of the three classes reduce ribonucleotides by an identical mechanism and critical, functionally involved cysteine residues are found in corresponding positions in the class I and II enzymes (4, 5). Furthermore the allosteric regulation of the substrate specificity of the enzymes is highly similar, suggesting that the different classes may in part have closely related tertiary structures in spite of the large divergence of their primary structures (1, 2).
For a long time the only characterized class II enzyme was the prototype from L. leichmannii, a eubacterium (6, 7). However, two recent studies reported the sequences and some properties of class II reductases from two archaebacteria, Pyrococcus furiosus (8) and Thermoplasma acidophilum (9). Both sequences show considerable homology not only to the L. leichmannii enzyme but also to class I reductases. In addition, at the N terminus ≈100 amino acids were homologous to the corresponding sequence of the E. coli class III reductase. This result is persuasive evidence that all three classes originated from a common ancestor.
Do the negative results with the Lactobacillus enzyme mean that its sequence has evolved so far that a common origin no longer is recognized? Knowledge of eubacterial class II ribonucleotide reductases is limited. We have now studied class II enzymes from some deeply rooted eubacteria (10, 11) and find that their sequences show a greater kinship to the enzymes from archaebacteria than to that from L. leichmannii. From a combination of sequence data of the class II enzymes with recent structural results from complexes between the E. coli class I reductase and allosteric effectors (12) we hypothethize that the two classes share an appreciable part of the tertiary structure involved in effector binding.
To facilitate the presentation and discussion of our work we suggest the name nrdJ for the gene of class II ribonucleotide reductases.
MATERIALS AND METHODS
Materials.
Thermotoga maritima (TM) strain MSB8 (DSM 3109 from the Deutsche Sammlung von Mikroorganismen und Zellkulturen, Braunschweig, Germany) and Deinococcus radiodurans R1 (ATCC 13939) (DR) were used for enzyme purification and PCR amplification of genomic DNA. Extracts from Chloroflexus aurantiacus J-10-fl (ATCC 29366), were used for enzyme assays. E. coli DH5αF′, and plasmids pBlueScript SK (+) (pBSK, Stratagene), pGEM-T (Promega) and pET22b (Novagen) were used for recombinant DNA techniques. Lysyl endopeptidase was from Wako Chemicals (Neuss, Germany).
Bacterial Growth and Enzyme Extraction.
Batch cultures of TM were grown under nitrogen for 12–14 h at 80°C in basal medium containing sodium sulfide (13), with the NaCl concentration adjusted to 20 g/liter and thiosulfate used as electron acceptor at 20 mM. After rapid cooling on ice the cells were sedimented at 8,000 × g and stored at −20°C under nitrogen until processed. Extraction of 8 g of packed cells was made in air at +4°C with 12 ml of 50 mM Tris⋅HCl, pH 7.5/50 mM KCl/1 mM EDTA/10 mM DTT/1 mM phenylmethanesulfonyl fluoride and 1 mg/ml egg white lysozyme by six consecutive cycles of freezing/thawing. A clear extract was obtained by centrifugation (45,000 rpm for l h in a Ty65 Beckman centrifuge).
DR was grown aerobically at 30°C in TGY medium (14) for 8 h until the end of the logarithmic growth phase. The cells were centrifuged, extracted in a French press in the buffer described for TM cells lacking KCl, and centrifuged at 45,000 rpm for 1 h to give a clear extract. Chloroflexus was grown anaerobically with light (900 lux) at 55°C in DG medium (15) for 48 h. The cells were extracted in air by sonication and centrifuged as described for DR.
Enzyme Purification.
The TM extract was precipitated with streptomycin (final concentration, 1%). After removal of the precipitate the TM reductase was precipitated with ammonium sulfate (final saturation, 45%), centrifuged and dialyzed against buffer A (50 mM Tris⋅HCl, pH 7.5/10 mM DTT/1 mM EDTA/0.1 mM phenylmethanesulfonyl fluoride). The enzyme was chromatographed on a Mono Q HR 5/5 column on a Pharmacia fast protein liquid chromatography (FPLC) machine with a 0–0.4 M KCl gradient in buffer A. The enzyme eluted at around 0.3 M KCl. The sample was concentrated by ultrafiltration in Centriprep 30 (Amicon) tubes and adsorbed to a column of dATP-Sepharose (0.2 ml/mg protein) in 30 mM Tris⋅HCl, pH7.5/10 mM CaCl2/2 mM DTT (buffer B)/0.5 M KCl. The column was washed successively with buffer B, buffer B/1 mM ATP, buffer B again, and finally 0.5 M ammonia. The last eluate contained the reductase. The enzyme was immediately concentrated and ammonia was removed by centrifugation in a Centricon 30 tube.
