

Abstract
Muscular dystrophy refers to a group of genetic diseases that cause severe muscle weakness and loss of skeletal muscle mass. Although research has helped understanding the molecular basis of muscular dystrophy, there is still no cure for this devastating disorder. Numerous lines of investigation suggest that the primary deficiency of specific proteins causes aberrant activation of several cell signaling pathways in skeletal and cardiac muscle leading to the pathogenesis of muscular dystrophy. Studies using genetic mouse models and pharmacological approaches have provided strong evidence that the modulation of the activity of specific cell signaling pathways has enormous potential to improving the quality of life and extending the life expectancy in muscular dystrophy patients. In this article, we have outlined the current understanding regarding the role of different cell signaling pathways in disease progression with particular reference to different models of muscular dystrophy and the development of therapy.
Keywords: Muscular dystrophy, Signaling, NF-κB, MAPK, Akt, calcineurin/NFAT
1. Introduction
Muscular dystrophies are devastating genetic disorders which cause progressive degeneration of skeletal muscle fibers leading to severe pain, disability, and eventually death (1). The primary cause for various forms of muscular dystrophies is the mutations in individual genes that encode a wide variety of proteins, including extracellular matrix proteins, transmembrane and membrane-associated proteins, cytoplasmic enzymes, and nuclear matrix proteins (2, 3).
Duchenne muscular dystrophy (DMD) is one of the most prevalent forms of muscular dystrophies afflicting 1 out of every 3500 male births (1). DMD results due to total or partial deficiency of functional dystrophin protein, a component of dystrophin-glycoprotein complex (DGC), which links the cytoskeleton of the muscle fibers to the extracellular matrix (4). In the absence of dystrophin, the DGC is functionally impaired, and the mechanical stress associated with contraction leads to the degeneration of skeletal muscle fibers, muscle-wasting, impairment of movements, and ultimately death of the affected boys due to respiratory and/or cardiac failure (5, 6). It is now established that the primary deficiency of dystrophin in skeletal and cardiac muscle results in the activation of several secondary processes including inflammation, extracellular matrix degradation, chronic degeneration and regeneration of fibers, and interstitial fibrosis, which exacerbate disease progression in DMD (5, 7–10).
Accumulating evidence suggests that besides acting as a molecular scaffold serving mechanical function, DGC also has an important signaling role in cardiac and skeletal muscle (5). Loss of dystrophin or other components of DGC causes significant mechanical instability of the sarcolemma leading to the activation of different intracellular signaling pathways in skeletal muscle (11, 12). Interestingly, abnormal myogenic signaling also occurs in other forms of muscular dystrophies that result due to loss of nuclear membrane protein (e.g. lamins A/C, Emerin) or cytoplasmic enzymes (e.g. calpain-3) suggesting that signaling defects may be common to all types of muscular dystrophies. Because the activation of various cell signaling pathways results in altered gene expression, aberrant activation of one or more pathways could be critical to the onset and perpetuation of pathogenesis in muscular dystrophy. In this article, we have discussed the most direct emerging evidence relating atypical myogenic signaling to the disease progression and the prospect of some of these pathways to be used as a drug target in muscular dystrophy.
2. DGC: A major signalosome in skeletal muscle
DGC present on the sarcolemma is multimeric structure composed of transmembrane (e.g. α, β, γ, and δ-sarcoglycans, β-dystroglycan, caveolin-3, and sarcospan), cytoplasmic (dystrophin, nNOS, α1 and β1-syntrophins, α-dystrobrevin), and extracellular proteins (e.g. α-dystroglycan and laminin) (Figure 1). Although none of the components of DGC has intrinsic kinase or phosphatase activities, they promote cellular signaling by providing an important link between extracellular signals to the intracellular signaling pathways. The signaling role of DGC is supported by the observations that calmodulin, a calcium-binding protein and a major transducer of calcium signals, binds to both dystrophin and syntrophin and calmodulin (CaM)-regulated activities are reduced in dystrophin-deficient myofibers (13, 14). CaM-dependent protein kinase II (CaMKII) which interacts and phosphorylates several DGC proteins not only regulate protein-protein interactions among DGC proteins but may also affect the activation of several downstream signal transduction pathways such as phosphatidylinositide-3 kinase (PI3K)/Akt which are involved in the regulation of muscle cell survival (5). Disruption in the interaction of extracellular matrix and cytoskeletal protein α-dystroglycan results in skeletal muscle cell death through the inhibition of PI3K/Akt pathway and the activation of caspases (15). In addition to CaM and CaMKII, many other signaling molecules including stress-activated protein kinase-3 (16), neuronal nitric oxide synthase (nNOS) (17), voltage-gated sodium channels (18), and phosphatidylinositol 4,5-bisphosphate (19) interact with DGC proteins (Figure 1).
FIGURE 1. Interactions of different signaling proteins with dystrophin-glycoprotein complex (DGC).
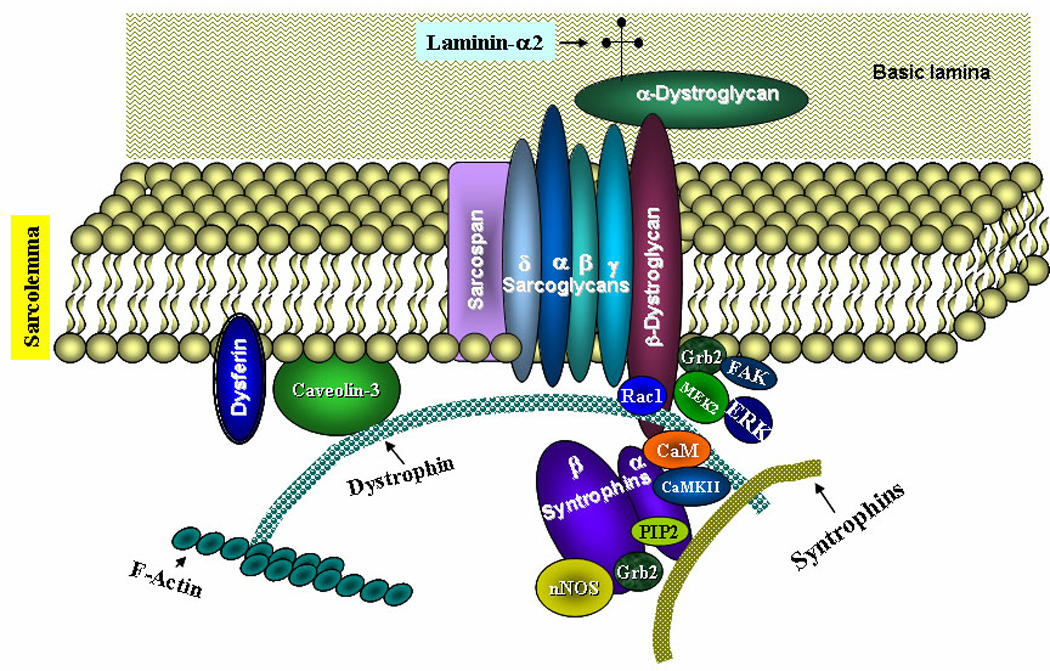
Diagrammatic representation of the DGC proteins and their physical interaction with different signaling proteins is shown. CaM, calmodulin; CaMKII, CaM kinase II; FAK, focal adhesion kinase, MEK2, mitogen-activated protein kinase kinase 2; nNOS, neuronal nitric oxide synthase; PIP2, phosphatidylinositol 4,5-bisphosphate.
Signal transducer and adaptor protein Grb2 plays a critical role in receptor tyrosine kinases and Ras signaling leading to the activation of MAPK. Grb2 interacts with proline-rich region within β-dystroglycan and mediates the interaction of β-dystroglycan from brain synaptosomes with focal adhesion kinase (FAK), a non-receptor tyrosine kinase, involved in cell signaling especially in mechanosensing and skeletal muscle contraction (20). Grb2 also physically associate with alpha1-syntrophin (21). Similarly, MAPK kinase 2 (MEK2) and extracellular signal-regulated kinase (ERK) have been found to interact with β-dystroglycan in mammalian cells suggesting that DGC protein β-dystroglycan is a multifunctional adaptor protein capable of interacting with multiple proteins of the mitogen-activated protein kinase (MAPK) cascade (22). Furthermore, it has been found that laminin-binding causes syntrophin to recruit Rac1, a Rho family small G-protein, in rabbit skeletal muscle providing additional evidence that DGC is an important signalosome in skeletal muscle (23).
Besides having a direct role in myogenic signaling, the disruption of DGC may lead to biophysical changes and activation of various ion channels resulting in disruption of physiological signaling. Indeed, dysregulation of stretch-activated ion channels (SAC) has been suggested as one of the important pathways leading to myofiber damage in DMD (24). It is also noteworthy that α1-syntrophin interacts with TRPC1 (transient receptor potential canonical 1) to form SAC further signifying the potential role of DGC in the regulation of SAC in skeletal muscle (25). Increased activity of SAC causes the influx of Ca2+, which might be critical for the activation of various intracellular signaling pathways in dystrophic muscle (12).
Although the pathogenesis of muscular dystrophy might involve coordinated activation of multiple cell signaling pathways, there has been strong evidence about the perturbation of nuclear factor-kappa B (NF-κB), MAPK, phosphatidylinositol 3-kinase (PI3K)/Akt, and calcineurin/NFAT signaling pathways in various types of muscular dystrophies (Figure 2). In the following sections, we have provided a succinct review of literature how the activation of some of these signaling pathways is regulated and their emerging role in the disease progression in muscular dystrophies.
