

Abstract
Recent studies have found that liver X receptors (LXRs) agonists decrease inflammation and possess neuroprotective properties. The aim of this study was to examine the mechanisms of liver X receptor agonist GW3965 on brain injury following global cerebral ischemia in the rat. The 48 male SD rats were randomly partitioned into three groups: sham, global ischemia (4-vessel occlusion for 15 minutes; 4VO) treated with vehicle and global ischemia treated with GW3965 (20mg/kg, via i.p at 10 minutes after reperfusion). The functional outcome was determined by neurological evaluation at 24 hours post ischemia and by testing rats in T maze at 3 and 7 days after reperfusion. The rats' daily body weight, incidence of seizures and 72 hours mortality were also determined. After Nissl staining and TUNEL in coronal brain sections, the numbers of intact and damaged cells were counted in the CA1 sector of the hippocampus. The expression of phosphorylated inhibitor of κB (p-IκBα), Nuclear Factor-κB (NF-κB) subunit p65, and cyclo-oxygenase-2 (COX-2) were analyzed with Western blot at 12 hours after reperfusion. GW3965 tended to reduce 72 hours mortality and the incidence of post-ischemic seizures. GW3965-treated rats showed an improved neuronal survivability in CA1 and a significant increase in the percentage of spontaneous alternations detected in T-maze on day 7 after ischemia.
GW3965-induced neuroprotection was associated with a significant reduction in nuclear translocation of NF-kB p65 subunit and a decrease in the hippocampal expression of NF-kB target gene, COX-2. LXR receptor agonist protects against neuronal damage following global cerebral ischemia. The mechanism of neuroprotection may include blockade of NF-κB activation and the subsequent suppression of COX-2 in the post ischemic brain.
Keywords: Global Cerebral Ischemia, Liver X Receptors, GW3965, NF-κB, Cyclo-oxygenase-2, Rats
Introduction
Global cerebral ischemia continues to be an important clinical problem with limited treatment options (Popp and Bottiger, 2006). It is a consequence of severe arrhythmia, cardiac arrest and hemorrhagic shock, leading to a delayed neuronal damage in the hippocampus and the cerebral cortex (Block, 1999). Global brain ischemia occurring intraoperatively is a major cause of cognitive and memory dysfunction (Fujioka et al., 2000). The inflammatory response, orchestrated by NF-κB and COX-2, is a crucial component of post-ischemic brain injury. In spite of the growing understanding of its role in delayed cell death, brain inflammation (Green and Shuaib, 2006) has been considerably under studied in global cerebral ischemia than in the focal ischemic stroke.
Liver X receptors (LXRs), LXRα and LXRβ, are nuclear receptors that belong to the large family of transcription factors. Considerable evidence has identified Liver X receptors as regulators of the inflammatory response both in human and rodents (Gabbi et al., 2009). LXR activation represses a set of inflammatory genes triggered by inflammatory (with LPS) and ischemic stimulation (Morales et al., 2008;Ninomiya et al., 2007;Wang et al., 2009). Amongst these genes, COX-2 appears to play a major role in neuronal damage after global cerebral ischemia (Xiang et al., 2007). Recent findings point towards Liver X receptors (LXR) as promising pharmacological targets for the treatment of focal ischemic stroke (Morales et al., 2008;Sironi et al., 2008). Two nonsteroidal synthetic compounds, GW3965 and T0314407, have been identified as potent and orally active agonists for both α and β LXRs (Collins et al., 2002;Schultz et al., 2000), expressed in cerebral tissues (Repa and Mangelsdorf, 2000). Both LXRα and LXRβ are expressed in the rat brain. LXRβ is particularly abundant in the hippocampus, including CA1 (Kainu et al., 1996;Whitney et al., 2002).
Although evidence suggest that LXR stimulation can combat brain inflammation involved in the pathophysiology of ischemic cell death, the treatment with LXR agonists has not been investigated in global cerebral ischemia. Studies showed that anti-inflammatory effect of LXR activation is mediated by transrepression of nuclear factor-κB (NF-κB) (Ghisletti et al., 2007;Joseph et al., 2003;Pascual et al., 2005). NF-κB governs the transcription of inflammatory genes after global cerebral ischemia (Ueno et al., 2001). Peroxisome proliferator-activated receptor (PPAR) agonists, which share many similarities with LXRs in intracellular signaling, can also effectively reduce NF-κB activation (Bright et al., 2008). It has been demonstrated in vitro that LXR agonists can reduce COX-2 expression via blocking NF-κB activation (Joseph et al., 2003). We hypothesized that the agonist of the LXR nuclear receptor GW3965 will deactivate NF-κB and thereby will prevent COX-2 upregulation to reduce neuronal damage following global cerebral ischemia.