The DR reductase was purified by a similar procedure omitting the FPLC chromatography. The dATP-Sepharose step was slightly different in that the enzyme was eluted with 1 mM dATP instead of ammonia.
Enzyme Assays.
Under standard conditions the TM reductase was incubated aerobically at 90°C for 20 min with 0.5 mM [3H]CDP or [3H]CTP (19 cpm/pmol), 20–40 mM DTT, 15 μM adenosylcobalamin, 0.3 mM dATP, 10 mM MgCl2 or CaCl2, and 50 mM Tris⋅HCl (pH 8). The DR and Chloroflexus enzymes were assayed similarly except for a temperature of 30°C for DR and 50°C for Chloroflexus. After acid hydrolysis the amount of [3H]dCMP formed was determined (16). One enzyme unit corresponds to 1 nmol dCMP/min. Specific activity is units/mg protein.
Isolation of nrdJ from TM by PCR and Single Specific Primer-PCR.
On gel electrophoresis the TM enzyme gave a single band at 90 kDa, the DR enzyme gave several bands, but with a dominating, rather diffuse band at 85 kDa. These bands were used for the determination of the N-terminal sequence as well as internal sequences from peptides obtained by in-gel digestion with lysyl endopeptidase (17) (Table 1). TM peptides 1 and 4 were used to design the following two degenerate primers according to the TM codon usage (18): PrA [5′-GA(C/T)AG(A/G)TA(C/T)TT(C/T)ATGAA-3′; peptide 1] and PrB [5′-TC(C/T)CTGTA(A/C)AC(A/G/C/T)GT(A/G/T)AT-3′; peptide 4, antisense]. Genomic DNA from TM (19) was amplified by PCR with the two primers by Taq polymerase at 50°C for annealing. The 1.8-kb amplification product was cloned in pGEM-T, and by sequencing it was found to contain the central part of nrdJ.
Table 1.
Peptide sequences
| No. | Location | Sequence |
|---|---|---|
| T. maritima | ||
| 1 | N terminal | M1KLSDLISRWIDVEPSKNAQIILRDRYFMK30 |
| 2 | Internal | K158VGGGVGSNFSELRPK173 |
| 3 | Internal | K523EPLLYVNQVLREK536 |
| 4 | Internal | K597TINMPQSATVDDVLNVYLEALRTNVRxIT- VYRDGSLQTQVL638 |
| D. radiodurans | ||
| 1 | N terminal | x2xAPDRSA9 |
| 2 | Internal | K15saQHFDENAQH26 |
| 3 | Internal | K382xFPVPQEGEYAGTFPEL399 |
| 4 | Internal | k1012FVELLESYPAPK1024 |
| 5 | Internal | K1111SAYEEAYRTGxK1123 |
| 6 | Internal | K15xxQHFDENAQ25 |
| 7 | Internal | K118LALVTK124 |
The numbers at the beginning and at the end of each sequence indicate their positions in the predicted translated sequence of the TM nrdJ gene obtained in this work and the nrdJ gene sequence found in the DR genome database. Lowercase letters are used when discrepancies exist. Peptides 1–5 from DR have been obtained from the 85-kDa band, and peptides 6 and 7 from the 105-kDa band. The intein present in the DR-translated product is included in the sequence; the total size of the unprocessed protein is 1,358 amino acids. No peptides were found corresponding to the intein region (positions 465–830).