FIGURE 2. Myogenic signaling in pathogenesis of muscular dystrophy.
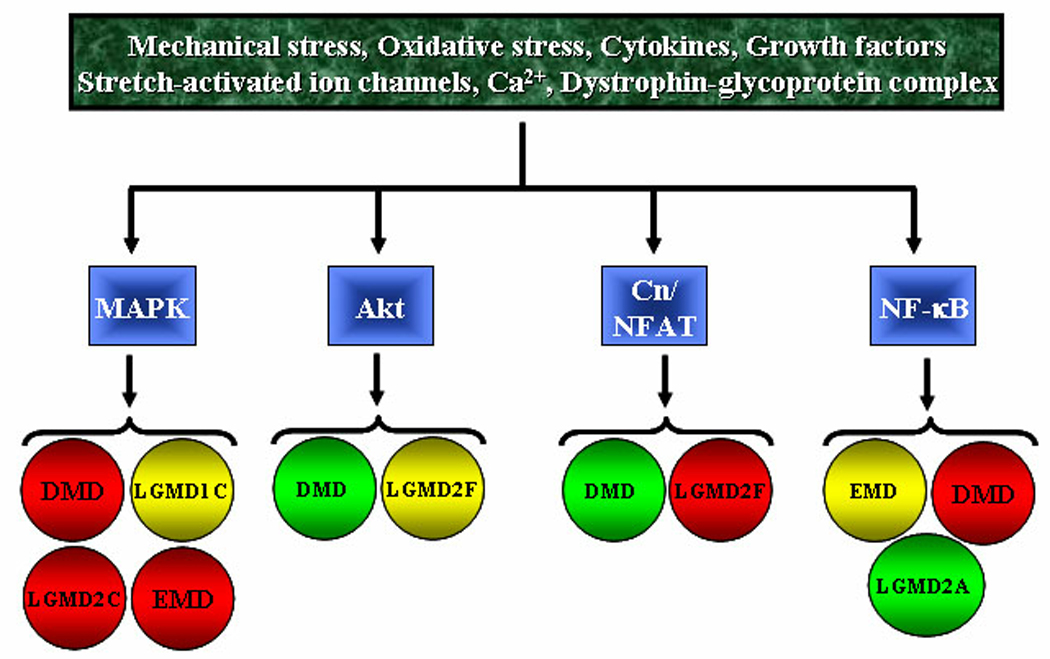
A number of intrinsic and/or extrinsic stimuli lead to the activation of specific cell signaling pathways in skeletal muscle in muscular dystrophy. Depending on the type of muscular dystrophy, activation of a specific pathway may ameliorate or exacerbate disease progression. Red: exaggeration in pathogenesis; green: amelioration of pathogenesis; yellow: role not yet known. Cn, Calcineurin; DMD, Duchenne muscular dystrophy; EMD, Emery-Dreifuss muscular dystrophy; LGMD, Limb girdle muscular dystrophy.
3. NF-κB in Muscular Dystrophy
NF-κB is a major nuclear transcription factor which regulates the expression of a plethora of genes involved in diverse biological responses including the development of immune system, inflammation, acute stress responses, cellular proliferation, and protection against cell death (26, 27). NF-κB family consists of five members: p50, p52, p65 (RelA), c-Rel, and RelB, which form various homo- and heterodimers. Most of the NF-κB dimers are retained in the cytoplasm by binding to specific inhibitors of NF-κB (i.e. IκBs). The interaction with IκBs masks the nuclear localization sequence in the NF-κB complex, preventing its activation and nuclear translocation thus maintaining it in an inactive state in the cytoplasmic compartment of the cell (26). NF-κB can be activated through either canonical or alternative signaling pathways. The canonical pathway involves the upstream activation of inhibitors of kappa B (IκB) kinase-β (IKKβ) and subsequent phosphorylation and degradation of IκB proteins (28). In contrast, the activation of NF-κB alternative pathway requires the upstream activation of NF-κB-inducing kinase and IKKα and the proteolytic processing of p100 subunit into p52 (28). Once activated, NF-κB translocates to the nucleus where it binds to the promoter/enhancer region of the genes having consensus NF-κB-binding sequences.
Because inflammation is a prominent pathological feature in muscular dystrophy (29–33) and NF-κB is a major proinflammatory transcription factor (27), in an earlier study, we investigated how the activation of NF-κB is regulated in skeletal muscle of mdx mice (a mouse model of DMD). The activation of NF-κB and the expression of NF-κB-regulated muscle catabolic proinflammatory cytokines TNF-α and IL-1β were found to be drastically up-regulated in the skeletal muscle of mdx mice (11, 34). Interestingly, myofiber-specific activation of NF-κB precedes the onset of muscle degeneration in mdx mice (11). Furthermore, we found that the application of mechanical stretch dramatically increased the activation of NF-κB in diaphragm of mdx mice suggesting that mechanical stress could be an important trigger for NF-κB activation in dystrophic muscle (11). Elevated levels of activated NF-κB have now been reported in skeletal muscle of different animal models of DMD (35–39) and Emery-Dreifuss muscular dystrophy (40). Furthermore, increased activation of NF-κB has been observed in muscle biopsies of DMD patients (29, 36, 41) and in inflammatory myopathies (42, 43).
Although NF-κB is also required for cell survival, in adults, NF-κB is a negative regulator of skeletal muscle mass (44). Activation of NF-κB causes the loss of skeletal muscle mass through at least three mechanisms: (a) NF-κB augments the expression of several proteins involved in the ubiquitin-proteasome system including E3 ubiquitin ligase MuRF1, which stimulates the loss of skeletal muscle mass in several unrelated conditions; (b) NF-κB also increases the expression of many proinflammatory cytokines, chemokines, cell adhesion molecules, and matrix-degrading enzymes which exacerbate skeletal muscle loss; (c) Finally activated NF-κB can also block the regeneration of myofibers in response to injury (44).
Direct evidence regarding the role of NF-κB in skeletal muscle homeostasis came from two recent studies employing genetic mouse models (45, 46). Skeletal muscle-restricted inhibition of NF-κB (through depletion of IKKβ) improved force production in normal muscle (45). In denervation model of muscle atrophy, inhibition of NF-κB maintained fiber type, preserved fiber size and strength, increased protein synthesis, and reduced protein degradation (45). Inhibition of NF-κB also augmented skeletal muscle regeneration through stimulating the satellite cell proliferation (45). Similarly, skeletal-muscle restricted overexpression of a degradation resistant mutant of IκBα protein (an inhibitor of NF-κB) has been found to rescue the loss of skeletal muscle mass in settings of denervation and tumor loads (46).
Even though the role of ubiquitin-proteasome system (especially MuRF1) in muscular dystrophy is less clear, there is significant amount of literature suggesting that the expression of several proinflammatory molecules is drastically increased in dystrophic muscle of animal models and in muscle biopsies of muscular dystrophy patients (29–33). Importantly, many of the inflammatory molecules expressed in dystrophic muscle contain consensus NF-κB binding sequence in their promoter/enhancer regions (47). In future studies, it will be of interest to know which genes NF-κB regulates in muscular dystrophy. Since NF-κB also interferes with skeletal muscle regeneration, activation of NF-κB may also be an important cause for insufficient myofiber regeneration commonly observed in muscular dystrophy (47).
3.1. Targeting NF-κB in DMD
The role of NF-κB in skeletal muscle pathogenesis of DMD has now been elucidated using genetic and pharmacological approaches (Figure 3). Deletion of single allele of NF-κB subunit p65 (RelA) was sufficient to considerably reduce the infiltration of macrophages, necrosis, and calcification in dystrophic muscle of mdx mice (36). Interestingly, inhibition of NF-κB also augmented the regeneration of myofibers in mdx mice (36). These observations are consistent with other published reports demonstrating that activated NF-κB inhibits the differentiation of muscle progenitor cells into multinucleated myotubes (44, 48, 49). Similar results were obtained when NF-κB was inhibited through intraperitoneal injection of NF-κB inhibitory peptide NBD (NF-κB Essential MOdulator (NEMO) Binding Domain) in mdx mice (36), which further supports the inference that the activation of NF-κB contributes to dystrophic phenotypes.
FIGURE 3. Schematic representation of the evidences about therapeutic targeting of signaling proteins in different types of muscular dystrophies.
