

Abstract
Ulcerative colitis (UC) is a dynamic, chronic inflammatory condition associated with an increased colon cancer risk. Inflammatory cell apoptosis is a key mechanism regulating UC. American ginseng (AG) is a putative anti-oxidant that can suppress hyperactive immune cells. We have recently shown that AG can prevent and treat mouse colitis. Because p53 levels are elevated in inflammatory cells in both mouse and human colitis, we tested the hypothesis that AG protects from colitis by driving inflammatory cell apoptosis through a p53 mechanism. We used isogenic p53+/+ and p53−/− inflammatory cell lines, as well as primary CD4+/CD25− effector T cells from p53+/+ and p53−/− mice to show that AG drives apoptosis in a p53-dependent manner. Moreover, we used a dextran sulfate sodium (DSS) model of colitis in C57BL/6 p53+/+ and p53−/− mice to test whether the protective effect of AG against colitis is p53-dependent. Data indicate AG induces apoptosis in p53+/+, but not in isogenic p53−/− cells in vitro. In vivo, C57BL/6 p53+/+ mice are responsive to the protective effects of AG against DSS-induced colitis, while AG fails to protect from colitis in p53−/− mice. Furthermore, TUNEL labeling of inflammatory cells within the colonic mesenteric lymph nodes is elevated in p53+/+ mice consuming DSS + AG, but not in p53−/− mice consuming DSS + AG. Results are consistent with our in vitro data, and with the hypothesis that AG drives inflammatory cell apoptosis in vivo, providing a mechanism by which AG protects from colitis in this DSS mouse model.
Keywords: p53, Inflammation, Ginseng, Colitis, Colon Cancer
Introduction
Dysfunction of the intestinal immune system has been implicated as a major mechanism by which chronic inflammation occurs in colitis. Immune responses are initiated when either cytotoxic T lymphocyte CD8+ cells or CD4+ T helper cells in the intestinal lumen recognize a foreign antigen. This initiates the immunological cascade responsible for eradicating the antigenic material. This includes the production of pro-inflammatory and anti-inflammatory cytokines and the infiltration of macrophages and neutrophils. Once the antigen has been eradicated, T lymphocytes of the intestinal mucosa require a method to attenuate the local immune response. Failure to regulate T-cell responses in the intestinal or colonic mucosa leads to an inappropriate, and sustained injurious immunologic reaction (1, 2). A key mechanism for immune suppression is apoptosis of overly aggressive effector T cells and defects in mucosal T-cell apoptosis are likely to be critical in the pathogenesis of colitis.
Apoptosis can proceed through two major pathways. There is an intrinsic (mitochondrial-mediated) pathway and an extrinsic (death receptor-mediated) pathway (3). A key molecule involved in apoptosis (mainly in the intrinsic pathway) is p53, which is activated in epithelial and inflammatory cells during the process of colitis. This is well documented by others and ourselves (4–8). Mechanistically, levels are elevated due to either a stabilization of the wild-type form of p53 by post-translational modification (often phosphorylation at Serine-15) (6) or through p53 mutation, leading to abnormally elevated levels of an inactivated form (4, 5). Consistent with this hypothesis is that patients with UC have an elevated p53 mutation load in non-cancerous colon epithelial cells (9, 10).
The finding that p53 levels are elevated in inflammatory cells during colitis (7, 8) is interesting and significant, because it identifies a potential molecular target for apoptosis by drugs and/or complementary and alternative medicines, such as AG. Although the mechanisms for the increased p53 observed in inflammatory cells are not fully delineated, many key inflammatory players have been shown to induce p53. A non-exhaustive list includes reactive nitrogen species (6, 11), reactive oxygen species (12), TNF-α (13), and other cytokines (14).
There are several different species of ginseng: 2 of the most commonly used are Panax ginseng (Asian ginseng) and Panax quinquefolius (American ginseng) (15). Here, we use Panax quinquefolius (American ginseng, AG) (root, harvested at 3–5 years, dried and ground to a powder). AG, although less studied, has similar ginsenosides as the Asian ginseng. Studies indicate that ginseng can improve endpoints associated with prevalent diseases in Western society such as cardiovascular disease and cancer; as well as autoimmune diseases such as diabetes [reviewed in: (16–18)]. Mechanisms include inhibition of DNA damage, induction of apoptosis, inhibition of cell proliferation, and inhibition of immune cell activity (19–21). Interestingly, ginseng has not been documented as a standard complementary and alternative medicine in patients with inflammatory bowel diseases. In addition to massage, meditation and Tai chi, pro-biotics such as acidophilus, and herbal therapies such as flax seed, aloe vera and garlic are common alternatives used in this population.
There is extensive evidence that: (1) Ginseng induces p53 (22–24); (2) p53 is elevated in activated inflammatory cells and plays a role in their apoptosis; (3) p53 is activated and activated in inflammatory cells during colitis (4–8); and (4) p53 protects from colitis-driven colon cancer (25, 26). In addition, AG induces apoptosis in inflammatory cells. For these reasons, here, we tested the hypothesis that AG protects from mouse colitis through a p53 mechanism that is involved in driving apoptosis of inflammatory cells. We describe results consistent with this hypothesis.
Materials and Methods
American ginseng extract
The Panax quinquefolus (American ginseng) extract has been described previously in detail by our laboratory (7). Briefly, American ginseng extract was purchased from the National Research Council (NRC) of Canada. This extract was cultivated by Chai-Na-Ta Farms Ltd. (Kamloops, British Columbia, Canada) and processed by Canadian Phytopharmaceuticals Corporation (Richmond, British Columbia, Canada). Following grinding to pass 80 mesh, 35 kg of the root material was extracted with aqueous ethanol (75% ethanol 25% water) in a re-circulating filter extraction system for 4 hours at a temperature of 60°C under vacuum. The ratio of solvent to root was 8:1 (v/w). After extraction, the filtrate was partially dried in-vacuo to yield a concentrated extract. 2.8 kg of maltodextrin (40% of final weight) was then blended as a support and the resultant slurry spray dried to yield 7 kg of free flowing powder. Analysis by Canadian Phytopharmaceuticals Corporation by HPLC-UV against pure standards determined the total ginsenoside content (as the sum of: Rg1, Re, Rb1, Rc, Rb2 and Rd) of the finished material to be 10.1% (w/w), and confirmed by HPLC-MS at the NRC of Cancada. The final, powder form of American ginseng extract also contained 2% additional ginsenosides (made up of F11, Ro, isomers of Rd, and traces of malonyl ginsenosides), and 40% of maltodextrin derived from hydrolysed corn starch. The remaining 48% of the powder was made up of ginseng root derived polysaccharides/ ligosaccharides and proteins and up to 5% of moisture. The lot used was screened and found to be free of heavy metals and contaminants. It should be noted here, that regular AIN-93M chow fed to mice contains 12.5000% maltodextrin. The addition of 75 ppm American ginseng in the chow, equates to 30 mg/kg final concentration of maltodextrin added to 12.5000% already in the chow. Therefore, there is 12.5000% maltodextrin in the AIN-93M chow and 12.5003% of maltodextrin in the AIN-93M chow supplemented with 75 ppm American ginseng extract.