The cloning of the two extremes of the gene was made with single specific primer-PCR (20). After Southern blot hybridization of the TM genome with the 1.8-kb fragment as a probe we deduced that the 5′ extreme would be included in a 1.4-kb XhoI fragment and the 3′ extreme in a 1.6-kb ClaI fragment. To clone the 5′ end XhoI-digested chromosomal DNA was ligated to XhoI-digested pBSK plasmid and PCR amplified with primer PrC (5′-CTTCGAAAATCTCTTCAATGC-3′) as specific primer and PrFor (pUC/M13 forward) as generic primer (see Fig. 1). Similarly, the 3′ extreme was obtained by amplifying ClaI-digested chromosomal DNA ligated to ClaI-digested pBSK plasmid with PrD (5′-TACGTGAACCAGGT-3′) as specific and PrRev (pUC/M13 reverse) as generic primers. The resulting products were cloned into pGEM-T and sequenced in both strands from several independent clones. From these sequences and their overlap with the 1.8-kb sequence a definite sequence of 3,256 bp between the restriction sites XhoI and ClaI (Fig. 1) was obtained and deposited in the GenBank database (accession no Y12877).
Figure 1.
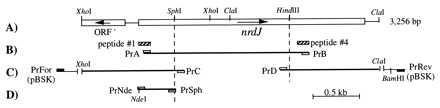
(A) Physical map of the 3,256-bp sequenced fragment containing the TM nrdJ gene with restriction sites mentioned in the text. (B) PCR amplification of an internal fragment with primers derived from peptides 1 and 4 (Table 1). (C) Single specific primer-PCR for cloning the 5′ and 3′ extremes of nrdJ. (D) Generation of an NdeI restriction site in the translation start codon. The entire gene was obtained by joining the different fragments via the SphI and HindIII sites.
Expression of TM nrdJ in E. coli.
An NdeI site and the entire reconstructed gene were introduced into pET22b plasmid, downstream the T7 promoter and ribosome binding site. Primers PrNde (5′-AGGAGGGAGCATATGAATTGTCCG-3′, NdeI site underlined, start codon in bold letters) and PrSph (5′-CGAAGCATGCGGAGAGCATGTGAAGATG-3′, antisense, SphI site underlined) were used to amplify the 5′ extreme of the gene. The 418-bp product was used together with the fragments generated with the amplifications PrA-PrB and PrD-PrRev to reconstruct the entire gene by using the unique SphI and HindIII sites (Fig. 1). The entire gene was obtained by digestion with NdeI–BamHI and cloned into pET22b. The resulting plasmid (pUA724) was transformed into E. coli BL21(DE3). nrdJ was expressed by addition of 1 mM isopropyl β-d-thiogalactoside to an exponential culture (OD550 of 0.6) and cells were harvested after 4 h. For denaturation of mesophilic host cells extracts were heated to 85°C for 15 min.
PCR Amplification of an Internal nrdJ Fragment of DR.
DR peptide 3 (Table 1) was used for the construction of primer 5′-GGCGA(A/G)TACGCCGGCAC(G/C)TTCCC-3′ and peptide 4 for primer 5′-CTTGGG(G/C)GC(G/C)GGGTAGCT(T/C)TC-3′ (antisense) for PCR amplification of a plate colony of DR with an annealing temperature of 64°C. An amplification product of 1.9 kb was obtained, cloned, and sequenced.
RESULTS
Enzyme Purification.
When CTP reduction was determined in extracts from TM, DR, and Chloroflexus under the conditions given in Materials and Methods, the extracts gave specific activities of 1.8, 0.6, and 0.4, respectively. Omission of adenosylcobalamin completely abolished the reaction with the TM and DR enzymes but left some activity in the Chloroflexus extract. Enzyme activity also depended on adenosylcobalamin when enzyme extraction and assay were made anaerobically excluding the additional activity of a class III enzyme in the anaerobes TM and Chloroflexus. The enzymes from TM and DR were then purified aerobically, as described in Materials and Methods. The purified TM enzyme had a specific activity of 1,100 with CDP as substrate. On gel electrophoresis it gave a single band of ≈90 kDa. During chromatography on a column of Superdex 200 at room temperature the protein eluted very close to the void volume, corresponding to a molecular mass of more than 500 kDa, indicating aggregation. The purified DR enzyme was not homogeneous on gel electrophoresis. It gave a minor sharp band at 105 kDa and a strong, rather diffuse band at 85 kDa, as well as additional minor bands of lower molecular mass. The 105- and 85-kDa bands were used for peptide analyses (Table 1). The deduced amino acid sequence [The Institute for Genomic Research (TIGR); personal communication] suggests a molecular mass of 107-kDa and the 85-kDa band may therefore correspond to degradation products. The specific enzyme activity with CDP as substrate was 150–200.