Experimental procedures
Animal groups and the model of global cerebral ischemia
A total of forty eight male Sprague Dawley rats (300-350g, Harlan, Indianapolis, IN) were partitioned into three groups including sham surgery (n = 15) and two global ischemic groups: global ischemia treated with vehicle (GI, n = 23) or with GW3965 (GI+GW3965; n=10). Rats in global ischemic group were euthanized at 12 hours, and at 3 and 7 days after ischemia. The animals in global ischemia +GW3965 group were euthanized at 12 hours and 7days after the induction of ischemia. All surgical and euthanization procedures were performed under anesthesia with Ketamine (100 mg/kg) and Xylazine (10 mg/kg) administered via i.p. injection. The animals were intubated and mechanically ventilated during surgery. Atropine at a dose 0.05 mg/kg s.c. was given preoperatively to reduce secretion in the respiratory tract. The 4VO (Pulsinelli and Brierley, 1979) with one stage anterior approach established in our laboratory, was used to induce global cerebral ischemia (Ostrowski et al., 2008;Yamaguchi et al., 2005). Briefly, the skin was incised on the neck and the subcutaneous connective tissue and muscles were gently pulled aside. The trachea and the esophagus were gently retracted to the right side. Cervical vertebral bodies were exposed and both vertebral arteries were permanently occluded between the second and third transverse processes using electrosurgical coagulator. Next, both common carotid arteries (CCA) were occluded with microvascular clips for a period of 15 minutes. During surgery and for 2 hours into the recovery period rectal temperature was monitored and maintained at 36.9–37.2 °C by means of a heating lamp. Sham surgery included an exposure of common carotid and vertebral arteries. All animal procedures complied with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee (IACUC) at Loma Linda University.
Treatment with LXR agonist GW3965
Ten minutes after CCA reperfusion rats received intraperitoneal injection of GW3965 (20 mg/kg; n= 10) dissolved in dimethyl sulfoxide (10% DMSO in saline) used as a vehicle. Rats in the untreated global ischemic group received only the equivalent volume of vehicle i.p. Sham-operated animals also received i.p. vehicle injection after surgical exposure of the arteries.
Perfusion fixation and Nissl staining
For all histological studies, rats were perfused intracardially with 200 ml of cold PBS followed by 300 ml of cold buffered 10% formalin. Brains were post fixed for more than 7days in formalin at 4 °C, then cryoprotected in 30% sucrose/PBS until they sank(Guerra-Crespo et al., 2009). Frozen brain tissue block encompassing dorsal hippocampus was cut from Bregma − 3.3 mm to − 4.5 mm into 10μm thick sections as described previously (Ostrowski et al., 2005). To visualize neurons with Nissl staining, brain sections were dried, rehydrated and immersed in 0.5% cresyl violet for 2 minutes. After washing in water, the sections were dehydrated in graded alcohols, cleared in Xylenes and cover-slipped with Permount (Titova et al., 2008).
TUNEL
Brain sections were immersed in citric buffer, pH 6.0, heated with microwave oven for 15 minutes, and labeled with an In Situ Cell Death Detection Kit (Roche). A mixture of FITC-labeled nucleotides and terminal deoxynucleotidyl transferase was applied onto sections for 60 minutes at 37 °C in a dark humidified chamber as previously described (Matchett et al., 2007;Ostrowski et al., 2008). Incubation with labeling solution without the enzyme served as a negative control.
Quantitative histology
On days 3 and 7 rats were euthanized, the brains were collected and processed for Nissl staining and TUNEL. A total of 20 Nissl photomicrographs of CA1 per animal were taken at 100× for the Nissl cell counts. Eight photomicrographs of CA1 per animal were used for counting TUNEL-positive cells. The numbers of visual fields selected for cell counting were derived from the previously published procedures (Matchett et al., 2007). All histological preparations were analyzed by a blinded investigator with the aid of ImageJ software (NIH)24.
Neurobehavioral testing
Prior to euthanization on day 3 and 7 rats were tested for spontaneous alternation behavior on T-maze (Gerlai, 2001;Hughes, 2004) as previously described (Matchett et al., 2007). Tested animal was initially placed in the maze and allowed to explore it over the period of 1 minute. Then the animal was placed in the stem of T-maze for a total of 10 trials. The number of left and right choices was recorded. Based on the sequence of choices the rate of spontaneous alternations was calculated and expressed as a percent of alternations that were maximally achievable. The baseline of alternation was set at the level of 50%. To evaluate sensorimotor deficits at 24 hours after global ischemia, we used Garcia scoring system, modified for the evaluation of symmetrical neurologic deficits (Garcia et al., 1995;Kusaka et al., 2004).