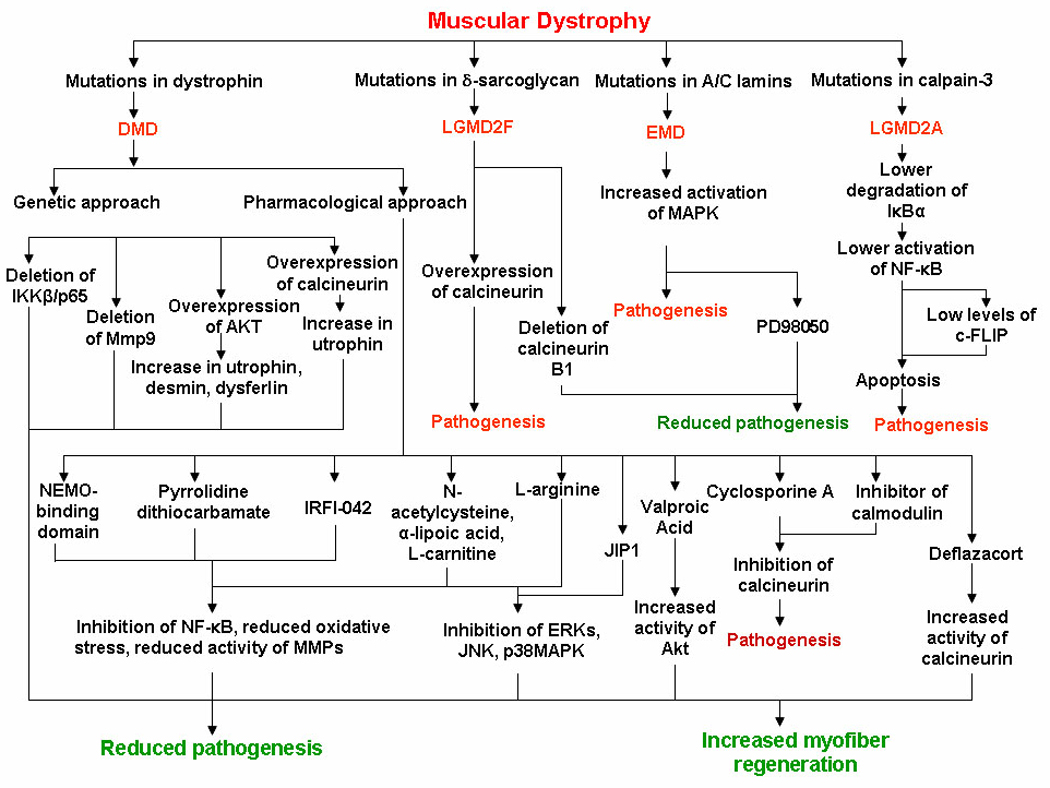
DMD, Duchenne muscular dystrophy; EMD, Emery-Dreifuss muscular dystrophy; LGMD, Limb girdle muscular dystrophy. JIP1, JNK inhibitory protein 1.
Although the exact mechanisms which lead to the activation of NF-κB in DMD remain unknown, several recent studies have indicated that the increased oxidative stress could be one of the potential reasons for the activation of NF-κB in dystrophic muscles (Figure 3). Carlson et al. evaluated the effect of chronic administration of pyrrolidine dithiocarbamate (PDTC), a potent antioxidant and inhibitor of NF-κB in mdx mice (50). PDTC increased skeletal muscle mass and functions (50). Similar beneficial effects of PDTC were also reported by Messina et al (51) where the authors found that PDTC treatment considerably reduces myofiber necrosis and improves muscle strength in mdx mice (51). IRFI-042, a potent antioxidant and inhibitor of lipid peroxidation, has also been found to rescue muscle pathology, augment myofiber regeneration, and reduce muscle fatigue in mdx mice (38). IRFI-042 blocked the activation of NF-κB and the expression of TNF-α indicating that oxidative stress and the consequent activation of NF-κB regulate the pathogenesis in myofibers of mdx mice (38).
The role of oxidative stress and NF-κB in skeletal muscle pathogenesis was also investigated using N-acetylcysteine (NAC), a free-radical scavenger (52). Treatment of mdx mice with NAC in drinking water inhibited NF-κB and improved sarcolemmal integrity and muscle functions (52). On the similar lines, it was also recently reported that free radical scavengers such as α-lipoic acid (ALA) and l-carnitine (L-Car) attenuate the pathogenesis by blocking the activation of NF-κB and the expression of matrix metalloproteinase (MMP)−2 and −9 in mdx mice (53).
Whereas the above studies clearly suggest that the activation of NF-κB deteriorates skeletal muscle pathology in mdx mice, the mechanisms by which activated NF-κB induces pathogenesis is not clearly known. We recently reported that one of the important mechanisms by which NF-κB exacerbates dystrophinopathy is through augmenting the expression of MMP-9 (54). MMP-9 is one of the major extracellular proteases, the increased expression of which causes inflammation, fibrosis, and remodeling of extracellular matrix (55). Interestingly, MMP-9 regulates its own expression through a positive feed-back mechanism that involves the activation of NF-κB in dystrophic muscle (54). The collaborative interaction between NF-κB and MMP-9 in mdx pathology is also supported by several parallel observations relating MMP-9 to NF-κB: (a) NF-κB activation and expression of MMP-9 follow similar pattern in skeletal muscle of mdx mice. The activation of NF-κB and levels of MMP-9 protein remain elevated after 2 weeks (11, 36, 54); (b) Genetic or pharmacological inhibition of either NF-κB or MMP-9 improved sarcolemmal integrity and attenuated inflammation (36, 54); (c) Inhibition of either NF-κB or MMP-9 augments skeletal muscle regeneration in mdx mice (36, 51, 54).
Though NF-κB seems to play a central role in muscle pathogenesis in DMD, it is unlikely that the mere activation of NF-κB is sufficient for transcriptional activation or induction of any single NF-κB target gene that is involved in the perpetuation of inflammatory responses. For most promoters, NF-κB requires assistance from other sequence specific transcription factors (44, 47) such as members of activator protein (AP)-1 family whose activity in turn is dependent on their phosphorylation by MAPK (56).
3.2 Targeting NF-κB in LGMD2A
While the inhibition of NF-κB seems to be a promising approach for treatment of DMD, it is important to recognize that blocking NF-κB might not be effective in all types of muscular dystrophies. It is possible that a certain level of NF-κB activity is required to protect the myofibers from undergoing apoptosis whereas constitutive activation of NF-κB leads to inflammation and other dystrophic features. LGMD2A is a recessive genetic disorder caused by mutations in the cysteine protease calpain-3 (1). Loss of functional calpain-3 leads to apoptosis in skeletal muscle of LGMD2A patients and in calpain-3 knockout mice (1). Calpain-3 is required for the degradation of IκBα protein and hence the activation of NF-κB and suppression of apoptosis in skeletal muscle (57, 58). The pro-survival function of NF-κB in muscular dystrophy is not very startling because NF-κB is also known to induce the expression of several anti-apoptotic genes (28, 47). It was recently reported that the expression of cellular-FLICE inhibitory protein (c-FLIP), a NF-κB-regulated cell survival protein, is down-regulated in LGMD2A muscle biopsies (59) (Figure 3). Though the reduced activity of NF-κB appears to be a reason for the pathogenesis of LGMD2A, it remains to be investigated whether specific activation/expression of NF-κB can rescue skeletal muscle pathogenesis in LGMD2A patients.
4. MAPK and AP-1 in Muscular Dystrophy
The MAPK constitute a family of serine/threonine kinases which mediate the transduction of external stimuli into intracellular signals that regulate cellular responses including proliferation, differentiation, self-renewal, and survival. In mammalian cells, three parallel MAPK pathways have been described: extracellular signal-related kinase (ERK1/2), c-Jun-N-terminal kinases (JNKs), and p38 MAPK (60). Activation of MAPKs requires phosphorylation on both a threonine and tyrosine residue and thus need the activity of dual specificity kinases, which are known as MAPK kinases (MEKs). MAPK acts via regulating the activity of many downstream transcription factors, including AP-1, c-myc, Elk1, and CCAAT/enhancer binding protein-β (60).
To understand the biochemical mechanisms responsible for muscle pathogenesis in muscular dystrophy, in a previous study, we investigated the activation of MAPK in skeletal muscle of mdx mice (12). To exclude the possible role of secondary mediators that may compound the activation of MAPK during active muscle degeneration, we employed diaphragm muscle from pre-necrotic mdx mice. Interestingly, the activation of ERK1/2 was noticeably up-regulated in diaphragm of prenecrotic mdx mice compared to age-matched normal mice. Although the activation of ERK1/2 was also increased in diaphragm of normal mice, the effect of mechanical stretch on the activation of ERK1/2 was dramatic in diaphragm of mdx mice (12). The activation of ERK1/2 involved the influx of Ca2+ ions through stretch-activated ion channels (12). Furthermore, we found that the basal level and mechanical stretch-induced activation of AP-1 were considerably higher in diaphragm of mdx mice signifying the potential role of physical stress and MAPK/AP-1 signaling in dystrophinopathy (12). Similar to our findings, there are also other reports demonstrating increased activation of ERK1/2, JNK1 and p38 MAPK in skeletal and cardiac muscle in animal models of DMD (37, 61–63).
Deficiency of DGC protein γ-sarcoglycan (γSG) leads to a particular form of muscular dystrophy referred to as limb-girdle muscular dystrophy 2C (LGMD2C) (1). Griffin et al have shown that myotubes from γSG-deficient mice show increased apoptosis with concomitant activation of ERK1/2 compared to the myotubes prepared from wild-type mice (64). The activation of ERK1/2 was further increased upon application of mechanical stretch (64). This phenomenon is quite similar to what we observed with pre-necrotic mdx mice (12). Because γSG is an important component of DGC, it is likely that the disruption of DGC, due to deficiency of any of its components, is sufficient for the hyper activation of MAPK cascades in skeletal muscle.