Properties of the TM Reaction.
The temperature optimum for the reaction was ≈80°C. Both Mg2+ and Ca2+ stimulated 3- to 4-fold, with Ca2+ giving a slightly higher temperature optimum (data not shown). The homogeneous enzyme was active with both CDP and CTP. Comparing the two substrates in a time curve (Fig. 2) we found that CTP was essentially not used as substrate during the first minutes, whereas CDP was reduced immediately. This finding suggests that the activity of CTP depended on its dephosphorylation to CDP at the high temperature of the reaction. In support of this we found a time-dependent transformation of CTP to CDP during incubation (Fig. 2).
Figure 2.
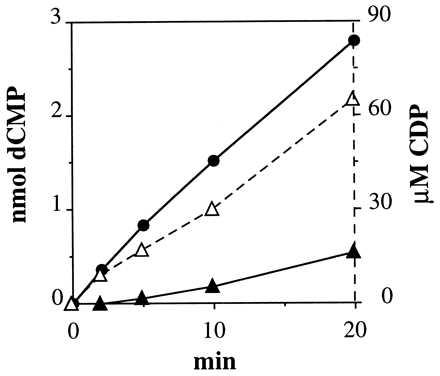
Comparison of CDP and CTP as substrates for the TM reductase. Assays were made under standard conditions with MgCl2 and 0.13 μg of enzyme and the amount of dCMP formed was determined at the indicated time points (CDP, •; CTP, ▴). In the CTP experiment, the concentration of CDP (▵) caused by degradation of CTP was also determined.
The reduction of CDP is influenced by allosteric effectors, with dATP and ATP stimulating and dTTP and dGTP inhibiting the reaction (data not shown). The extent of stimulation by dATP depended on temperature. At both 80°C and 90°C dATP strongly stimulated CDP reduction, whereas it had a marginal effect at 70°C (Fig. 3).
Figure 3.
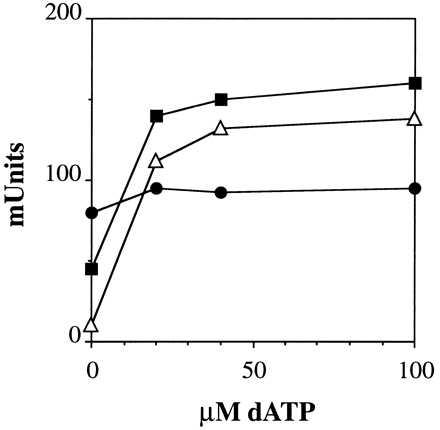
Dependence of the allosteric stimulation of the TM enzyme (0.13 μg) by dATP on temperature (70°C •; 80°C, ▪; 90°C ▵) in the presence of MgCl2.
Properties of the DR Reaction.
All experiments were done at 30°C. Again, both Mg2+ and Ca2+ stimulated the reaction and the enzyme reduced both CTP and CDP. Also in this case a time curve suggested CDP to be the preferred substrate (data not shown). The allosteric regulation was similar to that of the TM enzyme with dATP and ATP stimulating and dTTP and dGTP inhibiting the reaction (data not shown).
Cloning and Sequencing of the TM and DR nrdJ Genes.
The complete TM nrdJ gene was cloned by PCR and single specific primer-PCR as described in Materials and Methods. It is composed of 2,484 bp, encoding a putative protein of 827 residues with a predicted molecular mass of 94,018, lacking inteins. A hypothetical ribosome binding site (AGGAGG) complementary to the 3′ end of the TM 16S rRNA (21) is located 7 nt upstream of the ATG start codon for the first methionine. The methionine is not processed from the mature protein (Table 1). No definitive promoter or terminator sequences were identified. The 5′ end (467 bp) of an hypothetical ORF with a transcriptional sense opposed to the nrdJ gene is present upstream the nrdJ gene. It shows significant homologies with two ORFs of unknown function present in the Methanococcus jannaschii (22) and Synechocystis sp. (23) genomes.