Chemicals and reagents
Dextran Sulfate Sodium (DSS) was purchased from MP Biomedicals (Solon, OH; m.w. 36,000–50,000). Concanavalin A (Con-A, 2.5 μg/ml) was purchased from Sigma-Aldrich (St. Louis, MO). The following antibodies were used in these studies: anti-PARP (Cell Signaling, polyclonal, 1:500, Cat# 9542), anti-caspase-3 (Cell Signaling, polyclonal, 1:500, Cat# 9662), anti-caspase-7 (Calbiochem, polyclonal Ab1, 1:1000, Cat #PC334), anti-p53 (Calbiochem, monoclonal, Ab 1, 1:500, Clone PAb 421), anti-p53-Serine-15 phosphorylation (Cell Signaling, polyclonal, 1:500, Cat #9284), anti-bax (Cell Signaling, polyclonal, 1:500, Cat #2772), anti-caspase-9 (Cell Signaling, polyclonal, 1:500, Cat #9502), and anti-Actin (Calbiochem, monoclonal, Clone 1A4, 1:2000).
Cells
TK6 (p53+/+) and NH32 (p53−/−) were a kind gift from Curtis Harris (NCI), originally derived from Dr. William Thilly’s and Howard Liber’s labs. TK6 cells are a lymphobloastoid cell line derived from the spleen over 30 years ago (27). NH32 cells are an isogenic derivative of TK6 cells in which both alleles of the p53 gene were knocked out (28). Jurkat cells are an immortalized line of T lymphocyte cells derived in the late 1970s from the peripheral blood of a 14 year old boy with T cell leukemia (29). Jurkat cells have a defective p53 pathway, due to a mutation in the C-terminal domain responsible for transactivation (30, 31). CD4+/CD25− cells from C57BL/6 p53+/+ or C57BL/6 p53−/− mice were purified from the spleens using nylon wool columns (Polysciences, Warrington, PA) followed by depletion of B cells and macrophages. The purity of T cells was 90% as determined by flow cytometry (Cytomics FC 500, Beckman Coulter). CD4+/CD25− T cells were then isolated using a MACS mini separator and CD4 & CD25 micro-beads according to manufacturer’s instructions (Miltenyi BioTec, Germany) by depletion of CD4−CD25+ T cells (negative selection). 1 × 106 CD4+/CD25− effector T cells were cultured in six-well plates overnight, followed by experimentation as indicated. All cells were maintained in exponentially growing suspension culture at 37°C in a humidified, 5% CO2 atmosphere in RPMI 1640 supplemented with 10% heat-inactivated calf serum, 100 units/ml penicillin, 100 μg/ml streptomycin, and 2 mM L-glutamine.
Flow cytometry analysis for Annexin V
Cells were seeded at 1 × 106 cells/well into 6-well dishes for 24 hours. Following this, fresh media or media containing Con-A (2.5 μg/ml) was added to cells, and cultured for 12 hours. Con-A was then washed off, and fresh media, or fresh media containing freshly dissolved indicated concentrations of AG (0 – 1000 μg/ml) was added for 24–72 hours, as indicated. Cells were then harvested for Annexin V according to instructions provided by the kit manufacturer Annexin V (BD Bioscience, San Jose, CA). Annexin V/ propridium iodide (PI) staining was examined using a Beckman Coulter Cytomics FC500 flow cytometer.
DSS mouse model of colitis
We have described the DSS mouse model of colitis previously (7). Both p53−/− and p53+/+ were of C57BL/6 background. All males were used in the present study, because we have found that there are rarely females in p53−/− litters. Briefly, eight week-old C57BL/6 mice received either water ad libitum or 1% DSS. All mice were on anAIN76A high iron diet. American ginseng extract was mixed into the chow of indicated groups at 75 ppm (Research Diets, Inc. New Brunswick, NJ), which is the human equivalent dose of approximately 58 mg daily. Our calculation of the human equivalent amount of American ginseng consumed by mice uses the body surface area (BSA) normalization method (32), with the following assumptions: a typical mouse eats 3.5 g chow daily, and weighs 22 g; the average adult human weighs 60 kg. More specifically, here, chow contains 75 ppm American ginseng extract. This equates to 75 mg per kg of chow. A mouse consumes 3.5 g chow daily. Therefore, 75 mg/ 1000 g chow × 3.5 g chow/day = 0.2625 mg American ginseng extract daily. If a mouse weighs on average 22 g, then 0.2625 mg / 22 g × 1000 g/ 1 kg = 11.93 mg/kg daily. As discussed by Reagan-Shaw (32), the human equivalent dose (HED, mg/kg) = animal dose (mg/kg) × [Animal Km / Human Km]. As such, HED (mg/kg) for mouse = 11.93 mg/kg / [3/37] = 0.967 mg/kg. If an average human adult weighs 60 kg, this equates to 0.967 mg/kg × 60 kg = 58 mg daily for humans. Mice consumed the same amount of chow daily (on average 3.5 g) regardless of it containing American ginseng extract (data not shown).
To determine whether American ginseng extract prevents colitis onset, it was given to indicated groups of mice 1 week prior to and during DSS or water treatment. Organs were harvested from the treated mice after either 2½ cycles, where each cycle in the DSS groups consisted of 1% DSS in drinking water for 7 days, followed by a 7-day interval with normal drinking water. Mice were then euthanized at 1 cycle intervals (another 7 days water then 7 days DSS). For pathology and immunohistochemistry, colon tissue samples were washed with phosphate-buffered saline (PBS; Mediatech, Inc., Herndon, VA), cut longitudinally, swiss-roled, then formalin fixed and paraffin embedded.
Disease Activity Index (DAI)
The DAI, which monitors weight loss, stool consistency, and blood in the stool as a measure of disease severity, was scored for each animal every other day throughout the experiment. The DAI was calculated for each animal as described by Maines et al. (33) according to the system described in Table 1. With this scoring system, the DAI is calculated by scoring each animal for weight loss, stool consistency, and blood in the stool and then dividing the total score by three. For example, an animal that lost 12% of its body weight (score of 3) with evidence of loose stool (score of 2) plus gross rectal bleeding (score of 4) would have a calculated DAI of 3.