Nuclear and cytoplasmic protein extraction
Animals were euthanized under general anesthesia and perfused with ice-cold PBS (200ml) via trans-cardiac approach at 12 hours after ischemia. The syringe perfusion took less than 30 seconds, and the aorta was clamped during the entire procedure. Even though protein alterations are unlikely to occur within such short period of time, ice cold PBS was used to further make sure that protein integrity was maintained. Brains were removed and the hippocampus was rapidly isolated on ice, snap frozen in liquid nitrogen and kept at -80°C until analyzed. Nuclear proteins were extracted with nuclear extraction kit (Millipore) as described (Choi et al., 2006). Briefly the tissue was cut up into very small pieces, homogenized in cytoplasmic lysis buffer, and spun at 250 × g for 5 minutes at 4°C. Cell pellet was resuspended in ice cold Cytoplasmic Lysis Buffer containing 0.5mM DTT and protease inhibitor cocktail (diluted 1/1000), incubated on ice for 15 minutes and centrifuged at 250 × g for 5 minutes at 4°C. After discarding supe rnatant cell pellet was resuspended in Cytoplasmic Lysis Buffer and centrifuged at 8,000 × g for 20 minutes at 4°C. The supernatant (cytosolic portion of the cell lysate) was aliquoted, snap-frozen and stored at -80°C. Nuclear pellet was resuspended in ice cold Nuclear Extraction Buffer containing 0.5mM DTT and 1/1000 protease inhibitor cocktail. Nuclear suspension was agitated at 4°C for 30-60 minutes and centrifuged a t 16,000 × g for 5 minutes at 4°C. The resultant supernatant (nuclear extract) was divided into aliquots and snap-frozen, then stored at −80°C until analysis.
Western blot analysis
Western blotting procedure followed standard protocols (Shimamura et al., 2006). Equal amounts (30 μg) of total protein were separated in 10% SDS–polyacrylamide gel electrophoresis (PAGE) and blotted onto cellulose membranes. Antibodies included goat p-IκB-α (Santa Cruz Biotech, diluted 1:200), rabbit anti-NF-κB p65 (Millipore AB1604, diluted 1:2000), goat anti-β actin (Santa Cruz Biotech, SC-1615, diluted 1:2000), anti-Histone H1 (Millipore, 05-457, diluted 1:1000) and appropriate secondary antibodies (1:2000). Protein bands were detected with chemiluminescent kit (Amersham Bioscience, NJ) and exposed to X-ray film (Kodak, Rochester, NY). Optical density of the bands was quantified in Image J software, and normalized with respect to beta actin or Histone H1 as well as to sham (Yatsushige et al., 2007).
Statistical analysis
Data are expressed as mean ± SEM. One-way ANOVA followed by Tukey test was used to analyze inter-group differences of means except for the neurobehavioral scores, analyzed with Kruskal Wallis ANOVA and Dunn's test. Chi square test was used to analyze mortality rates and the incidence of seizures. P level <0.05 was considered statistically significant.
Results
Body weight, mortality and neurobehavioral scores
All groups of rats demonstrated body weight loss during the initial two days (Figure 1A). Body weight in global ischemia +GW3965 group showed a tendency for greater decrease and sooner recovery than in the vehicle-treated global ischemic group.
Figure 1.

(A) A decrease in body weight was noted in each group (sham n=11; global ischemia n = 10, global ischemia + GW3965 n = 5). While this decrease was equivalent in all groups, the body weight returned to preischemic levels only in the sham group on day 7. (B) The incidence of post ischemic seizures tended to be attenuated with GW3965 treatment. (C) The mortality showed a tendency toward a reduction amongst rats treated with GW3965.
The incidence of seizures in global ischemia group without treatment was 21.05% (4/19), while it was 16.67% (1/6) in the treatment group (non-significant, chi-square; Figure 2B). Sham-operated animals did not exhibit seizure activity, which in the affected rats presented as fully developed tonic-clonic seizures.
Figure 2.
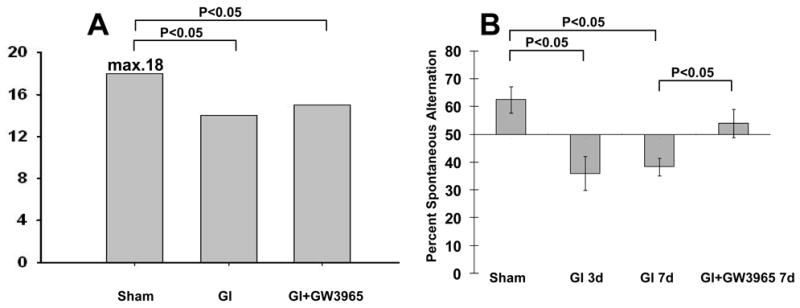
(A) A slight but significant deterioration of neurological score was detected at 24 hours after global ischemia, regardless with or without treatment (p<0.05) (sham n=11; global ischemia = 10, global ischemia + GW3965 n = 5). (B) T-maze test demonstrated worse performance of untreated rats compared with GW3965-treated rats at 3 and 7 days after ischemia. GW3965 treatment resulted in a recovery of normal alternating behavior at 7 days after ischemia (sham animals on day 3: n = 4; global ischemia on day 3: n = 6; global ischemia on day 7: n = 5; global ischemia with GW3965 treatment on day 3: n = 5).