The complete TM nrdJ gene was cloned into pET22b to give plasmid pUA724 as described in Materials and Methods. After transformation of E. coli BL21(DE3) with pUA724 expression of the nrdJ gene could be induced with isopropyl β-d-thiogalactoside resulting in a moderate production of the NrdJ protein. Gels of extracts from induced bacteria did not show overproduction of a specific band, but enzyme assays demonstrated the presence of an adenosylcobalamin-dependent, heat-stable reductase activity. The specific activity of the recombinant enzyme in the crude extract was increased from 5 to 150 by heating at 85°C. The poor overproduction is probably caused by differences in the codon usage of TM and E. coli (18). Nevertheless pUA724 should be a useful tool for further work.
The DR nrdJ gene was only partially cloned because in the course of our work the TIGR group released the provisional and uncompleted sequence of the DR genome, including the nrdJ gene. Our work is presented here briefly, because it confirms and slightly extends the other results. The amino acid sequence deduced from the nucleotide sequence of the 1.9-kb product cloned by us was in complete agreement with the reported sequence. All the DR peptides of Table 1 obtained from either the 85-kDa and 105-kDa bands are present in the sequence. The sequence of peptide 1 suggests that the N-terminal methionine is processed. The sequence contains one intein that is located in the same position as the second intein of the P. furiosus enzyme (8) and also is of similar length. In the position of the first Pyrococcus intein, the DR sequence contains an insertion relative to the TM sequence that does not possess the elements of an intein (Fig. 4).
Figure 4.
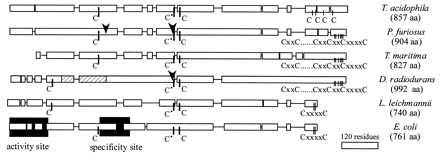
Schematic alignment of class II reductases with the class I E. coli R1 protein. The alignment was generated with the clustal w version 1.7 (24) program. Single lines show larger gaps introduced in the sequences. The lowest bar gives the length of 120 residues. The arrows show the positions of inteins in the P. furiosus and DR enzymes. The striped area of the DR structure indicates the insertion referred to in the text. The shaded areas show the locations of the two allosteric sites of the E. coli enzyme identified by crystallography (12).
DISCUSSION
Species belonging to three of the most deeply rooted eubacteria groups (10, 11) are here shown to use a class II reductase for deoxyribonucleotide synthesis. The genome of DR contains sequences characteristic of both a class Ib and a class II reductase (TIGR, personal communication). Our work shows that it is the latter that provides an active enzyme under normal growth conditions. At least two of the investigated enzymes were found to be ribonucleoside diphosphate reductases, as are two earlier investigated class II enzymes from archaebacteria (8, 9). They differ from the L. leichmannii enzyme that reduces triphosphates (6). Clearly a distinction between class I and II enzymes can no longer be made on the basis of the level of phosphorylation of the substrate. TM is an extreme thermophile (25) and the TM reductase accordingly requires temperatures above 60°C for activity, with an optimum between 80°C and 90°C. All ribonucleotide reductases are allosteric enzymes (1, 2, 5) whose substrate specificity is regulated by binding of effectors to a specific site (specificity site). Reduction of CDP is stimulated by binding of ATP or dATP to this site. A group of class I reductases, among them the E. coli enzyme, has a second site (activity site). Binding of dATP there strongly inhibits all enzyme activity. Reduction of CDP by the reductases from the two ancient eubacteria investigated here are stimulated, not inhibited, by dATP, suggesting the absence of an activity site. With the TM enzyme a clear stimulation by dATP is only seen at high temperatures, even though the nucleotide binds to the protein in the cold room (unpublished data). This result suggests that the protein at low temperature does not have the required flexibility to transmit the message from the allosteric to the catalytic site.