Table 1.
Scoring system for DAI
| Score | Weight Loss | Stool Consistency | Blood in Stool |
|---|---|---|---|
| 0 | None | Normal | Negative |
| 1 | 1–5% | ||
| 2 | 6–10% | Loose Stool | Positive |
| 3 | 11–15% | ||
| 4 | >15% | Diarrhea | Gross Rectal Bleeding |
Quantification of inflammation
The colon was transected, pinned open, and rinsed with phosphate-buffered saline (PBS), and the colon length was recorded. The tissue was then Swiss-Rolled, and fixed in formalin overnight, followed by embedding in paraffin, sectioning, and staining with hematoxylin and eosin (H&E). The sections were microscopically examined for histopathologic changes using the following scoring system: Histology score was determined by multiplying the percent involvement for each of the three following histological features by the percent area of involvement (33, 34): Inflammation severity (0: None; 1: Minimal; 2: Moderate; 3: Severe), Inflammation extent (0: None; 1: Mucosa; 2: Mucosa and submucosa; 3: Transmural), Crypt Damage (0: None; 1: 1/3 of Crypt Damaged; 2: 2/3 of Crypt Damaged; 3: Crypts Lost, Surface Epithelium Intact; 4: Crypts Lost; Surface Epithelium Lost), Percent Area Involvement (0: 0%; 1: 1–25%; 2: 26–50%; 3: 51–75%; 4: 76–100%). Therefore, the minimal score is 0 and the maximal score is 40.
TUNEL assay
A TUNEL assay was carried out to assess apoptosis in vivo, according to the manufacturer’s directions (DeadEnd™ Colirimetric TUNEL System; Promega, Madison, WI). Briefly, this is a modified TUNEL Assay designed to provide simple, accurate and rapid detection of apoptotic cells in situ at the single-cell level. The system measures nuclear DNA fragmentation, an important biochemical indicator of apoptosis. It end-labels the fragmented DNA of apoptotic cells using a modified TUNEL (TdT-mediated dUTP Nick-End Labeling) assay. Biotinylated nucleotide is incorporated at the 3′-OH DNA ends using the enzyme Terminal Deoxynucleotidyl Transferase (TdT). Horseradish-peroxidase-labeled streptavidin (Streptavidin HRP) is then bound to these biotinylated nucleotides, which are detected using the peroxidase substrate, hydrogen peroxide, and the stable chromogen, diaminobenzidine (DAB). Using this procedure, apoptotic nuclei are stained dark brown. The counterstain was CAT Hematoxylin (Biocare Medical, Concord, CA). Labeling was carried out on serial sections to that we used to score inflammatory index (Figure 3). TUNEL labeling in 10 separate sections from 10 individual mice was quantified in both the epithelial areas, and the mesenteric lymph nodes (MLNs). For the epithelial areas, 10 random fields were evaluated per slide. Because there are relatively fewer MLNs, we evaluated each MLN for TUNEL labeling. Labeled tissues were examined for intensity of staining using a method similar to that previously described (35). Briefly, intensity of staining was evaluated independently by blinded investigators. For each tissue section, the percentage of positive cells in either the epithelium, or in the MLNs, was scored on a scale of 0 to 4 for the percentage of tissue stained: 0 (0% positive cells), 1 (<10%), 2 (11% to 50%), 3 (51% to 80%), or 4 (> 80%). Staining intensity was scored on a scale of 0 to 3: 0-negative staining, 1-weak staining, 2-moderate staining, or 3-strong staining. The two scores were multiplied resulting in an immunoreactivity score (IRS) value ranging from 0 to 12.
Fig. 3.

Effects of AG on the colon histology score in the acute DSS-colitis model. 10 mice from each group described in Fig. 2 were sacrificed on day 35, and the colon was harvested from each animal and the histology score was determined. Values represent the mean ± standard error of the mean. Representative H&E stained colons are shown for each group. Significant differences are indicated (p < 0.05).
Statistical analysis
Statistical analysis was performed using one-way ANOVA with Scheffe’s post hoc test for TUNEL scores or the Kruskal-Wallis test when comparing histology scores. A two-way ANOVA for repeated measures was used to test for group and time effects on clinical data (e.g. disease activity index) over successive days of observation. The P-value chosen for significance in this study was 0.05.
Results
American Ginseng (AG) causes apoptosis in inflammatory cells in a p53-dependent manner
We had recently shown that AG prevents and treats mouse colitis (7). As well, we and other have shown that p53 levels are elevated in inflammatory cells in mouse and human colitis (6–8). Still others have shown that AG can drive apoptosis and induce p53 in leukemia cells (36, 37). We therefore decided to test the hypothesis that AG drives apoptosis of inflammatory cells in a p53-dependent manner.
We initially used the isogenic lymphoblostoid cell lines, TK6 and NH32. TK6 cells are a lymphobloastoid cell line and NH32 cells are an isogenic derivative of TK6 cells in which both alleles of the p53 gene were knocked out (28). Table 2 and Supplementary Figure 1 show that AG induces apoptosis (identified as Annexin V positive/ PI negative cells) in TK6 cells, but not in NH32 cells. As well, we were unable to induce significant apoptosis in Jurkat T cells, which have a mutant and dysfunctional p53 pathway (Table 3). A minimum of 10,000 cells were counted from three separate plates for each time and dose indicated. Representative experiments are shown.
Table 2.
Percentage of cells from a representative experiment staining positive for Annexin V and negative for PI (early apoptotic cells) in TK6 (p53+/+) and NH32 (p53−/−) isogenic lymphoblastoid cells.
| Dose | TK6 (p53+/+) | NH32 (p53−/−) | ||||
|---|---|---|---|---|---|---|
| AG (μg/ml) | 24 hrs | 48 hrs | 72 hrs | 24 hrs | 48 hrs | 72 hrs |
| 0 | 3.20% | 3.20% | 2.40% | 1.30% | 1.50% | 1.00% |
| 50 | ND | 3.50% | 2.80% | ND | 1.30% | 1.30% |
| 100 | 3.70% | 3.60% | 3.20% | 1.50% | 1.40% | 1.40% |
| 200 | ND | 4.20% | 4.40% | ND | 1.20% | 1.30% |
| 300 | 4.20% | 4.70% | 5.50% | 1.60% | 1.00% | 1.50% |
| 500 | 4.70% | 5.30% | 8.20% | 1.70% | 1.20% | 2.20% |
| 800 | 5.10% | ND | ND | 1.50% | ND | ND |
| 1000 | 6.60% | ND | ND | 1.60% | ND | ND |
ND, indicates not done
Table 3.