The mortality at 72 hours was 0% (0/15) in the sham group, 26.31% (5/19) in the global ischemia group, and 16.07% (1/6) in the global ischemia +GW3965 treatment group (Figure 1C). Most deaths occurred within the first 48 hours after ischemic insult. Chi-square, however, did not detect significant differences in mortality rates between analyzed groups of animals.
The results of neurological testing are presented as median scores (Figure 2A). Sham animals showed no detectable neurological deficit during whole observation period. The neurological scores after global ischemia were significantly lower compared with the score in the sham group (p<0.05). In the global ischemia group median neurological score was 14 while it was 15 in the treatment group. The difference between these groups, however, did not reach statistical significance.
During testing on T-maze the rats showed a significant decrease in percent spontaneous alternations; to 36.00±6.00% on day 3 after global ischemia, and 38.33±3.07% on day 7, compared to the sham operated controls; 62.5± 4.79% (Figure 2B). GW3965-treated rats showed significant improvement in the percent of spontaneous alternations on day 7 (54.00 ±5.10%) compared with vehicle-injected rats. This rate of percent alternations was statistically equivalent with that in the sham group.
Nissl stain and TUNEL
Nissl staining demonstrated the presence of intact cells in the hippocampus of sham operated rats (Figure 3A1). The numerous dark-stained neurons, displaying shrunken nucleus and condensed cytoplasm, were observed in CA1 on day 3 after vehicle-treated ischemia (Figure 3A2). On day 7 the neuronal damage was more severe compared to day 3 after untreated global ischemia (Figure 3A3). At this time point cell loss and ghost cells were noted in CA1 hippocampal sector. Treatment with GW3965 attenuated CA1 cell death on day 7 compared with vehicle treatment (Figure 3A4). The multiple TUNEL-positive cells were present in CA1 on day 3 (Figure 4A2) and day 7 (Figure 4A3) after ischemia. Treatment with GW3965 reduced the extent of TUNEL positivity in the CA1 sector of the hippocampus (Figure 4A4).
Figure 3.
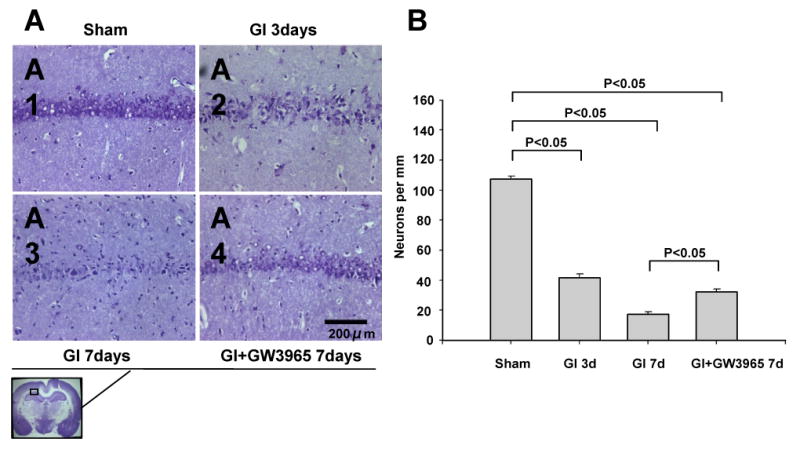
(A) Nissl stain revealed a neuronal damage present in the CA1 region progressive over 3-7 days after occlusion. The protection of CA1 neurons could be seen with GW3965 treatment on day 7. Scale bar indicates 200 μm. (B) Counting intact CA-1 neurons revealed a significantly higher neuronal survivability of hippocampal neurons in global ischemia +GW3965 group than in the global ischemia group on day 7 (day 3: sham n = 4, global ischemia = 5; day 7: global ischemia = 5, global ischemia + GW3965 n = 5).
Figure 4.
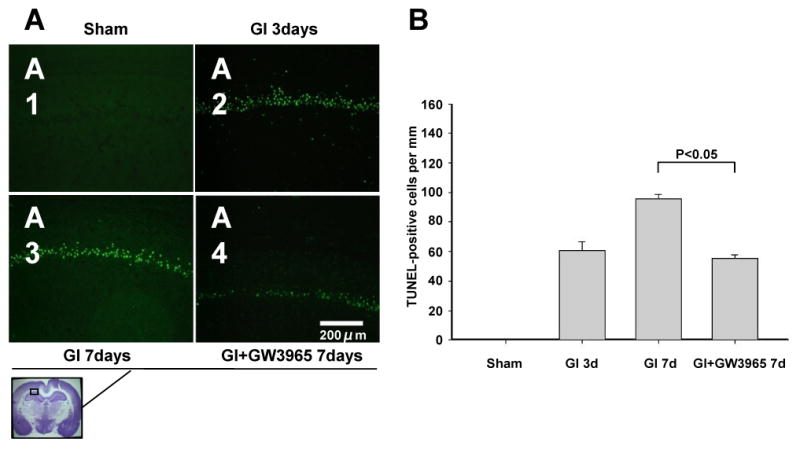
(A) Microphotographs of TUNEL preparations demonstrate that the neurons in the pyramidal layer of CA1 undergo apoptosis, present both on day 3 and 7 after ischemia. GW3965 effectively reduced apoptotic cell death in hippocampal sector. Scale bar indicates 200 μm. (B) Quantitative cell count confirmed that treatment with GW3965 reduced number of TUNEL positive cells in CA1 on day 7 after global cerebral ischemia (day 3: sham n = 4, global ischemia = 5; day 7: global ischemia = 5, global ischemia + GW3965 n = 5).