Fig. 4 gives an overview of the general construction of the TM and DR enzymes, comparing them with the two archaeal class II reductases, the Lactobacillus enzyme and the R1 protein of the E. coli class Ia reductase. The latter two enzymes use three active cysteines with specific functions for the same free radical-based chemistry (4, 5). In Fig. 4 these cysteines can be identified in all sequences. Although located at slightly different positions, their relative order is the same. The first redox-active cysteine is located closer to the N terminus in the Lactobacillus and Deinococcus enzymes than in the other organisms. This result created difficulties in sequence alignments. The Deinococcus sequence was, however, quite homologous with the Thermotoga sequence, which could form a bridge to the other sequences. The redox-active cysteine pair at the C terminus of the E. coli and Lactobacillus enzymes have been shown to transfer electrons from thioredoxin to the cysteine pair in the catalytic site (4, 5). The Thermoplasma enzyme lacks such a pair, whereas the Pyrococcus, TM and DR enzymes have three. It is not clear how this affects transthiolation. The TM enzyme lacks the first 100 amino acids present in the others and has instead an extended C terminus.
Fig. 5 shows a computer alignment of the sequences of the TM reductase with those of the Pyrococcus and E. coli enzymes. The alignment of the Deinococcus enzyme whose preliminary sequence was reported by the TIGR group cannot be included, but it also shows retention of crucial residues. The three catalytically active cysteines are found in identical positions, which gives confidence to the correctness of the alignments and also suggests that the tertiary structure of the enzymes around the catalytic site is similar. In the E. coli enzyme the activity site is located at the N terminus of R1, the specificity site at the interface between the two subunits of R1 after C225 in the primary structure. Specific amino acids involved in effector binding have been identified and are shaded in Fig. 5 (12). B12 enzymes do not contain an activity site. The Thermotoga enzyme lacks the first 60 residues at the N terminus of the E. coli enzyme responsible for the activity site, which explains the absence of this site. The signature sequence VXKRDG at the N terminus involved in effector binding is found in Pyrococcus and Thermoplasma, but not in Deinococcus. However, all three sequences lack most other binding residues of the activity site (Fig. 5). In the specificity site, D232, H-bonded to the 3′-OH group of the effector, and R262, binding the γ-phosphate, are two key amino acids present in all class I enzymes. These residues are also present in the TM and Pyrococcus class II enzymes (Fig. 5). The other binding residues vary slightly between various class I reductases. Thus L234 is I in the mouse and M in class Ib, I268 is V in Synechocystis (23) and C292 is V or I in some eukaryotes. In all but one case the corresponding residues of the B12 enzymes fall within these variations. Also the enzymes from Thermoplasma and Deinococcus, as well as two sequences from Archaeoglobus fulgidus and Mycobacterium tuberculosis reported by genome sequencing (TIGR, personal communication), conform completely to this pattern (data not shown). Thus the binding residues for the allosteric effector in the specificity site are identical in class I reductases and six B12 enzymes, three from eubacteria and three from archaea. This finding together with retention of the catalytically active cysteines strongly suggests that the tertiary structure of the catalytic and allosteric sites, i.e., the 10-stranded β/α-barrel, is present also in class II reductases.
Figure 5.

Alignment of the N-terminal 378 residues of the T. maritima ribonucleotide reductase (Tmar) with the reductase from P. furiosus (Pfur) (8) and the R1 protein from E. coli (Ecol) (26). The remaining TM sequence is shown by itself. The cysteines corresponding to those in the active site of E. coli (C225, C439, and C462) as well as the C-terminal cysteines hypothetically involved in transthiolation are shaded lightly. Residues involved in effector binding at the activity (N terminal) and specificity sites of E. coli are shaded as well as the conserved counterparts of the class II enzymes. The specificity site residues also carry a ▾. Consensus among the two class II proteins is shown by lowercase letters; consensus among all three enzymes is shown by capital letters. The numbering of the three sequences is given at the end of each line. The alignment was generated with the pileup program of the GCG (Genetic Computer Group, University of Wisconsin, version 9.0-UNIX) package.
The allosteric regulation of the L. leichmannii enzyme is identical to that of the other B12 enzymes (unpublished data), indicating that also this enzyme may contain the barrel structure. One complication is that the Lactobacillus enzyme is believed to be a monomer (6), whereas the allosteric E. coli site is located between the polypeptide chains of a dimer.
At present the sequences of 8 class II enzymes are available. Fig. 6 shows an unrooted phylogenetic tree of the deduced amino acid sequences. The main finding is that the TM and DR enzymes are closely related to the archeal reductases, whereas the L. leichmannii enzyme is more distant. Interestingly, also the M. tuberculosis class II sequence is more related to the archeal enzymes. In contrast to the DR case, a class Ib reductase is the active enzyme in extracts from M. tuberculosis (28).