Percentage of cells from a representative experiment staining positive for Annexin V and negative for PI (early apoptotic cells) Jurkat T cells, which have a dysfunctional p53 pathway.
| Dose | Jurkat T cells | |
|---|---|---|
| AG (ug/ml) (72 hrs) | No Con A | Con A |
| 0 | 1.40% | 1.80% |
| 50 | 2.30% | 2.40% |
| 100 | 2.50% | 3.00% |
| 200 | 2.60% | 3.00% |
| 300 | 3.00% | 3.20% |
| 500 | 3.30% | 3.50% |
A key mechanism for immune suppression is apoptosis of overly aggressive effector T cells and defects in mucosal T-cell apoptosis are likely to be critical in the pathogenesis of colitis (1, 2). Therefore, as a penultimate measure that p53 is key to the AG-driven apoptosis of inflammatory cells that drive colitis, we isolated CD4+/CD25− effector T cells from p53+/+ and p53−/− mice. Four mice from each genotype were used. Table 4 and Supplementary Figure 2 shows that following exposure to AG for 24 hours, CD4+/CD25− effector T cells from p53+/+ undergo apoptosis. However, CD4+/CD25− effector T cells from p53−/− are resistant to apoptosis.
Table 4.
Percentage of cells from a representative experiment staining positive for Annexin V and negative for PI (early apoptotic cells) in CD4+/CD25− effector T cells from p53+/+ and p53−/− C57BL/6 mice.
| AG (ug/ml) | p53+/+ | p53−/− | ||
|---|---|---|---|---|
| (24 hrs) | No Con A | Con A | No Con A | Con A |
| 0 | 1.50% | 0.90% | 2.60% | 3.70% |
| 50 | 4.60% | 4.30% | 2.30% | 2.30% |
| 100 | 3.90% | 6.50% | 2.70% | 2.40% |
| 200 | 9.00% | 29.20% | 3.40% | 3.40% |
| 300 | 12.70% | 52.20% | 3.90% | 4.60% |
AG prevents mouse colitis in p53+/+ mice, but not in p53−/− mice
We have previously shown that AG can be used to prevent and treat mouse colitis (7). Here, we repeated such experiments, but concomitantly tested the ability of AG (again 75 ppm in chow) to prevent colitis in p53−/− mice. The DAI, which monitors weight loss, stool consistency, and blood in the stool as a measure of disease severity, was scored for each animal every other day. DAI increased in both p53+/+ and p53−/− consuming DSS and chow without AG. However, only p53+/+ mice consuming AG had a significantly lower DAI. Significance (p < 0.05) was reached at Day 21, and continued until the end of the experiment (Figure 2).
Fig. 2.

Effects of AG on the Disease Activity Index (DAI) in p53+/+ and p53−/− mice in the chronic dextran sulfate sodium (DSS)-colitis model. Mice received 2½ (7 days DSS followed by 7 days water per cycle) of DSS with and without AG in chow, as indicated. (◦, indicates p53−/− mice on DSS and on chow without AG; ●, indicates p53−/− mice on DSS and on chow with 75 ppm AG; □, indicates p53+/+ mice on DSS and on chow without AG; ■, indicates p53+/+ mice on DSS and on chow with 75 ppm AG). *, indicates significant difference from other groups (p < 0.005) from day 21 to the end of the experiment.
On day 35, the animals were sacrificed by cervical dislocation, and the entire colon was Swiss-Roled, formalin-fixed, sectioned, and examined histologically on a blinded basis. Histological examination of colon sections from the various treatment groups were consistent with the DAI endpoint, revealing marked damage in the DSS-alone groups (both p53+/+ and p53−/− mice). Figure 3 shows representative sections from each group. Importantly, it shows that AG protected from DSS-induced colitis in the p53+/+ mice, but not in the p53−/− mice. The panel shows a severely inflamed and damaged colon from DSS-treated animals (p53+/+ and p53−/− as indicated). Numerous neutrophils were present throughout the section, along with severely damaged crypts, and moderate to severe inflammatory infiltration with submucosal edema. A section from p53+/+ mice treated with AG + DSS shows a healthier colon, with minimal infiltrating inflammatory cells and largely intact crypts. However, AG was not protective in this way in p53−/− mice. Figure 3 also shows quantitative data reflecting these observations. The colons were graded for their histology score, which is based on inflammation severity, inflammation extent, crypt damage, and percentage of surface area demonstrating the characteristic. These morphologies were scored on a blinded basis by two separate investigators. As indicated in Figure 3, p53+/+ and p53−/− animals receiving DSS in their drinking water had substantially higher histology scores (representing moderate-to-severe IBD) than animals receiving normal drinking water. Protection by AG from damage was significant only in the p53+/+ genotype, but no in the p53−/− genotype.
AG stimulates apoptosis of lymphocytes in vivo
To examine the effects of AG on apoptosis in vivo, we carried out a TUNEL assay on serial sections used for quantifying inflammation (Figure 3). As shown in Figure 4, for the epithelial cells, there is a significantly higher IRS (i.e. TUNEL label) in both the p53+/+ and p53−/− mice treated with DSS, than in mice treated with water. However, the IRS in the epithelial cells only decreased in the p53+/+, but not the p53−/− mice when they were treated with both DSS + AG. This observation is consistent with the data from the inflammatory index (Figure 3), indicating that AG protects epithelial cells from DNA damage in vivo only when p53 is present. However, this observation was the opposite in the lymphocytes in the MLNs. Although there was an increase in IRS in the MLNs in both the p53+/+ and p53−/− mice when treated with DSS alone, AG exacerbated the apoptotic response only in the p53+/+ mice. Such results are consistent with our in vitro data, and with the hypothesis that AG drives apoptosis in inflammatory cells in vivo, providing a mechanism by which AG protects from colitis in this DSS mouse model.
Fig. 4.

Effects of AG on apoptosis in cells of the epithelium and the MLNs. (A) IRS (TUNEL staining) in epithelial cells of indicated groups. (B) IRS (TUNEL staining) in MLNs of indicated groups. (C) Representative staining in the epithelium of indicated groups. (D) Representative staining of MLNs in indicated groups.