The potential role of MAPK in pathogenesis of muscular dystrophy has also been highlighted in caveolin-3 knockout mice. Caveolin-3 interacts with dystrophin but it is not an integral component of the DGC. Skeletal muscle fibers from caveolin-3 knockout mice show a number of myopathic changes, consistent with a mild-to-moderate muscular dystrophic phenotype (65). Studies conducted on caveolin-3 knockout mice have revealed that MAPK are highly activated in their cardiac muscle (65).
4.1. Targeting MAPK in DMD
The potential role of MAPK has been partially investigated in DMD. While we did not find any significant difference in the level of activation of JNK1 in skeletal muscle of pre-necrotic (2.5 week) mdx mice (12), Kolodziejczyk et al have reported that JNK1 is highly activated in skeletal and cardiac muscle of 5- and 12-months old mdx and mdx:MyoD−/− mice (61). Interestingly, intramuscular injection of an adenoviral construct expressing the JNK1 inhibitory protein, JIP1, attenuated myofiber destruction providing initial evidence that the activation of JNK1 contributes to muscle pathogenesis in mdx mice (61). Since ERK1/2 are activated early (12) and the activation of JNK1 was more pronounced in older mdx mice (61), these observations suggest that specific MAPK are activated at different stages of disease progression and the coordinated activation of different MAPKs may be responsible for the onset and/or perpetuation of dystrophic phenotypes in DMD.
Accumulating evidence suggests that the cell death in muscular dystrophies may be caused due to free radicals-mediated injury (66–68). In addition to NF-κB, oxidative stress is also considered as one of the potent activators of different MAPK in mammalian cells and tissues (69). It was recently reported that free-radical scavenger α-lipoic acid (ALA) and l-carnitine (l-Car) restores antioxidant enzymes (superoxide dismutase, catalase, and glutathione peroxidase) activity, reduces lipid peroxidation (thiobarbituric acid reactive substances) and serum creatine kinase levels, and improves the dystrophic pattern in mdx diaphragm muscle (53). More importantly, ALA/l-Car also significantly blocked the activation of ERK1/2, JNK1, and p38MAPK in skeletal muscle of mdx mice. Furthermore, the levels of β-dystroglycan, a major DGC protein capable of interacting with components of the ERK-MAPK cascade (22), were also found to be increased after treatment with ALA/L-Car (53) implicating oxidative stress and cell signaling defects in dystrophin-deficient muscle via the β-dystroglycan-MAPK interaction and that the interruption of oxidative stress and the inflammatory cascade might have therapeutic potential in DMD.
4.2. Targeting MAPK in Emery-Dreifuss muscular dystrophy (EMD)
Mutations in genes encoding inner nuclear membrane protein A-type lamins (LMNA) and emerin cause cardiomyopathy and muscular dystrophy (1). Lack of A-type lamins (Lamin A/C) leads to increased nuclear fragility and eventual nuclear disruption in tissues subjected to mechanical strain. Although the causative genetic mutations have now been identified, cellular mechanisms linking mutations in LMNA gene to cardiomyopathy are unknown. It has been proposed that mutations in A-type lamins lead to anomalous tissue-specific gene regulation which is based on the findings that A-type lamins and emerin and associated proteins bind to chromatin and transcriptional regulators. Increased activation of MAPK has also been observed in LMNA-null mice (70) indicating that spurious activation of MAPK by mutant A-type lamins could be a major reason for the development of cardiomyopathy in autosomal dominant EMD. A recent study employing array approach has demonstrated that the expression and phosphorylation levels of a number of signaling proteins including ERK1/2, JNK1, and Elk1 are considerably increased in heart and cultured cardiomyocytes of LMNA-deficient mice compared to wild-type mice (71). More importantly, pharmacological inhibition of ERK1/2 prevented cardiomyopathy (72) further suggesting that MAPK are important target for the pharmacological treatment of conditions which result due to mutations in the genes encoding emerin and A-type lamins (Figure 2 and Figure 3).
5. PI3K/Akt in Muscular Dystrophy
The phosphatidylinositol 3-kinase (PI3K)/Akt is an important signaling pathway that is involved in regulation of cell viability and protein synthesis (73, 74). Akt, the central kinase of PI3K/Akt, is a serine/threonine kinase that belongs to the AMP-dependant protein kinase A/ protein kinase G/ protein kinase C (AGC) super family of protein kinases (75). Akt is activated in response to a wide variety of stimuli via mechanisms involving PI3K and phosphoinositide-dependent kinase-1. Activation of Akt causes the phosphorylation of cytoplasmic and nuclear target proteins, notably glycogen synthase kinase-3β, p27Kip, mammalian target of rapamycin (mTOR), p70S6 kinase, and forkhead transcription factors (73, 75).
Activation of Akt causes muscle hypertrophy in vivo by stimulating protein synthesis via the kinase mTOR (reviewed in (76)). Akt also blocks protein degradation by inhibiting Foxo family transcription factors, which regulate ubiquitin-proteasome pathway (76). Activation of Akt has been consistently observed in animal models of muscular dystrophies though it appears to be insufficient to prevent muscle degeneration. Barton et al provided initial evidence that the Akt activation is significantly increased in mdx mice upon expression of insulin growth factor-1 (IGF-1) in skeletal muscle (77). Consistent with the established role of IGF-1 in activation of Akt pathway, transgenic overexpression of IGF-1 resulted in hyperplasia and hypertrophy in mdx mice (77). We also noticed increased levels of total and phosphorylated Akt in skeletal muscle of mdx mice at both pre-necrotic and necrotic stages (34). Similarly, Peter and Crosbie demonstrated that the activation of Akt is increased at very early stages of disease progression, and the maximal activation occurs during peak stages of muscle hypertrophy though the members of Akt signaling pathway do not directly interact with DGC proteins (78). Furthermore, like mdx mice, the increased activation of Akt is also observed in skeletal muscle of δ-sarcoglycan-deficient mice (a mouse model of LGMD2F) (78).
5.1. Targeting PI3K/Akt in DMD
The role of Akt in pathogenesis of mdx mice has been recently investigated. Peter et al reported an increase in skeletal muscle regeneration, muscle hypertrophy, and decreased sarcolemmal fragility in transgenic mdx mice expressing Akt compared to control mdx mice suggesting that activation of Akt can attenuate disease progression in DMD (79). Akt increased the expression of utrophin, a homologue of dystrophin, which partially compensates for dystrophin-deficiency in mdx mice (80). Their findings suggest that the overexpression and extra-synaptic localization of the utrophin-glycoprotein complex and integrin compensate for the disruption of DGC in Akt-transgenic mdx mice (80).
Using similar inducible muscle-specific Akt transgenic model, Blaauw et al investigated whether Akt activation can compensate for the loss in force production induced by lengthening contractions in mdx skeletal muscle. Interestingly, the force drop in whole muscles and also in skinned fibers isolated from mdx muscles exposed to eccentric contractions in vivo were significantly prevented by the expression of activated Akt in skeletal muscle (81). Expression of Akt induced hypertrophy and increased the protein levels of utrophin, desmin, and dysferlin (81). Furthermore, the transcript levels of several genes associated with Z-disks, costameres, anti-oxidant, and chaperone function were also elevated upon expression of activated Akt kinase (81).
Similar to transgenic approaches, a recent study by Gurpur et al showed that valproic acid (VPA) induces skeletal muscle hypertrophy and inhibits apoptosis in cultured myotubes by activating the Akt/mTOR/p70S6K pathway (82). Daily intraperitoneal injection of VPA in mdx/utrophin double knockout (dko) mice inhibited inflammation and fibrosis in dystrophic muscle, improved sarcolemmal structure, and ameliorated hind limb contractures (Figure 3). The improved muscle pathology was associated with increased levels of phosphorylated Akt in skeletal muscle of VPA-treated dko mice (82). These finding suggests that the activation of Akt improves skeletal muscle pathology and stimulation of Akt can be an important approach to enhancing sarcolemmal integrity and muscle strength in DMD.
6. Calcineurin (Cn)/NFAT in Muscular dystrophy
Nuclear factor of activated T-cells (NFAT) is a multigene family of an inducible nuclear transcription factor containing five members: NFATc1, NFATc2, NFATc3, NFATc4, and NFAT5. NFAT family members (except NFAT5) are regulated by the calcium-activated protein phosphatase calcineurin (83). Phosphorylation of serine residues on NFAT results in intramolecular masking of its nuclear localization sequence resulting in cytoplasmic sequestration of the transcription factor (83). In response to signals that increase intracellular calcium, NFAT is dephosphorylated by the calcium- and calmodulin-dependent phosphatase calcineurin, resulting in nuclear import of the transcription factor. When calcium signaling is terminated, NFAT can be rephosphorylated by several protein kinases such as glycogen synthase kinase-3, protein kinase A, casein kinase 1 (CK1), p38 MAP kinase (p38), and c-Jun NH2-terminal kinase (JNK), which facilitate its nuclear export (83, 84). Calcineurin/NFAT activation is important for skeletal muscle differentiation, fiber type specialization, and hypertrophy (85, 86).
Because calmodulin (CaM) and calcium/calmodulin-dependent protein kinase II (CaMKII) interact with DGC and these molecules regulate the activation of calcineurin/NFAT pathway, it is conceivable that this pathway may be regulated in DGC-related muscular dystrophy. Indeed, there are reports demonstrating increased activation of calcineurin signal transduction pathway in skeletal muscle of mdx mice (87–89). Nakamura et al studied the activation of calcineurin/NFAT pathway in cardiac muscle of mdx/utrophin double knockout (dko) mice (63). The expression of calcineurin was found to be dramatically increased in dko mice. Although the levels of calcineurin in mdx mice were not different from control mice, physical exercise increased the activation of calcineurin in hearts of mdx but not control mice (62) indicating that the increased expression and activation of calcineurin could possibly be related to hypertrophy and dilated cardiomyopathy in these mice.