Quantitative cell count
In the sham group the number of CA1 neurons per mm was 107.17±18.57 (Figure 3B). Marked reduction in numbers of intact neurons was found on day 3 and day 7 after ischemia, 41.42 ± 22.22 and 17.39 ± 13.94, respectively (p<0.05). On day 7 in the treatment group 32.04 ± 19.15 neurons per mm were found. This level was significantly higher than in the vehicle-treated global ischemia group (by 83.19 per cent), although still significantly lower than in the sham group (29.90% of sham level).
In the sham group, TUNEL positive cells were not detectable (Figure 4B). On day 3 after global ischemia a number of 63.45 ± 37.29 TUNEL-positive cells per mm was calculated. The number of TUNEL positive cells per mm increased to 90.98 ± 23.10 on day 7. Treatment with GW3965 reduced the number of TUNEL positive cells by 37.81%, to the level of 56.58 ± 30.05 cells per mm of CA1 length.
Levels of pIκBα and subcellular localization of p65 subunit of NF-κB
Western blot analysis showed that cytoplasmic p65 decreased 4.38 fold in hippocampus at 12 hours after global ischemia (Figure 5A). GW3965 treated animals, however, demonstrated p65 level equivalent with that of sham animals. In the nuclear fraction, p65 significantly increased after untreated global ischemia, by 7.75-fold (Figure 5B). GW3965 attenuated this increase, by 57.55% so that the level of p65 in the nucleus was 3.86-fold higher than in the sham group (p<0.05).
Figure 5.
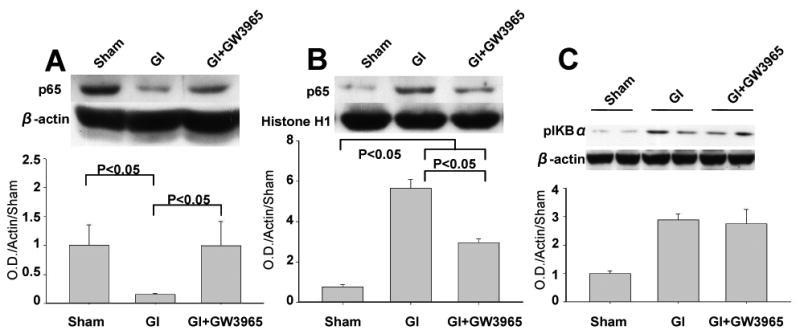
(A) The level of NF-κB p65 in the cytoplasm dissipated at 12 hours after global brain ischemia but was maintained in the global ischemia + GW3965 group (sham n=4; global ischemia = 4, global ischemia + GW3965 n=4). (B) The postichemic increase in p65 level was noted in the nucleus. The attenuated increase of p65 level was detected in global ischemia +GW3965 group compared with global ischemia group (sham n=4; global ischemia = 4, global ischemia + GW3965 n=4). (C) The levels of pIκBα tended to increase after ischemia irrespective of GW3965 treatment (sham n=4; global ischemia = 4, global ischemia + GW3965 n= 4).
Immunoblot analysis of pIKBα showed a trend towards increase compared with sham group, which was equivalent between the global ischemia and global ischemia +GW3965 treatment groups at 12 hours after ischemia.
COX-2 protein expression
COX-2 protein expression increased 4.29-fold after ischemia compared to COX-2 level in the sham group at 12 hours (Figure 6). The increase in COX-2 was reduced by 81.28 % with GW3965 treatment so that the reduced level of COX-2 was statistically equivalent with the level in the sham group.
Figure 6.