Discussion
In this current study, we have identified the p53 pathway as key to the ability of AG to suppress colitis. Previous studies have shown that p53 is mutated in non-cancerous and pre-cancerous epithelial lesions of patients with chronic Ulcerative Colitis (UC) (9, 10, 38, 39). Studies performed on p53+/+ vs. p53−/− mice have shown that although the intermediate inflammatory index (i.e. colitis) is not significantly different between these two genotypes (26) (consistent with our findings; Figure 3), colitis-associated neoplasias are significantly elevated in p53−/− mice vs. p53+/+ mice (25, 26). Although this indicates that the level of colon inflammation may not play a key role in driving colon carcinogenesis in such models, we are currently examining whether AG has different capabilities to prevent colon cancer associated with colitis in p53+/+ vs. p53−/− mice.
Because of the complicated nature of AG, it is difficult to pin down the specific ingredients within the extract capable of interacting with p53. We feel it is important to present our present results on the entire AG extract. However, although beyond the scope of this current study, we are currently trying to identify possible mechanisms of p53 activation by AG, including the possibility of minor, targeted DNA damage, the induction of hypoxia, altered glucose metabolism, thymidylate synthase inhibition, interference with ribosome biogenesis, and cytokine stimulation (40, 41). Also, because the activation of p53 is associated with an increase in reactive oxygen species, facilitating the ability to selectively kill off p53 over-expressing cells (42, 43), it is possible that AG not only increases p53 in itself, but selectively kills off CD4+/CD25− effector T cells by facilitating the release of ROS from p53-over-expressing activated CD4+/CD25− effector T cells. Other future studies we are carrying out are bio-assay guided fractionation of AG to delineate ingredients active against colitis and capable of interacting with p53. In Human Arterial Smooth Muscle Cells, it has been demonstrated that Rg1 induces p53 transcription and increases p53 protein levels, thought to play a role in the inhibition of proliferation of such cells (44). Similarly, mRg2, a mixture of ginsenosides containing 60% of Rg2, was capable of inducing p53 and p21 in response to UVB exposure in NIH 3T3 cells, thereby protecting such cells from genotoxicity (45). Ming et al (24) showed that 0-O-(β-D-glucopyranosyl)-20(S)-protopanaxadiol (IH-901), a novel intestinal bacterial metabolite of ginseng protopanaxadiol saponins, up-regulates p53 and drives apoptosis through a mitochondrial pathway by increasing the levels of the pro-apoptotic molecule, bax, in hepatocellular carcinoma cells. Similarly, Huang et al (23) showed that a new triterpenoid isolated from Panax Ginseng, was potent in stimulating p53-mediated cell cycle arrest, leading to apoptosis via activation of the caspase signaling pathway in HepG2 cells. As well, ginsenosides -Rs4 and –Rs3, isolated from Panax ginseng can selectively elevate protein levels of p53 and p21WAF1 and induce apoptosis in SK-HEP-1 Human Hepatoma cells (46, 47). More recently, Wang et al. (22) showed that steaming AG increased Rg3 and Rh2 content, and AG was capable of inducing apoptosis of colon cancer cells (SW-480) though the mitochondrial pathway, involving the activation of p53. The finding that Rh2 isolated from AG was potent in the induction of apoptosis of colon cancer cells was confirmatory to a previous study showing similar findings (48).
Although others have reported an apoptotic effect of ginseng on blood cells (36), ours is the first study reporting a p53-dependent induction of apoptosis in isogenic lymphoblastoid and primary CD4+/CD25− primary T cells. Indeed, although there is relatively little induction of apoptosis in the lymphoblastoid cells (Table 2), these data provided useful initial data indicating a p53-dependent mechanism to apoptosis by AG. We recognized that the more relevant experiment is that which is presented in Table 4. Importantly, AG not only induces apoptosis in CD4+/CD25− effector T cells from p53+/+ mice, but preferentially induces apoptosis in stimulated CD4+/CD25− effector T cells from p53+/+ mice. It is recognized that CD4+ cells play a critical role in driving colitis in a DSS model (49), and that the lack of a capability to control apoptosis of CD4+/CD25− T cells is thought to play an intricate role in colitis (2). Because p53 plays a key role in apoptosis, it is reasonable that the ability of AG to drive apoptosis in activated p53+/+ CD4+/CD25− effector T cells in vitro, is a key mechanism towards its ability to protect from colitis in this genotype. Results shown here are consistent with this hypothesis. Importantly, we also show that AG drives apoptosis of inflammatory cells of the MLNs in vivo (Figure 4). The finding that DSS induces apoptosis of WT mice in the epithelium is consistent with other studies (50–52), and the understanding that DSS causes DNA damage to epithelial cells. The finding that apoptosis is reduced in the epithelium by AG only in the WT mice, is confirmatory that AG protects the epithelial tissues in a p53-dependent manner. Based on our in vitro data (Tables 2–4; Supplemental Figures 1 and 2), and our in vivo data (Figure 4), we suggest that this protection is at least in part due to the ability of AG to target the apoptosis of inflammatory cells.
Overall, we have presented data underpinning a key role for the p53 pathway in the protection from colitis by AG. Although p53 is mutated and the pathway is dysfunctional in patients with long-standing colitis, it is not completely clear whether this mutation is in epithelial cells and/or in inflammatory cells. A functional p53 pathway in inflammatory cells appears to be a necessary pre-requisite to the ability of AG to protect from colitis. In this regard, p53 levels are induced under inflammatory stress (7), as well as by exposure to AG under non-stressed conditions (Figure 1). In inflammatory conditions, we suggest AG has the capability to facilitate p53-driven apoptosis in inflammatory cells, thereby killing them off. Although we have no evidence to date, it would also be interesting to test the ability of AG to protect from p53 mutation in epithelial cells, thereby reducing the risk of colon cancer associated with colitis.
Fig. 1.