6.1. Targeting Cn/NFAT in DMD
The activation of calcineurin is important for successful regeneration of limb muscles of mdx mice because the inhibition of this pathway using cyclosporine A exaggerates skeletal muscle degeneration, impairs regeneration, and reduces skeletal muscle force-producing capacity (87). The most important target of calcineurin/NFAT pathway in skeletal muscle could be the cytoskeleton protein utrophin, which compensates for dystrophin-deficiency in skeletal muscle (Figure 3). Transgenic mice over-expressing calcineurin display elevated levels of utrophin around their sarcolemma (90). Furthermore, transgenic expression of calcineurin in skeletal muscle resulted in increased nuclear localization of NFATc1, fiber type shifting towards a slower phenotype, increased levels of utrophin, and improved sarcolemmal integrity (91). These findings are also supported by a recent report demonstrating transgenic overexpression of a small peptide inhibitor for calmodulin, an upstream activator of calcineurin, represses the levels of utrophin, and exacerbates dystrophic phenotype in mdx slow-type fibers (92).
Interestingly, it has been found that one of the mechanisms by which glucocorticoid deflazacort attenuates disease progression in DMD is through augmenting the activation of calcineurin leading to activation of NFATc1-dependent gene expression (93). It is also of interest to note that in comparison to mdx mice, serum level of calcineurin is significantly lower in DMD patients (94) which may account for the impairment of muscle regeneration, insufficient utrophin expression, and severe pathology in DMD patients.
Whereas the above findings signify calcineurin or its downstream effector NFAT as a promising therapy for DMD, the potential of damaging side-effects caused by stimulating calcineurin-NFAT signaling in other tissues must not be overlooked. For example, cardiac hypertrophy is known to be induced by calcineurin (95) and, thus, systemic stimulation of calcineurin signaling can further aggravate the incidence of heart failure in DMD patients.
6.2. Targeting Cn/NFAT in LGMD2F
Like many other intracellular signaling pathways, it appears that the activation of calcineurin/NFAT pathway may benefit only select types of muscular dystrophies. This is because transgenic overexpression of calcineurin using a muscle-specific promoter has been found to increase muscle fiber loss and cardiac fibrosis and reduced cardiac ventricular shortening in δ-sarcoglycan-deficient mouse model of limb-girdle muscular dystrophy 2F (LGMD2F) (96). Conversely, muscle-specific deletion of calcineurin B1 (CnB1) gene reduced skeletal muscle degeneration and histopathology in these mice (96) indicating that calcineurin activation has deteriorating effects in LGMD2F. The potential reason why calcineurin improves muscle pathology in mdx mice but deteriorates in δ-sarcoglycan-deficient mice could be due to inherent differences in these two mouse models themselves. Besides primary genetic defect, there is significant difference in onset and severity of disease progression in these two types of mice (97).
7. Conclusions and Future Perspectives
From the above discussion, it is clear that substantial progress has been made towards identifying intracellular signaling pathways that may regulate myopathy in different types of muscular dystrophies. However, there are still many outstanding questions that need to be addressed. First of all, it is critical to investigate how the loss of a single protein leads to the activation of multiple signaling pathways in dystrophic muscles? Additional studies are required to understand the cross-talk between different pathways and whether the activation of other cell signaling pathways (e.g. JAK-STAT, Wnt, and TGF-β/SMAD, etc.) is also perturbed in dystrophic muscles in animal models.
Furthermore, why the activation of a specific signaling pathway produces antagonizing effects in different types of muscular dystrophies needs to be investigated. For example, whereas the activation of Akt leads to the improvement of muscle pathology in animal models of DMD, Akt also contributes to the activation of NF-κB which causes muscle degeneration in DMD. Understanding what parameter of muscle pathology is affected by the activation or inhibition of a specific signaling pathway may help understand this phenomenon. Indeed, this has been partly elucidated by the observations that apoptosis is a common feature in LGMD 2A and in this type of muscular dystrophy; the role of NF-κB appears to be protective for myofibers, which is consistent with the anti-apoptotic actions of NF-κB. However, the primary mechanism of muscle cell death is necrosis in DMD, where the activation of NF-κB perpetuates inflammation and stimulates muscle degeneration.
There is also a need to determine to what extent the pathogenic symptoms can be improved by regulation of a specific signaling pathway. Although modulation of the activities of various signaling pathways was found to be beneficial in mouse models, it did not restore full function in them. More experiments are needed to determine whether simultaneous intonation of multiple signaling pathways can be more effective to improving muscle pathogenesis and whether long-term studies in higher organisms such as golden retriever muscular dystrophy model will prove successful. Furthermore, it is crucial to determine at what stage of disease progression, systemic intervention of a specific pathway via drug therapy will be most effective. It is also noteworthy that some of these signaling pathways may be non-specifically activated as a result of disease progression or due to compensatory mechanisms in muscular dystrophy. Similar signaling pathways have also been found to be activated in several other chronic diseases such as muscle deconditioning, atrophy, sepsis, ventilation induced diaphragm dysfunction, chronic obstructive pulmonary disease, cystic fibrosis, and cancer where oxidative stress play an important role in muscular dysfunction. Indeed, there is sufficient literature suggesting that blocking the activity of different signaling pathways also improves myopathy and muscle functions in chronic diseases. Since gene therapy is unlikely in near future, modulation of different cellular pathways may be used in multiple therapeutic approaches to attenuate the common pathophysiological events in genetic disorders including muscular dystrophy.
While offering great clinical potential for treatment of muscular disorders, it should also be taken into consideration that continued inhibition/activation of some of these signaling pathways in vivo could also be detrimental. This is because many of these signaling pathways play an essential role in normal homeostasis, including development and functioning of immune system and cellular proliferation, self-renewal, and survival. It may also be helpful to identify the genes, which are targeted by specific signaling pathways in dystrophic muscles. Modulation of the expression and/or activity of those target genes can be used as a potential alternative therapeutic approach to attenuating disease progression in muscular dystrophies. The balance between therapeutic benefits and potential changes in normal cellular function will be one of the most important challenges in future clinical studies. Nevertheless, research in the past decade has made a strong case that proper regulation of the activity of various signaling pathways could be an important approach to attenuating disease progression in muscular dystrophy patients.
Acknowledgements
We would like to apologize to the many researchers whose contributions were not cited due to our oversight or space limitation. This work was supported by a RO1 grant AG029623 from National Institute of Health, USA (to AK).
Abbreviations
- AP-1
activator protein-1
- CaM
calmodulin
- DGC
dystrophin-glycoprotein complex
- DMD
Duchenne muscular dystrophy
- EMD
Emery-Dreifuss muscular dystrophy
- IκB
inhibitor of kappa B
- IKK
IκB kinase
- JNK
c-Jun-N-terminal kinase
- LGMD
limb-girdle muscular dystrophy
- MAPK
Mitogen activated protein kinase
- NEMO
NF-κB Essential Modulator NBD, NEMO-binding domain
- NFAT
nuclear factor of activated T-cells
- NF-κB
nuclear factor-kappa B
- PDTC
pyrrolidine dithiocarbamate
- PI3K
phosphatidylinositol 3-kinase
- SAC
stretch-activated channels.