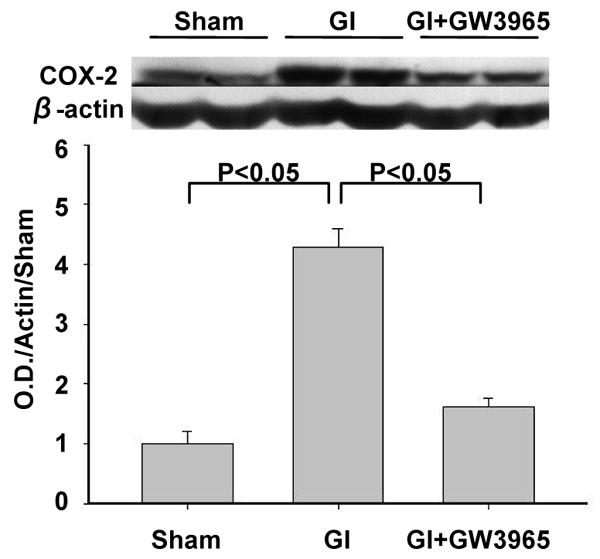
Global ischemia markedly elevated COX-2 protein level in the hippocampus at 12 hours after ischemia. GW3965 treatment reduced postischemic elevation of COX-2 level (sham n=4; global ischemia n = global ischemia, global ischemia + GW3965 n = 4).
Discussion
The 24quantitation of Nissl staining and TUNEL showed that GW3965 provides a robust histological protection of selectively vulnerable CA1 hippocampus region following global cerebral ischemia. Prior to reducing neuronal damage, LXR agonist prevented nuclear translocation of NF-κB subunit p65. The reduced activation of NF-κB was associated with a decreased expression of its target gene, COX-2.
GW3965 tended to decrease the mortality and incidence of epilepsy following global cerebral ischemia in the rat. GW3956 had no detectable effect of on neurological scores however it improved functional performance on T-maze compared with the vehicle-treated group. This finding indicates that GW3965 may protect against deterioration of working or short memory, caused by global cerebral ischemia (Matchett et al., 2007).
We selected the dose of LXR agonist based on the results of the previous study employing GW3965 in the treatment of focal ischemic stroke (Morales et al., 2008). In that study experimenters found that the dose of 20 mg/kg was associated with the greatest reduction of histological injury following MCAO. For this reason we adopted the same dose in our study. It is has been recently demonstrated that GW3965 has a high specificity for LXRs (Wang et al., 2009).
Morales et al. were first to report about effects and mechanism of LXR agonists (GW3965 and TO901317) in the rat and mouse middle cerebral artery occlusion models (Morales et al., 2008). The authors demonstrated that both agonists provided histological protection against focal cerebral ischemia. However, the use of GW3965 against global ischemia identifies several points of novelty in this present study. First, it is important in the translational research that investigational stroke treatments are tested with use of different species and different models of stroke. The consistent results coming from several laboratories also strengthen the evaluation of investigational drugs. Second, this study for the first time reveals that GW3965 treatment improves neurocognitive outcomes after cerebral ischemia. Third, we were able to determine that NF-κB suppression with GW3965 treatment holds several characteristics specific for global ischemia setting. Forth, this study also shows that COX-2 can be regulated by LXR agonism after global brain insult. This may point toward usability of this class of agents as an alternative anti-inflammatory treatment for brain injury after global ischemia.
We used the four-vessel occlusion rat model using one-stage anterior approach, established in our laboratory (Yamaguchi et al., 2005). We found robust neurobehavioral deficits and morphological changes in the cerebral cortex, white matter, thalamus, CA1, CA2, CA3, and dentate gyrus. Studies have shown that selective neuronal damage develops between 48-72 hours following global brain ischemia and that the destruction of CA1 pyramidal cells is complete or near complete after 7 days (Lipton, 1999). Therefore we imply that our study design adheres to the timing of neuronal injury following global brain ischemia (Block, 1999). The ischemic changes in our modified global ischemic model are consistent with the lesions produced by the original 4VO (Pulsinelli and Brierley, 1979). The occurrence of delayed apoptotic cell death in the CA1 sector of the hippocampus is also in accordance with earlier studies of global ischemic insult (Magnoni et al., 2004). Therefore we wish to imply that a significant reduction of brain injury and functional improvement in the treatment group were caused by GW3965 treatment. Overall our data may suggest that intraperitoneally administered GW3965 reduced NF-κB activation which diminished inflammatory gene expression in the brain and thereby led to reduced neuronal damage.
It has been demonstrated that the anti-inflammatory action of LXR agonists may be mediated by transrepression of NF-κB (Ghisletti et al., 2007;Joseph et al., 2003). The nonsteroidal GW3965, a LXR full agonist on both LXRα and LXRβ, (Collins et al., 2002) can cross the blood-brain barrier and exert specific actions on LXR receptors. In this study we therefore investigated whether LXR agonist can protect the brain through reducing NF-κB, activated after global ischemic insult (Ji et al., 2008;Rehni et al., 2009;Webster et al., 2009). We have found that LXR agonist has no effect on the phosphorylated IκBα, the major inhibitor of NF-κB (Zhang et al., 2005). Instead we have found that GW3965 blocked postischemic reduction of p65 in the cytoplasm and attenuated the increase of p65 level in the nucleus. This indicates that LXR agonist prevented translocation of p65 from the cytoplasm to the nucleus (Collino et al., 2006) and thereby reduced NF-κB activation. With this respect the mechanism of NF-κB inhibition appears to be distinct from that found in focal cerebral ischemia. In the latter, GW3965 did not affect p65 translocation although it reduced levels of IκBα, which is the NF-κB inhibitor and also its target gene (Morales et al., 2008). Based on this finding we suggested that LXR-induced neuroprotection is accounted for by the inhibition of NF-κB transcriptional activity. Other studies however suggest that LXR agonists can inhibit the nuclear translocation of p65 and/or enhance p65 nuclear export (Chang et al., 2007).