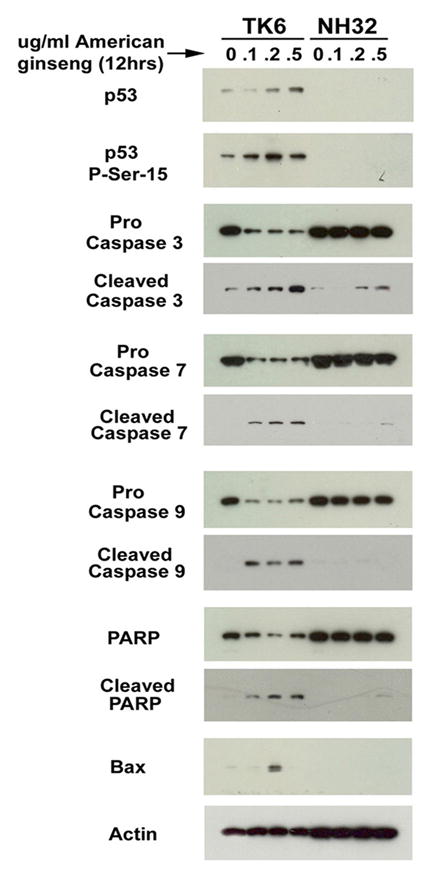
AG drives apoptosis of lymphoblasts through the p53 pathway. The TK6 human lymphoblast line, and NH32 isogenic p53 knock out cell line were cultured in RPMI + 10% FCS. Cells were exposed to indicated doses of AG for 12 hours, then harvested for protein. Following separation by 10% SDS-Page electrophoresis, gels were blotted onto nitrocellulose, then probed with the indicated antibodies. Results indicate that caspase 3, 7, 9 and PARP cleavage, and induction of Bax by AG are p53 dependent. p53 and phosphorylated p53 are also shown, indicating that p53 is activated by AG.
Supplementary Material
Supplementary Fig. 1. TK6 (p53+/+) but not NH32 (p53−/−) cells exposed to AG undergo apoptosis in vitro. TK6 and NH32 cells were exposed to vehicle (media) or indicated doses of AG dissolved in media for 24, 48, 72 hours. Annexin V assays were performed to determine apoptosis post treatments and analyzed using flow cytometry. TK6 (p53+/+) with AG had increased apoptosis compared with NH32 (p53−/−), indicating apoptosis driven by AG is p53-dependent. A minimum of 10,000 cells were counted from three separate plates for each time and dose indicated.
Supplementary Fig. 2. Primary CD4+/CD25− T-lymphocytes from p53+/+ mice exposed to AG undergo apoptosis (assessed by Annexin V staining). Primary CD4+/CD25− T-lymphocytes from p53−/− mice exposed to AG are resistant to apoptosis. CD4+/CD25− T-lymphocytes were isolated from the spleens of four different p53+/+ C57BL/6 mice or four different p53−/− C57BL/6 mice. Purified (>90% T-cell purity) non-activated or Con A-activated T lymphocytes (1 × 106) were exposed to vehicle (media) or indicated doses of AG dissolved in media for 24 hours. After culture for 24 hours, Annexin V assays were performed to determine apoptosis post treatments and analyzed using flow cytometry. Percentage of apoptotic cells are depicted in each histogram. CD4+/CD25− T cells from spleens of wild type C57BL/6 mice with AG had increased apoptosis compared with CD4+/CD25 T cells of p53−/− C57BL/6 mice, indicating p53 drives apoptosis of CD4+CD25− effector T cells in vitro. A minimum of 10,000 cells were counted from three separate plates for each time and dose indicated.
Acknowledgments
This work was supported by the Center for CAM Research on Autoimmune and Inflammatory Diseases, NIH grant 1P01AT003961-01A1 (PN, LJH, MN), and the COBRE funded University of South Carolina Center for Colon Cancer Research, NIH grant P20RR17698-01 (Franklin Berger, Director). Thanks also to the P20RR17698-01 Statistical Core (Dr. Edsel Pena, Director), and 1P01AT003961-01A1 Immunotoxicology Core (Dr. Narendra Singh, Director), P20RR17698-01 Mouse Core (Dr. Marj Pena, Director) and P20RR17698-01 Imaging/Histology Core.
References
- 1.Sartor RB. Mechanisms of disease: pathogenesis of Crohn’s disease and ulcerative colitis. Nat Clin Pract Gastroenterol Hepatol. 2006;3:390–407. doi: 10.1038/ncpgasthep0528. [DOI] [PubMed] [Google Scholar]
- 2.Neuman MG. Immune dysfunction in inflammatory bowel disease. Transl Res. 2007;149:173–186. doi: 10.1016/j.trsl.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 3.Hofseth LJ, Hussain SP, Harris CC. p53: 25 years after its discovery. Trends Pharmacol Sci. 2004;25:177–181. doi: 10.1016/j.tips.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 4.Lashner BA, Bauer WM, Rybicki LA, Goldblum JR. Abnormal p53 immunohistochemistry is associated with an increased colorectal cancer-related mortality in patients with ulcerative colitis. Am J Gastroenterol. 2003;98:1423–1427. doi: 10.1111/j.1572-0241.2003.07573.x. [DOI] [PubMed] [Google Scholar]
- 5.Alkim C, Savas B, Ensari A, et al. Expression of p53, VEGF, Microvessel Density, and Cyclin-D1 in Noncancerous Tissue of Inflammatory Bowel Disease. Dig Dis Sci. 2008;26:26. doi: 10.1007/s10620-008-0554-x. [DOI] [PubMed] [Google Scholar]
- 6.Hofseth LJ, Saito S, Hussain SP, et al. Nitric oxide-induced cellular stress and p53 activation in chronic inflammation. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:143–148. doi: 10.1073/pnas.0237083100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jin Y, Kotakadi VS, Ying L, et al. American ginseng suppresses inflammation and DNA damage associated with mouse colitis. Carcinogenesis. 2008;29:2351–2359. doi: 10.1093/carcin/bgn211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kotakadi VS, Jin Y, Hofseth AB, et al. Ginkgo biloba extract EGb 761 has anti-inflammatory properties and ameliorates colitis in mice by driving effector T cell apoptosis. Carcinogenesis. 2008;29:1799–1806. doi: 10.1093/carcin/bgn143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hussain SP, Amstad P, Raja K, et al. Increased p53 mutation load in noncancerous colon tissue from ulcerative colitis: a cancer-prone chronic inflammatory disease. Cancer Res. 2000;60:3333–3337. [PubMed] [Google Scholar]
- 10.Yoshida T, Mikami T, Mitomi H, Okayasu I. Diverse p53 alterations in ulcerative colitis-associated low-grade dysplasia: full-length gene sequencing in microdissected single crypts. J Pathol. 2003;199:166–175. doi: 10.1002/path.1264. [DOI] [PubMed] [Google Scholar]
- 11.Brockhaus F, Brune B. p53 accumulation in apoptotic macrophages is an energy demanding process that precedes cytochrome c release in response to nitric oxide. Oncogene. 1999;18:6403–6410. doi: 10.1038/sj.onc.1203058. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Y, Fong CC, Wong MS, et al. Molecular mechanisms of survival and apoptosis in RAW 264.7 macrophages under oxidative stress. Apoptosis. 2005;10:545–556. doi: 10.1007/s10495-005-1885-0. [DOI] [PubMed] [Google Scholar]