References
- 1.Emery AE. The muscular dystrophies. Lancet. 2002;359:687–695. doi: 10.1016/S0140-6736(02)07815-7. [DOI] [PubMed] [Google Scholar]
- 2.Blake DJ, Weir A, Newey SE, Davies KE. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev. 2002;82:291–329. doi: 10.1152/physrev.00028.2001. [DOI] [PubMed] [Google Scholar]
- 3.Campbell KP. Three muscular dystrophies: loss of cytoskeleton-extracellular matrix linkage. Cell. 1995;80:675–679. doi: 10.1016/0092-8674(95)90344-5. [DOI] [PubMed] [Google Scholar]
- 4.Hoffman EP, Brown RH, Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 5.Rando TA. The dystrophin-glycoprotein complex, cellular signaling, and the regulation of cell survival in the muscular dystrophies. Muscle Nerve. 2001;24:1575–1594. doi: 10.1002/mus.1192. [DOI] [PubMed] [Google Scholar]
- 6.Petrof BJ, Shrager JB, Stedman HH, Kelly AM, Sweeney HL. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci U S A. 1993;90:3710–3714. doi: 10.1073/pnas.90.8.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Engvall E, Wewer UM. The new frontier in muscular dystrophy research: booster genes. FASEB J. 2003;17:1579–1584. doi: 10.1096/fj.02-1215rev. [DOI] [PubMed] [Google Scholar]
- 8.Khurana TS, Davies KE. Pharmacological strategies for muscular dystrophy. Nat Rev Drug Discov. 2003;2:379–390. doi: 10.1038/nrd1085. [DOI] [PubMed] [Google Scholar]
- 9.Chakkalakal JV, Thompson J, Parks RJ, Jasmin BJ. Molecular, cellular, and pharmacological therapies for Duchenne/Becker muscular dystrophies. FASEB J. 2005;19:880–891. doi: 10.1096/fj.04-1956rev. [DOI] [PubMed] [Google Scholar]
- 10.Spencer MJ, Tidball JG. Do immune cells promote the pathology of dystrophin-deficient myopathies? Neuromuscul Disord. 2001;11:556–564. doi: 10.1016/s0960-8966(01)00198-5. [DOI] [PubMed] [Google Scholar]
- 11.Kumar A, Boriek AM. Mechanical stress activates the nuclear factor-kappaB pathway in skeletal muscle fibers: a possible role in Duchenne muscular dystrophy. FASEB J. 2003;17:386–396. doi: 10.1096/fj.02-0542com. [DOI] [PubMed] [Google Scholar]
- 12.Kumar A, Khandelwal N, Malya R, Reid MB, Boriek AM. Loss of dystrophin causes aberrant mechanotransduction in skeletal muscle fibers. FASEB J. 2004;18:102–113. doi: 10.1096/fj.03-0453com. [DOI] [PubMed] [Google Scholar]
- 13.Madhavan R, Massom LR, Jarrett HW. Calmodulin specifically binds three proteins of the dystrophin-glycoprotein complex. Biochem Biophys Res Commun. 1992;185:753–759. doi: 10.1016/0006-291x(92)91690-r. [DOI] [PubMed] [Google Scholar]
- 14.Madhavan R, Jarrett HW. Phosphorylation of dystrophin and alpha-syntrophin by Ca(2+)-calmodulin dependent protein kinase II. Biochim Biophys Acta. 1999;1434:260–274. doi: 10.1016/s0167-4838(99)00193-4. [DOI] [PubMed] [Google Scholar]
- 15.Langenbach KJ, Rando TA. Inhibition of dystroglycan binding to laminin disrupts the PI3K/AKT pathway and survival signaling in muscle cells. Muscle Nerve. 2002;26:644–653. doi: 10.1002/mus.10258. [DOI] [PubMed] [Google Scholar]
- 16.Hasegawa M, Cuenda A, Spillantini MG, Thomas GM, Buee-Scherrer V, Cohen P, Goedert M. Stress-activated protein kinase-3 interacts with the PDZ domain of alpha1-syntrophin. A mechanism for specific substrate recognition. J Biol Chem. 1999;274:12626–12631. doi: 10.1074/jbc.274.18.12626. [DOI] [PubMed] [Google Scholar]
- 17.Adams ME, Dwyer TM, Dowler LL, White RA, Froehner SC. Mouse alpha 1- and beta 2-syntrophin gene structure, chromosome localization, and homology with a discs large domain. J Biol Chem. 1995;270:25859–25865. doi: 10.1074/jbc.270.43.25859. [DOI] [PubMed] [Google Scholar]
- 18.Gee SH, Madhavan R, Levinson SR, Caldwell JH, Sealock R, Froehner SC. Interaction of muscle and brain sodium channels with multiple members of the syntrophin family of dystrophin-associated proteins. J Neurosci. 1998;18:128–137. doi: 10.1523/JNEUROSCI.18-01-00128.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chockalingam PS, Gee SH, Jarrett HW. Pleckstrin homology domain 1 of mouse alpha 1-syntrophin binds phosphatidylinositol 4,5-bisphosphate. Biochemistry. 1999;38:5596–5602. doi: 10.1021/bi982564+. [DOI] [PubMed] [Google Scholar]
- 20.Cavaldesi M, Macchia G, Barca S, Defilippi P, Tarone G, Petrucci TC. Association of the dystroglycan complex isolated from bovine brain synaptosomes with proteins involved in signal transduction. J Neurochem. 1999;72:1648–1655. doi: 10.1046/j.1471-4159.1999.721648.x. [DOI] [PubMed] [Google Scholar]
- 21.Oak SA, Russo K, Petrucci TC, Jarrett HW. Mouse alpha1-syntrophin binding to Grb2: further evidence of a role for syntrophin in cell signaling. Biochemistry. 2001;40:11270–11278. doi: 10.1021/bi010490n. [DOI] [PubMed] [Google Scholar]
- 22.Spence HJ, Dhillon AS, James M, Winder SJ. Dystroglycan, a scaffold for the ERK-MAP kinase cascade. EMBO Rep. 2004;5:484–489. doi: 10.1038/sj.embor.7400140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oak SA, Zhou YW, Jarrett HW. Skeletal muscle signaling pathway through the dystrophin glycoprotein complex and Rac1. J Biol Chem. 2003;278:39287–39295. doi: 10.1074/jbc.M305551200. [DOI] [PubMed] [Google Scholar]
- 24.Allen DG. Skeletal muscle function: role of ionic changes in fatigue, damage and disease. Clin Exp Pharmacol Physiol. 2004;31:485–493. doi: 10.1111/j.1440-1681.2004.04032.x. [DOI] [PubMed] [Google Scholar]
- 25.Vandebrouck A, Sabourin J, Rivet J, Balghi H, Sebille S, Kitzis A, Raymond G, Cognard C, Bourmeyster N, Constantin B. Regulation of capacitative calcium entries by alpha1-syntrophin: association of TRPC1 with dystrophin complex and the PDZ domain of alpha1-syntrophin. FASEB J. 2007;21:608–617. doi: 10.1096/fj.06-6683com. [DOI] [PubMed] [Google Scholar]
- 26.Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 27.Aggarwal BB. Nuclear factor-kappaB: the enemy within. Cancer Cell. 2004;6:203–208. doi: 10.1016/j.ccr.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 28.Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–288. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 29.Chen YW, Nagaraju K, Bakay M, McIntyre O, Rawat R, Shi R, Hoffman EP. Early onset of inflammation and later involvement of TGFbeta in Duchenne muscular dystrophy. Neurology. 2005;65:826–834. doi: 10.1212/01.wnl.0000173836.09176.c4. [DOI] [PubMed] [Google Scholar]
- 30.Pescatori M, Broccolini A, Minetti C, Bertini E, Bruno C, D'Amico A, Bernardini C, Mirabella M, Silvestri G, Giglio V, Modoni A, Pedemonte M, Tasca G, Galluzzi G, Mercuri E, Tonali PA, Ricci E. Gene expression profiling in the early phases of DMD: a constant molecular signature characterizes DMD muscle from early postnatal life throughout disease progression. FASEB J. 2007;21:1210–1226. doi: 10.1096/fj.06-7285com. [DOI] [PubMed] [Google Scholar]
- 31.Porter JD, Guo W, Merriam AP, Khanna S, Cheng G, Zhou X, Andrade FH, Richmonds C, Kaminski HJ. Persistent over-expression of specific CC class chemokines correlates with macrophage and T-cell recruitment in mdx skeletal muscle. Neuromuscul Disord. 2003;13:223–235. doi: 10.1016/s0960-8966(02)00242-0. [DOI] [PubMed] [Google Scholar]
- 32.Porter JD, Khanna S, Kaminski HJ, Rao JS, Merriam AP, Richmonds CR, Leahy P, Li J, Guo W, Andrade FH. A chronic inflammatory response dominates the skeletal muscle molecular signature in dystrophin-deficient mdx mice. Hum Mol Genet. 2002;11:263–272. doi: 10.1093/hmg/11.3.263. [DOI] [PubMed] [Google Scholar]
- 33.Porter JD, Merriam AP, Leahy P, Gong B, Feuerman J, Cheng G, Khanna S. Temporal gene expression profiling of dystrophin-deficient (mdx) mouse diaphragm identifies conserved and muscle group-specific mechanisms in the pathogenesis of muscular dystrophy. Hum Mol Genet. 2004;13:257–269. doi: 10.1093/hmg/ddh033. [DOI] [PubMed] [Google Scholar]
- 34.Dogra C, Changotra H, Wergedal JE, Kumar A. Regulation of phosphatidylinositol 3-kinase (PI3K)/Akt and nuclear factor-kappa B signaling pathways in dystrophin-deficient skeletal muscle in response to mechanical stretch. J Cell Physiol. 2006;208:575–585. doi: 10.1002/jcp.20696. [DOI] [PubMed] [Google Scholar]
- 35.Durham WJ, Arbogast S, Gerken E, Li YP, Reid MB. Progressive nuclear factor-kappaB activation resistant to inhibition by contraction and curcumin in mdx mice. Muscle Nerve. 2006;34:298–303. doi: 10.1002/mus.20579. [DOI] [PubMed] [Google Scholar]