Interestingly, GW3965 reduced nuclear localization of p65 relatively early (12 hours) after ischemia. One study by earlier authors found that the antioxidant-induced neuroprotection was associated with late (72 hours) but not early (24 hours) inhibition of NF-κB nuclear translocation (Clemens et al., 1998). This may indicate more potent and more specific impact of LXR agonists on NF-κB signaling upon cerebral ischemia. The reduced NF-κB activation in this present study might lead to a decrease in COX-2 expression and lessened brain inflammation. The early increase of COX-2 levels is consistent with reported upregulation of other NF-κB target genes at 3-24 hours after global ischemic insult (Zhu et al., 2006). Deleterious role of COX-2 in the global ischemic injury was demonstrated by Xiang and colleagues (Xiang et al., 2007) who found aggravated brain damage in mice carrying COX-2 transgene. COX-2 is a primary source of free radicals in ischemic brain (Candelario-Jalil et al., 2003). More importantly, the increase in the COX-2 product, PGE2 directly preceded the onset of histopathological changes in the hippocampus following global cerebral ischemia (Candelario-Jalil et al., 2003). It has been demonstrated that selective COX-2 inhibitors as well as COX-2 gene disruption reduced cell damage in CA1 after global ischemia (Sasaki et al., 2004). We therefore postulate that the reduction of COX-2 level, observed in our study, might contribute to GW3965-induced neuroprotection. Further studies are however needed to better understand effects of LXR activation on inflammatory response after global cerebral ischemia.
Generalized convulsive seizures observed in this study might induce hippocampal damage and thereby could influence the results. However, as GW3965 tended to exert a beneficial effect on seizure incidence we wanted to include this clinically relevant aspect of our study. We postulate that stroke research (especially employing global ischemia) should more commonly include epilepsy as an outcome measure. Although we did not use video monitoring for detection of seizures, the observational time gap spanned only over a couple of hours in the night. If animals convulsed at night the chips from cage bedding were invariably scattered on the floor around the cage. This “signs” although crude, has 100% sensitivity for generalized seizures (and specificity, considering no activity of animal care personnel at night). Therefore it was still possible to determine whether an individual exhibited nocturnal seizure activity. It needs to be mentioned that in the majority of experimental stroke studies video EEG is as yet rare. For this reason the reported seizure frequencies after stroke are though to be underestimated (Karhunen et al., 2005).
There are several limitations of this study. Theoretically the smaller sample size in the treatment group might negatively impact power of used statistical tests. However, we detected robust differences in the investigated parameters between groups and verified that statistical power was above the desired level of 0.8. In our analysis, we used SigmaStat software, which does not require equal sample sizes in the groups being compared.
Our study demonstrates that only one dose GW3965 treatment produced neuroprotection. The agonist was administered in a critical time for the development of cell death program, which starts acutely after global cerebral ischemia (Ostrowski et al., 2008). Therefore it appears that the application of a single dose of GW3965 in the acute post-ischemic phase may be both required and sufficient for the therapeutic effect.
Future studies should seek to define the effect of LXR agonists on non-neuronal cell survivability. Determination of the therapeutic window of opportunity for LXR agonists after global cerebral ischemia also warrants further invesigations. Morales et al. showed that LXR agonists administered 10 minutes or 1 hour after MCAO improved stroke outcomes (Morales et al., 2008). Global cerebral ischemia however may be associated with a relatively large therapeutic window due to prolonged brain injury maturation.
The anti-inflammatory profile and efficacy of LXR agonists should be further determined. In face of serious side effects associated with the use of COX inhibitors, LXR agonists may become alternative treatment of choice. Further studies are also needed to find out whether these compounds may combat negative effects of inflammation on neurogenesis (Ekdahl et al., 2003).
We conclude that GW3965 protects against neuronal damage following global cerebral ischemia and that the reduction of NF-κB activation may underlie this beneficial effect. Our results indicate that the administration of LXR agonists may become a novel therapeutic approach for global cerebral ischemia.
Acknowledgments
Work was partially supported by grants of NS43338 and NS53407 from NIH to JH Zhang.