Figure 6.
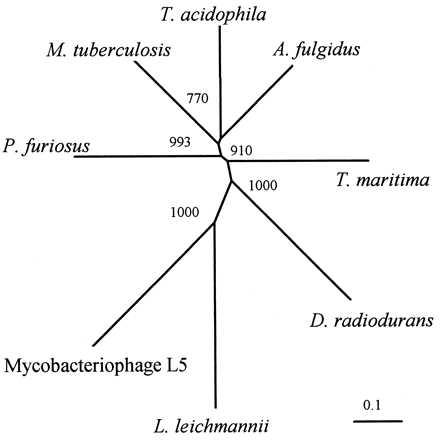
Unrooted phylogenetic tree of the deduced amino acid sequences from T. maritima, D. radiodurans (TIGR, personal communication), P. furiosus (8), T. acidophila (9), A. fulgidus (TIGR), M. tuberculosis (TIGR; partial sequence), L. leichmannii (7), and the mycobacteriophage L5 (27). The clustal w program was used for sequence alignments and to compute the phylogenetical tree. The significance of the branching order was evaluated by bootstrap analysis of 1,000 computer-generated trees. Bootstrap values are indicated. (Bar = 0.1 changes per site.)
This work and the appearance of additional genome sequences show that a large number of class II sequences are more related to the two reported archeal proteins than to the until now considered prototype from Lactobacillus. In the Lactobacillus sequence it is more difficult to recognize the structure of the specificity site, mainly because of the large distance existing between the cysteine residues homologous to C225 and C439. In DR where this distance also is large, one can recognize by comparison with the TM sequence a fragment that may have been inserted more recently during evolution. One can speculate that an intein originally was present in this position, as is the case in the Pyrococcus enzyme (Fig. 4).
Acknowledgments
We acknowledge TIGR for availability of sequence data before publication, and K. W. Minton, R. Sirevag, and J. Shiozawa for providing us with D. radiodurans and C. aurantiacus strains. A.J. was supported by fellowships from Margit and Folke Pehrzon Foundation and Axel Wenner-Grens Stiftelse, E.T. by a predoctoral fellowship from the Direccio General d’Universitats de la Generalitat de Catalunya and the European Molecular Biology Organization, I.G. and J.B. by grants from the Spanish Dirección General de Investigación Cientifica y Técnica and from the Comissionat per Universitats in Recerca de la Generalitat de Catalunya, P.R. by a grant from the Swedish Medical Research Council, and C.J. by grants from Centre National de la Recherche Scientifique and Fonds Structurel Européen (FEDER 5b).
ABBREVIATIONS
- TM
Thermotoga maritima
- DR
Deinococcus radiodurans
- TIGR
The Institute for Genomic Research
Footnotes
Data deposition: The sequence reported in this paper has been deposited in the GenBank database (accession no. Y12877).