- 36.Acharyya S, Villalta SA, Bakkar N, Bupha-Intr T, Janssen PM, Carathers M, Li ZW, Beg AA, Ghosh S, Sahenk Z, Weinstein M, Gardner KL, Rafael-Fortney JA, Karin M, Tidball JG, Baldwin AS, Guttridge DC. Interplay of IKK/NF-kappaB signaling in macrophages and myofibers promotes muscle degeneration in Duchenne muscular dystrophy. J Clin Invest. 2007;117:889–901. doi: 10.1172/JCI30556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hnia K, Gayraud J, Hugon G, Ramonatxo M, De La Porte S, Matecki S, Mornet D. L-arginine decreases inflammation and modulates the nuclear factor-kappaB/matrix metalloproteinase cascade in mdx muscle fibers. Am J Pathol. 2008;172:1509–1519. doi: 10.2353/ajpath.2008.071009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Messina S, Altavilla D, Aguennouz M, Seminara P, Minutoli L, Monici MC, Bitto A, Mazzeo A, Marini H, Squadrito F, Vita G. Lipid peroxidation inhibition blunts nuclear factor-kappaB activation, reduces skeletal muscle degeneration, and enhances muscle function in mdx mice. Am J Pathol. 2006;168:918–926. doi: 10.2353/ajpath.2006.050673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Siegel AL, Bledsoe C, Lavin J, Gatti F, Berge J, Millman G, Turin E, Winders WT, Rutter J, Palmeiri B, Carlson CG. Treatment with inhibitors of the NF-kappaB pathway improves whole body tension development in the mdx mouse. Neuromuscul Disord. 2009;19:131–139. doi: 10.1016/j.nmd.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 40.Lammerding J, Schulze PC, Takahashi T, Kozlov S, Sullivan T, Kamm RD, Stewart CL, Lee RT. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J Clin Invest. 2004;113:370–378. doi: 10.1172/JCI19670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Monici MC, Aguennouz M, Mazzeo A, Messina C, Vita G. Activation of nuclear factor-kappaB in inflammatory myopathies and Duchenne muscular dystrophy. Neurology. 2003;60:993–997. doi: 10.1212/01.wnl.0000049913.27181.51. [DOI] [PubMed] [Google Scholar]
- 42.Haslbeck KM, Friess U, Schleicher ED, Bierhaus A, Nawroth PP, Kirchner A, Pauli E, Neundorfer B, Heuss D. The RAGE pathway in inflammatory myopathies and limb girdle muscular dystrophy. Acta Neuropathol. 2005;110:247–254. doi: 10.1007/s00401-005-1043-3. [DOI] [PubMed] [Google Scholar]
- 43.Macaione V, Aguennouz M, Rodolico C, Mazzeo A, Patti A, Cannistraci E, Colantone L, Di Giorgio RM, De Luca G, Vita G. RAGE-NF-kappaB pathway activation in response to oxidative stress in facioscapulohumeral muscular dystrophy. Acta Neurol Scand. 2007;115:115–121. doi: 10.1111/j.1600-0404.2006.00724.x. [DOI] [PubMed] [Google Scholar]
- 44.Li H, Malhotra S, Kumar A. Nuclear factor-kappa B signaling in skeletal muscle atrophy. J Mol Med. 2008;86:1113–1126. doi: 10.1007/s00109-008-0373-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mourkioti F, Kratsios P, Luedde T, Song YH, Delafontaine P, Adami R, Parente V, Bottinelli R, Pasparakis M, Rosenthal N. Targeted ablation of IKK2 improves skeletal muscle strength, maintains mass, and promotes regeneration. J Clin Invest. 2006;116:2945–2954. doi: 10.1172/JCI28721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cai D, Frantz JD, Tawa NE, Jr, Melendez PA, Oh BC, Lidov HG, Hasselgren PO, Frontera WR, Lee J, Glass DJ, Shoelson SE. IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell. 2004;119:285–298. doi: 10.1016/j.cell.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 47.Kumar A, Takada Y, Boriek AM, Aggarwal BB. Nuclear factor-kappaB: its role in health and disease. J Mol Med. 2004;82:434–448. doi: 10.1007/s00109-004-0555-y. [DOI] [PubMed] [Google Scholar]
- 48.Bakkar N, Wang J, Ladner KJ, Wang H, Dahlman JM, Carathers M, Acharyya S, Rudnicki MA, Hollenbach AD, Guttridge DC. IKK/NF-kappaB regulates skeletal myogenesis via a signaling switch to inhibit differentiation and promote mitochondrial biogenesis. J Cell Biol. 2008;180:787–802. doi: 10.1083/jcb.200707179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guttridge DC, Mayo MW, Madrid LV, Wang CY, Baldwin AS., Jr NF-kappaB-induced loss of MyoD messenger RNA: possible role in muscle decay and cachexia. Science. 2000;289:2363–2366. doi: 10.1126/science.289.5488.2363. [DOI] [PubMed] [Google Scholar]
- 50.Carlson CG, Samadi A, Siegel A. Chronic treatment with agents that stabilize cytosolic IkappaB-alpha enhances survival and improves resting membrane potential in MDX muscle fibers subjected to chronic passive stretch. Neurobiol Dis. 2005;20:719–730. doi: 10.1016/j.nbd.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 51.Messina S, Bitto A, Aguennouz M, Minutoli L, Monici MC, Altavilla D, Squadrito F, Vita G. Nuclear factor kappa-B blockade reduces skeletal muscle degeneration and enhances muscle function in Mdx mice. Exp Neurol. 2006;198:234–241. doi: 10.1016/j.expneurol.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 52.Whitehead NP, Pham C, Gervasio OL, Allen DG. N-Acetylcysteine ameliorates skeletal muscle pathophysiology in mdx mice. J Physiol. 2008;586:2003–2014. doi: 10.1113/jphysiol.2007.148338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hnia K, Hugon G, Rivier F, Masmoudi A, Mercier J, Mornet D. Modulation of p38 mitogen-activated protein kinase cascade and metalloproteinase activity in diaphragm muscle in response to free radical scavenger administration in dystrophin-deficient Mdx mice. Am J Pathol. 2007;170:633–643. doi: 10.2353/ajpath.2007.060344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li H, Mittal A, Makonchuk DY, Bhatnagar S, Kumar A. Matrix metalloproteinase-9 inhibition ameliorates pathogenesis and improves skeletal muscle regeneration in muscular dystrophy. Hum Mol Genet. 2009;18:2584–2598. doi: 10.1093/hmg/ddp191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vu TH, Werb Z. Matrix metalloproteinases: effectors of development and normal physiology. Genes Dev. 2000;14:2123–2133. doi: 10.1101/gad.815400. [DOI] [PubMed] [Google Scholar]
- 56.Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. J Biol Chem. 1995;270:16483–16486. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- 57.Richard I, Roudaut C, Marchand S, Baghdiguian S, Herasse M, Stockholm D, Ono Y, Suel L, Bourg N, Sorimachi H, Lefranc G, Fardeau M, Sebille A, Beckmann JS. Loss of calpain 3 proteolytic activity leads to muscular dystrophy and to apoptosis-associated IkappaBalpha/nuclear factor kappaB pathway perturbation in mice. J Cell Biol. 2000;151:1583–1590. doi: 10.1083/jcb.151.7.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Baghdiguian S, Martin M, Richard I, Pons F, Astier C, Bourg N, Hay RT, Chemaly R, Halaby G, Loiselet J, Anderson LV, Lopez de Munain A, Fardeau M, Mangeat P, Beckmann JS, Lefranc G. Calpain 3 deficiency is associated with myonuclear apoptosis and profound perturbation of the IkappaB alpha/NF-kappaB pathway in limb-girdle muscular dystrophy type 2A. Nat Med. 1999;5:503–511. doi: 10.1038/8385. [DOI] [PubMed] [Google Scholar]
- 59.Benayoun B, Baghdiguian S, Lajmanovich A, Bartoli M, Daniele N, Gicquel E, Bourg N, Raynaud F, Pasquier MA, Suel L, Lochmuller H, Lefranc G, Richard I. NF-kappaB-dependent expression of the antiapoptotic factor c-FLIP is regulated by calpain 3, the protein involved in limb-girdle muscular dystrophy type 2A. FASEB J. 2008;22:1521–1529. doi: 10.1096/fj.07-8701com. [DOI] [PubMed] [Google Scholar]
- 60.Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 61.Kolodziejczyk SM, Walsh GS, Balazsi K, Seale P, Sandoz J, Hierlihy AM, Rudnicki MA, Chamberlain JS, Miller FD, Megeney LA. Activation of JNK1 contributes to dystrophic muscle pathogenesis. Curr Biol. 2001;11:1278–1282. doi: 10.1016/s0960-9822(01)00397-9. [DOI] [PubMed] [Google Scholar]
- 62.Nakamura A, Yoshida K, Takeda S, Dohi N, Ikeda S. Progression of dystrophic features and activation of mitogen-activated protein kinases and calcineurin by physical exercise, in hearts of mdx mice. FEBS Lett. 2002;520:18–24. doi: 10.1016/s0014-5793(02)02739-4. [DOI] [PubMed] [Google Scholar]
- 63.Nakamura A, Harrod GV, Davies KE. Activation of calcineurin and stress activated protein kinase/p38-mitogen activated protein kinase in hearts of utrophin-dystrophin knockout mice. Neuromuscul Disord. 2001;11:251–259. doi: 10.1016/s0960-8966(00)00201-7. [DOI] [PubMed] [Google Scholar]
- 64.Griffin MA, Feng H, Tewari M, Acosta P, Kawana M, Sweeney HL, Discher DE. gamma-Sarcoglycan deficiency increases cell contractility, apoptosis and MAPK pathway activation but does not affect adhesion. J Cell Sci. 2005;118:1405–1416. doi: 10.1242/jcs.01717. [DOI] [PubMed] [Google Scholar]