- 13.Sun L, Yang G, Zaidi M, Iqbal J. TNF-induced gene expression oscillates in time. Biochem Biophys Res Commun. 2008;371:900–905. doi: 10.1016/j.bbrc.2008.03.114. [DOI] [PubMed] [Google Scholar]
- 14.Choi KS, Song EK, Yim CY. Cytokines secreted by IL-2-activated lymphocytes induce endogenous nitric oxide synthesis and apoptosis in macrophages. J Leukoc Biol. 2008;83:1440–1450. doi: 10.1189/jlb.1007701. [DOI] [PubMed] [Google Scholar]
- 15.Kitts DD, Wijewickreme AN, Hu C. Antioxidant properties of a North American ginseng extract. Mol Cell Biochem. 2000;203:1–10. doi: 10.1023/a:1007078414639. [DOI] [PubMed] [Google Scholar]
- 16.Hofseth LJ. Ginseng and Cancer. Healthy Aging. 2006:53–58. [Google Scholar]
- 17.Hofseth LJ, Wargovich MJ. Inflammation, cancer, and targets of ginseng. J Nutr. 2007;137:183S–185S. doi: 10.1093/jn/137.1.183S. [DOI] [PubMed] [Google Scholar]
- 18.Hofseth LJ. Nitric oxide as a target of complementary and alternative medicines to prevent and treat inflammation and cancer. Cancer Lett. 2008;24:24. doi: 10.1016/j.canlet.2008.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park S, Yeo M, Jin JH, et al. Rescue of Helicobacter pylori-induced cytotoxicity by red ginseng. Dig Dis Sci. 2005;50:1218–1227. doi: 10.1007/s10620-005-2763-x. [DOI] [PubMed] [Google Scholar]
- 20.Volate SR, Davenport DM, Muga SJ, Wargovich MJ. Modulation of aberrant crypt foci and apoptosis by dietary herbal supplements (quercetin, curcumin, silymarin, ginseng and rutin) Carcinogenesis. 2005;26:1450–1456. doi: 10.1093/carcin/bgi089. [DOI] [PubMed] [Google Scholar]
- 21.Kang KA, Kim YW, Kim SU, et al. G1 phase arrest of the cell cycle by a ginseng metabolite, compound K, in U937 human monocytic leukamia cells. Arch Pharm Res. 2005;28:685–690. doi: 10.1007/BF02969359. [DOI] [PubMed] [Google Scholar]
- 22.Wang CZ, Li XL, Wang QF, et al. The mitochondrial pathway is involved in American ginseng-induced apoptosis of SW-480 colon cancer cells. Oncol Rep. 2009;21:577–584. doi: 10.3892/or_00000259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang J, Tang XH, Ikejima T, et al. A new triterpenoid from Panax ginseng exhibits cytotoxicity through p53 and the caspase signaling pathway in the HepG2 cell line. Arch Pharm Res. 2008;31:323–329. doi: 10.1007/s12272-001-1159-8. [DOI] [PubMed] [Google Scholar]
- 24.Ming YL, Song G, Chen LH, et al. Anti-proliferation and apoptosis induced by a novel intestinal metabolite of ginseng saponin in human hepatocellular carcinoma cells. Cell Biol Int. 2007;31:1265–1273. doi: 10.1016/j.cellbi.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 25.Fujii S, Fujimori T, Kawamata H, et al. Development of colonic neoplasia in p53 deficient mice with experimental colitis induced by dextran sulphate sodium. Gut. 2004;53:710–716. doi: 10.1136/gut.2003.028779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang WC, Coudry RA, Clapper ML, et al. Loss of p53 enhances the induction of colitis-associated neoplasia by dextran sulfate sodium. Carcinogenesis. 2007;28:2375–2381. doi: 10.1093/carcin/bgm134. [DOI] [PubMed] [Google Scholar]
- 27.Skopek TR, Liber HL, Penman BW, Thilly WG. Isolation of a human lymphoblastoid line heterozygous at the thymidine kinase locus: possibility for a rapid human cell mutation assay. Biochem Biophys Res Commun. 1978;84:411–416. doi: 10.1016/0006-291x(78)90185-7. [DOI] [PubMed] [Google Scholar]
- 28.Chuang YY, Chen Q, Brown JP, et al. Radiation-induced mutations at the autosomal thymidine kinase locus are not elevated in p53-null cells. Cancer Res. 1999;59:3073–3076. [PubMed] [Google Scholar]
- 29.Schneider U, Schwenk HU, Bornkamm G. Characterization of EBV-genome negative “null” and “T” cell lines derived from children with acute lymphoblastic leukemia and leukemic transformed non-Hodgkin lymphoma. Int J Cancer. 1977;19:621–626. doi: 10.1002/ijc.2910190505. [DOI] [PubMed] [Google Scholar]
- 30.Cheng J, Haas M. Frequent mutations in the p53 tumor suppressor gene in human leukemia T-cell lines. Mol Cell Biol. 1990;10:5502–5509. doi: 10.1128/mcb.10.10.5502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laumann R, Jucker M, Tesch H. Point mutations in the conserved regions of the p53 tumour suppressor gene do not account for the transforming process in the Jurkat acute lymphoblastic leukemia T-cells. Leukemia. 1992;6:227–228. [PubMed] [Google Scholar]
- 32.Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. Faseb J. 2007;17:17. doi: 10.1096/fj.07-9574LSF. [DOI] [PubMed] [Google Scholar]
- 33.Maines LW, Fitzpatrick LR, French KJ, et al. Suppression of ulcerative colitis in mice by orally available inhibitors of sphingosine kinase. Dig Dis Sci. 2008;53:997–1012. doi: 10.1007/s10620-007-0133-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krieglstein CF, Cerwinka WH, Laroux FS, et al. Role of appendix and spleen in experimental colitis. J Surg Res. 2001;101:166–175. doi: 10.1006/jsre.2001.6223. [DOI] [PubMed] [Google Scholar]
- 35.Denkert C, Koch I, von Keyserlingk N, et al. Expression of the ELAV-like protein HuR in human colon cancer: association with tumor stage and cyclooxygenase-2. Mod Pathol. 2006;19:1261–1269. doi: 10.1038/modpathol.3800645. [DOI] [PubMed] [Google Scholar]
- 36.Park SE, Park C, Kim SH, et al. Korean red ginseng extract induces apoptosis and decreases telomerase activity in human leukemia cells. J Ethnopharmacol. 2009;121:304–312. doi: 10.1016/j.jep.2008.10.038. [DOI] [PubMed] [Google Scholar]