References
- 1.Reichard P. Science. 1993;260:8383–8386. doi: 10.1126/science.8511586. [DOI] [PubMed] [Google Scholar]
- 2.Reichard P. Trends Biochem Sci. 1997;22:81–85. doi: 10.1016/s0968-0004(97)01003-7. [DOI] [PubMed] [Google Scholar]
- 3.Benner S A, Ellington A D, Tauer A. Proc Natl Acad Sci USA. 1989;86:7054–7058. doi: 10.1073/pnas.86.18.7054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stubbe J, van der Donk W A. Chem Biol. 1995;2:793–801. doi: 10.1016/1074-5521(95)90084-5. [DOI] [PubMed] [Google Scholar]
- 5.Sjöberg B-M. Struct Bonding. 1997;88:139–173. [Google Scholar]
- 6.Panagou D, Orr M D, Dunstone J R, Blakley R L. Biochemistry. 1972;11:2378–2388. doi: 10.1021/bi00762a025. [DOI] [PubMed] [Google Scholar]
- 7.Booker S, Stubbe J. Proc Natl Acad Sci USA. 1993;90:8352–8356. doi: 10.1073/pnas.90.18.8352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Riera J, Robb F T, Weiss R, Fontecave M. Proc Natl Acad Sci USA. 1997;94:475–478. doi: 10.1073/pnas.94.2.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tauer A, Benner S A. Proc Natl Acad Sci USA. 1997;94:53–58. doi: 10.1073/pnas.94.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Woese C R. Microbiol Rev. 1987;51:221–271. doi: 10.1128/mr.51.2.221-271.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stetter K O. FEMS Microbiol Rev. 1996;18:149–158. doi: 10.1111/j.1574-6976.2000.tb00562.x. [DOI] [PubMed] [Google Scholar]
- 12.Eriksson M, Uhlin U, Ramaswamy S, Ekberg M, Regnström K, Sjöberg B-M, Eklund H. Structure. 1997;5:1077–1092. doi: 10.1016/s0969-2126(97)00259-1. [DOI] [PubMed] [Google Scholar]
- 13.Ravot G, Ollivier B, Magot M, Patel B C, Crolet J-L, Fardeau M-L, Garcia J-L. Appl Environ Microbiol. 1995;61:2053–2055. doi: 10.1128/aem.61.5.2053-2055.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carrol J D, Daly M J, Minton K W. J Bacteriol. 1996;178:130–135. doi: 10.1128/jb.178.1.130-135.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pierson B K, Castenholz R W. In: The Prokaryotes: A Handbook on the Biology of Bacteria. Barlows A, Truper H G, Dworkin M, Harder W, Schleifer K-H, editors. Berlin: Springer; 1991. pp. 3754–3774. [Google Scholar]
- 16.Thelander L, Sjöberg B-M, Eriksson S. Methods Enzymol. 1978;51:227–237. doi: 10.1016/s0076-6879(78)51032-x. [DOI] [PubMed] [Google Scholar]
- 17.Hellman U. In: Protein Structure Analysis: Preparation, Characterization, and Microsequencing. Kamp R M, Choli-Papadopoulou T, Wittmann-Liebold B, editors. Berlin: Springer; 1997. pp. 97–104. [Google Scholar]
- 18.Kim C W, Markiewicz P, Lee J J, Schierle C F, Miller J H. J Mol Biol. 1993;231:960–981. doi: 10.1006/jmbi.1993.1345. [DOI] [PubMed] [Google Scholar]
- 19.Jeanthon C, Reysenbach A-L, L’Haridon S, Gambacorta A, Pace N, Prieur D. Arch Microbiol. 1995;164:91–97. [PubMed] [Google Scholar]
- 20.Shyamala V, Ames G F-L. Methods Mol Biol. 1993;15:339–348. doi: 10.1385/0-89603-244-2:339. [DOI] [PubMed] [Google Scholar]
- 21.Achenbach-Richter L, Gupta R, Stetter K O, Woese C R. Syst Appl Microbiol. 1987;9:34–39. doi: 10.1016/s0723-2020(87)80053-x. [DOI] [PubMed] [Google Scholar]
- 22.Bult C J, White O, Olsen G J, Zhou L, Fleischmann R D, et al. Science. 1996;273:1058–1073. doi: 10.1126/science.273.5278.1058. [DOI] [PubMed] [Google Scholar]
- 23.Kaneko T, Sato S, Kotani H, Tanaka A, Asamizu E, et al. DNA Res. 1996;3:109–136. doi: 10.1093/dnares/3.3.109. [DOI] [PubMed] [Google Scholar]
- 24.Thompson J D, Higgins D G, Gibson T J. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huber R, Langworthy T A, Konig H, Thomm M, Woese C R, Sleytr U B, Stetter K O. Arch Microbiol. 1986;144:324–333. [Google Scholar]
- 26.Nilsson O, Åberg A, Lundqvist T, Sjöberg B-M. Nucleic Acids Res. 1988;16:4174. doi: 10.1093/nar/16.9.4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hatfull G F, Sarkis G J. Mol Microbiol. 1993;7:395–405. doi: 10.1111/j.1365-2958.1993.tb01131.x. [DOI] [PubMed] [Google Scholar]
- 28.Yang F, Lu G, Rubin H. J Bacteriol. 1994;176:6738–6743. doi: 10.1128/jb.176.21.6738-6743.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]