- 65.Woodman SE, Park DS, Cohen AW, Cheung MW, Chandra M, Shirani J, Tang B, Jelicks LA, Kitsis RN, Christ GJ, Factor SM, Tanowitz HB, Lisanti MP. Caveolin-3 knock-out mice develop a progressive cardiomyopathy and show hyperactivation of the p42/44 MAPK cascade. J Biol Chem. 2002;277:38988–38997. doi: 10.1074/jbc.M205511200. [DOI] [PubMed] [Google Scholar]
- 66.Disatnik MH, Dhawan J, Yu Y, Beal MF, Whirl MM, Franco AA, Rando TA. Evidence of oxidative stress in mdx mouse muscle: studies of the pre-necrotic state. J Neurol Sci. 1998;161:77–84. doi: 10.1016/s0022-510x(98)00258-5. [DOI] [PubMed] [Google Scholar]
- 67.Murphy ME, Kehrer JP. Oxidative stress and muscular dystrophy. Chem Biol Interact. 1989;69:101–173. doi: 10.1016/0009-2797(89)90075-6. [DOI] [PubMed] [Google Scholar]
- 68.Rando TA, Disatnik MH, Yu Y, Franco A. Muscle cells from mdx mice have an increased susceptibility to oxidative stress. Neuromuscul Disord. 1998;8:14–21. doi: 10.1016/s0960-8966(97)00124-7. [DOI] [PubMed] [Google Scholar]
- 69.Kefaloyianni E, Gaitanaki C, Beis I. ERK1/2 and p38-MAPK signalling pathways, through MSK1, are involved in NF-kappaB transactivation during oxidative stress in skeletal myoblasts. Cell Signal. 2006;18:2238–2251. doi: 10.1016/j.cellsig.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 70.Muchir A, Pavlidis P, Bonne G, Hayashi YK, Worman HJ. Activation of MAPK in hearts of EMD null mice: similarities between mouse models of X-linked and autosomal dominant Emery Dreifuss muscular dystrophy. Hum Mol Genet. 2007;16:1884–1895. doi: 10.1093/hmg/ddm137. [DOI] [PubMed] [Google Scholar]
- 71.Muchir A, Pavlidis P, Decostre V, Herron AJ, Arimura T, Bonne G, Worman HJ. Activation of MAPK pathways links LMNA mutations to cardiomyopathy in Emery-Dreifuss muscular dystrophy. J Clin Invest. 2007;117:1282–1293. doi: 10.1172/JCI29042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Muchir A, Shan J, Bonne G, Lehnart SE, Worman HJ. Inhibition of extracellular signal-regulated kinase signaling to prevent cardiomyopathy caused by mutation in the gene encoding A-type lamins. Hum Mol Genet. 2009;18:241–247. doi: 10.1093/hmg/ddn343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 74.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 75.Lawlor MA, Alessi DR. PKB/Akt: a key mediator of cell proliferation, survival and insulin responses? J Cell Sci. 2001;114:2903–2910. doi: 10.1242/jcs.114.16.2903. [DOI] [PubMed] [Google Scholar]
- 76.Glass DJ. Signalling pathways that mediate skeletal muscle hypertrophy and atrophy. Nat Cell Biol. 2003;5:87–90. doi: 10.1038/ncb0203-87. [DOI] [PubMed] [Google Scholar]
- 77.Barton ER, Morris L, Musaro A, Rosenthal N, Sweeney HL. Muscle-specific expression of insulin-like growth factor I counters muscle decline in mdx mice. J Cell Biol. 2002;157:137–148. doi: 10.1083/jcb.200108071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Peter AK, Crosbie RH. Hypertrophic response of Duchenne and limb-girdle muscular dystrophies is associated with activation of Akt pathway. Exp Cell Res. 2006;312:2580–2591. doi: 10.1016/j.yexcr.2006.04.024. [DOI] [PubMed] [Google Scholar]
- 79.Peter AK, Ko CY, Kim MH, Hsu N, Ouchi N, Rhie S, Izumiya Y, Zeng L, Walsh K, Crosbie RH. Myogenic Akt signaling upregulates the utrophin-glycoprotein complex and promotes sarcolemma stability in muscular dystrophy. Hum Mol Genet. 2009;18:318–327. doi: 10.1093/hmg/ddn358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rafael JA, Tinsley JM, Potter AC, Deconinck AE, Davies KE. Skeletal muscle-specific expression of a utrophin transgene rescues utrophin-dystrophin deficient mice. Nat Genet. 1998;19:79–82. doi: 10.1038/ng0598-79. [DOI] [PubMed] [Google Scholar]
- 81.Blaauw B, Mammucari C, Toniolo L, Agatea L, Abraham R, Sandri M, Reggiani C, Schiaffino S. Akt activation prevents the force drop induced by eccentric contractions in dystrophin-deficient skeletal muscle. Hum Mol Genet. 2008;17:3686–3696. doi: 10.1093/hmg/ddn264. [DOI] [PubMed] [Google Scholar]
- 82.Gurpur PB, Liu J, Burkin DJ, Kaufman SJ. Valproic acid activates the PI3K/Akt/mTOR pathway in muscle and ameliorates pathology in a mouse model of Duchenne muscular dystrophy. Am J Pathol. 2009;174:999–1008. doi: 10.2353/ajpath.2009.080537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hogan PG, Chen L, Nardone J, Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003;17:2205–2232. doi: 10.1101/gad.1102703. [DOI] [PubMed] [Google Scholar]
- 84.Shen T, Liu Y, Cseresnyes Z, Hawkins A, Randall WR, Schneider MF. Activity- and calcineurin-independent nuclear shuttling of NFATc1, but not NFATc3, in adult skeletal muscle fibers. Mol Biol Cell. 2006;17:1570–1582. doi: 10.1091/mbc.E05-08-0780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schulz RA, Yutzey KE. Calcineurin signaling and NFAT activation in cardiovascular and skeletal muscle development. Dev Biol. 2004;266:1–16. doi: 10.1016/j.ydbio.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 86.Olson EN, Williams RS. Calcineurin signaling and muscle remodeling. Cell. 2000;101:689–692. doi: 10.1016/s0092-8674(00)80880-6. [DOI] [PubMed] [Google Scholar]
- 87.Stupka N, Gregorevic P, Plant DR, Lynch GS. The calcineurin signal transduction pathway is essential for successful muscle regeneration in mdx dystrophic mice. Acta Neuropathol. 2004;107:299–310. doi: 10.1007/s00401-003-0807-x. [DOI] [PubMed] [Google Scholar]
- 88.Stupka N, Schertzer JD, Bassel-Duby R, Olson EN, Lynch GS. Stimulation of calcineurin Aalpha activity attenuates muscle pathophysiology in mdx dystrophic mice. Am J Physiol Regul Integr Comp Physiol. 2008;294:R983–R992. doi: 10.1152/ajpregu.00375.2007. [DOI] [PubMed] [Google Scholar]
- 89.Stupka N, Michell BJ, Kemp BE, Lynch GS. Differential calcineurin signalling activity and regeneration efficacy in diaphragm and limb muscles of dystrophic mdx mice. Neuromuscul Disord. 2006;16:337–346. doi: 10.1016/j.nmd.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 90.Chakkalakal JV, Stocksley MA, Harrison MA, Angus LM, Deschenes-Furry J, St-Pierre S, Megeney LA, Chin ER, Michel RN, Jasmin BJ. Expression of utrophin A mRNA correlates with the oxidative capacity of skeletal muscle fiber types and is regulated by calcineurin/NFAT signaling. Proc Natl Acad Sci U S A. 2003;100:7791–7796. doi: 10.1073/pnas.0932671100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chakkalakal JV, Harrison MA, Carbonetto S, Chin E, Michel RN, Jasmin BJ. Stimulation of calcineurin signaling attenuates the dystrophic pathology in mdx mice. Hum Mol Genet. 2004;13:379–388. doi: 10.1093/hmg/ddh037. [DOI] [PubMed] [Google Scholar]
- 92.Chakkalakal JV, Michel SA, Chin ER, Michel RN, Jasmin BJ. Targeted inhibition of Ca2+ /calmodulin signaling exacerbates the dystrophic phenotype in mdx mouse muscle. Hum Mol Genet. 2006;15:1423–1435. doi: 10.1093/hmg/ddl065. [DOI] [PubMed] [Google Scholar]
- 93.St-Pierre SJ, Chakkalakal JV, Kolodziejczyk SM, Knudson JC, Jasmin BJ, Megeney LA. Glucocorticoid treatment alleviates dystrophic myofiber pathology by activation of the calcineurin/NF-AT pathway. FASEB J. 2004;18:1937–1939. doi: 10.1096/fj.04-1859fje. [DOI] [PubMed] [Google Scholar]
- 94.Sundaram JS, Rao VM, Meena AK, Anandaraj MP. Decreased calcineurin activity in circulation of Duchenne muscular dystrophy. Clin Biochem. 2007;40:443–446. doi: 10.1016/j.clinbiochem.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 95.Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J, Grant SR, Olson EN. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93:215–228. doi: 10.1016/s0092-8674(00)81573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Parsons SA, Millay DP, Sargent MA, Naya FJ, McNally EM, Sweeney HL, Molkentin JD. Genetic disruption of calcineurin improves skeletal muscle pathology and cardiac disease in a mouse model of limb-girdle muscular dystrophy. J Biol Chem. 2007;282:10068–10078. doi: 10.1074/jbc.M609368200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Durbeej M, Campbell KP. Muscular dystrophies involving the dystrophin-glycoprotein complex: an overview of current mouse models. Curr Opin Genet Dev. 2002;12:349–361. doi: 10.1016/s0959-437x(02)00309-x. [DOI] [PubMed] [Google Scholar]