- 37.Koo HN, Jeong HJ, Choi IY, et al. Mountain grown ginseng induces apoptosis in HL-60 cells and its mechanism have little relation with TNF-alpha production. Am J Chin Med. 2007;35:169–182. doi: 10.1142/S0192415X07004710. [DOI] [PubMed] [Google Scholar]
- 38.Takaku H, Ajioka Y, Watanabe H, et al. Mutations of p53 in morphologically non-neoplastic mucosa of long-standing ulcerative colitis. Jpn J Cancer Res. 2001;92:119–126. doi: 10.1111/j.1349-7006.2001.tb01073.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Holzmann K, Klump B, Borchard F, et al. Comparative analysis of histology, DNA content, p53 and Ki-ras mutations in colectomy specimens with long-standing ulcerative colitis. Int J Cancer. 1998;76:1–6. doi: 10.1002/(sici)1097-0215(19980330)76:1<1::aid-ijc1>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 40.Vousden KH. Outcomes of p53 activation--spoilt for choice. J Cell Sci. 2006;119:5015–5020. doi: 10.1242/jcs.03293. [DOI] [PubMed] [Google Scholar]
- 41.Lavin MF, Gueven N. The complexity of p53 stabilization and activation. Cell Death Differ. 2006;13:941–950. doi: 10.1038/sj.cdd.4401925. [DOI] [PubMed] [Google Scholar]
- 42.Polyak K, Xia Y, Zweier JL, et al. A model for p53-induced apoptosis. Nature. 1997;389:300–305. doi: 10.1038/38525. [DOI] [PubMed] [Google Scholar]
- 43.Hussain SP, Amstad P, He P, et al. p53-induced up-regulation of MnSOD and GPx but not catalase increases oxidative stress and apoptosis. Cancer Res. 2004;64:2350–2356. doi: 10.1158/0008-5472.can-2287-2. [DOI] [PubMed] [Google Scholar]
- 44.Zhang HS, Wang SQ. Ginsenoside Rg1 inhibits tumor necrosis factor-alpha (TNF-alpha)-induced human arterial smooth muscle cells (HASMCs) proliferation. J Cell Biochem. 2006;98:1471–1481. doi: 10.1002/jcb.20799. [DOI] [PubMed] [Google Scholar]
- 45.Jeong SJ, Han SH, Kim DY, et al. Effects of mRg2, a mixture of ginsenosides containing 60% Rg2, on the ultraviolet B-induced DNA repair synthesis and apoptosis in NIH3T3 cells. Int J Toxicol. 2007;26:151–158. doi: 10.1080/10915810701226370. [DOI] [PubMed] [Google Scholar]
- 46.Kim SE, Lee YH, Park JH, Lee SK. Ginsenoside-Rs4, a new type of ginseng saponin concurrently induces apoptosis and selectively elevates protein levels of p53 and p21WAF1 in human hepatoma SK-HEP-1 cells. Eur J Cancer. 1999;35:507–511. doi: 10.1016/s0959-8049(98)00415-8. [DOI] [PubMed] [Google Scholar]
- 47.Kim SE, Lee YH, Park JH, Lee SK. Ginsenoside-Rs3, a new diol-type ginseng saponin, selectively elevates protein levels of p53 and p21WAF1 leading to induction of apoptosis in SK-HEP-1 cells. Anticancer Res. 1999;19:487–491. [PubMed] [Google Scholar]
- 48.Popovich DG, Kitts DD. Mechanistic studies on protopanaxadiol, Rh2, and ginseng (Panax quinquefolius) extract induced cytotoxicity in intestinal Caco-2 cells. J Biochem Mol Toxicol. 2004;18:143–149. doi: 10.1002/jbt.20019. [DOI] [PubMed] [Google Scholar]
- 49.Shintani N, Nakajima T, Okamoto T, et al. Involvement of CD4+ T cells in the development of dextran sulfate sodium-induced experimental colitis and suppressive effect of IgG on their action. Gen Pharmacol. 1998;31:477–481. doi: 10.1016/s0306-3623(98)00004-4. [DOI] [PubMed] [Google Scholar]
- 50.Martinez JA, Williams CS, Amann JM, et al. Deletion of Mtgr1 sensitizes the colonic epithelium to dextran sodium sulfate-induced colitis. Gastroenterology. 2006;131:579–588. doi: 10.1053/j.gastro.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 51.Murai R, Kanbe T, Mukoyama T, et al. Effect of rectal administration of rebamipide on dextran sulfate sodium-induced colitis: role of hepatocyte growth factor. Inflamm Res. 2007;56:240–245. doi: 10.1007/s00011-007-6100-z. [DOI] [PubMed] [Google Scholar]
- 52.Nam SY, Kim JS, Kim JM, et al. DA-6034, a Derivative of Flavonoid, Prevents and Ameliorates Dextran Sulfate Sodium-Induced Colitis and Inhibits Colon Carcinogenesis. Exp Biol Med (Maywood) 2008;233:180–191. doi: 10.3181/0707-RM-186. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1. TK6 (p53+/+) but not NH32 (p53−/−) cells exposed to AG undergo apoptosis in vitro. TK6 and NH32 cells were exposed to vehicle (media) or indicated doses of AG dissolved in media for 24, 48, 72 hours. Annexin V assays were performed to determine apoptosis post treatments and analyzed using flow cytometry. TK6 (p53+/+) with AG had increased apoptosis compared with NH32 (p53−/−), indicating apoptosis driven by AG is p53-dependent. A minimum of 10,000 cells were counted from three separate plates for each time and dose indicated.
Supplementary Fig. 2. Primary CD4+/CD25− T-lymphocytes from p53+/+ mice exposed to AG undergo apoptosis (assessed by Annexin V staining). Primary CD4+/CD25− T-lymphocytes from p53−/− mice exposed to AG are resistant to apoptosis. CD4+/CD25− T-lymphocytes were isolated from the spleens of four different p53+/+ C57BL/6 mice or four different p53−/− C57BL/6 mice. Purified (>90% T-cell purity) non-activated or Con A-activated T lymphocytes (1 × 106) were exposed to vehicle (media) or indicated doses of AG dissolved in media for 24 hours. After culture for 24 hours, Annexin V assays were performed to determine apoptosis post treatments and analyzed using flow cytometry. Percentage of apoptotic cells are depicted in each histogram. CD4+/CD25− T cells from spleens of wild type C57BL/6 mice with AG had increased apoptosis compared with CD4+/CD25 T cells of p53−/− C57BL/6 mice, indicating p53 drives apoptosis of CD4+CD25− effector T cells in vitro. A minimum of 10,000 cells were counted from three separate plates for each time and dose indicated.