Comprehensive list of abbreviations
- 4-VO
four-vessel occlusion
- CA1
Cornu Ammonis area 1
- CCA
common carotid artery
- COX-2
cyclo-oxygenase-2
- DTT
1,4-dithio-DL-threitol
- LXRs
liver X receptors
- MCAO
middle cerebral artery occlusion
- NF-κB
nuclear factor-κB
- PAGE
polyacrylamide gel electrophoresis
- PBS
phosphate buffered saline
- PPARs
peroxisome proliferator-activated receptors
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
Footnotes
Conflicts of interest disclosure:
Oumei Cheng: None
Robert Ostrowski: None
John Zhang: Grant Support as mentioned above.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Block F. Global ischemia and behavioural deficits. Prog Neurobiol. 1999;58:279–295. doi: 10.1016/s0301-0082(98)00085-9. [DOI] [PubMed] [Google Scholar]
- Bright JJ, Kanakasabai S, Chearwae W, Chakraborty S. PPAR Regulation of Inflammatory Signaling in CNS Diseases. PPAR Res. 2008;2008:658520. doi: 10.1155/2008/658520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candelario-Jalil E, Gonzalez-Falcon A, Garcia-Cabrera M, Alvarez D, Al Dalain S, Martinez G, Leon OS, Springer JE. Assessment of the relative contribution of COX-1 and COX-2 isoforms to ischemia-induced oxidative damage and neurodegeneration following transient global cerebral ischemia. J Neurochem. 2003;86:545–555. doi: 10.1046/j.1471-4159.2003.01812.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L, Zhang Z, Li W, Dai J, Guan Y, Wang X. Liver-X-receptor activator prevents homocysteine-induced production of IgG antibodies from murine B lymphocytes via the ROS-NF-kappaB pathway. Biochem Biophys Res Commun. 2007;357:772–778. doi: 10.1016/j.bbrc.2007.04.016. [DOI] [PubMed] [Google Scholar]
- Choi SH, Langenbach R, Bosetti F. Cyclooxygenase-1 and -2 enzymes differentially regulate the brain upstream NF-kappa B pathway and downstream enzymes involved in prostaglandin biosynthesis. J Neurochem. 2006;98:801–811. doi: 10.1111/j.1471-4159.2006.03926.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens JA, Stephenson DT, Yin T, Smalstig EB, Panetta JA, Little SP. Drug-induced neuroprotection from global ischemia is associated with prevention of persistent but not transient activation of nuclear factor-kappaB in rats. Stroke. 1998;29:677–682. doi: 10.1161/01.str.29.3.677. [DOI] [PubMed] [Google Scholar]
- Collino M, Aragno M, Mastrocola R, Gallicchio M, Rosa AC, Dianzani C, Danni O, Thiemermann C, Fantozzi R. Modulation of the oxidative stress and inflammatory response by PPAR-gamma agonists in the hippocampus of rats exposed to cerebral ischemia/reperfusion. Eur J Pharmacol. 2006;530:70–80. doi: 10.1016/j.ejphar.2005.11.049. [DOI] [PubMed] [Google Scholar]
- Collins JL, Fivush AM, Watson MA, Galardi CM, Lewis MC, Moore LB, Parks DJ, Wilson JG, Tippin TK, Binz JG, Plunket KD, Morgan DG, Beaudet EJ, Whitney KD, Kliewer SA, Willson TM. Identification of a nonsteroidal liver X receptor agonist through parallel array synthesis of tertiary amines. J Med Chem. 2002;45:1963–1966. doi: 10.1021/jm0255116. [DOI] [PubMed] [Google Scholar]
- Ekdahl CT, Claasen JH, Bonde S, Kokaia Z, Lindvall O. Inflammation is detrimental for neurogenesis in adult brain. Proc Natl Acad Sci U S A. 2003;100:13632–13637. doi: 10.1073/pnas.2234031100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujioka M, Nishio K, Miyamoto S, Hiramatsu KI, Sakaki T, Okuchi K, Taoka T, Fujioka S. Hippocampal damage in the human brain after cardiac arrest. Cerebrovasc Dis. 2000;10:2–7. doi: 10.1159/000016018. [DOI] [PubMed] [Google Scholar]
- Gabbi C, Warner M, Gustafsson JA. Minireview: liver X receptor beta: emerging roles in physiology and diseases. Mol Endocrinol. 2009;23:129–136. doi: 10.1210/me.2008-0398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia JH, Wagner S, Liu KF, Hu XJ. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke. 1995;26:627–634. doi: 10.1161/01.str.26.4.627. [DOI] [PubMed] [Google Scholar]
- Gerlai R. Behavioral tests of hippocampal function: simple paradigms complex problems. Behav Brain Res. 2001;125:269–277. doi: 10.1016/s0166-4328(01)00296-0. [DOI] [PubMed] [Google Scholar]
- Ghisletti S, Huang W, Ogawa S, Pascual G, Lin ME, Willson TM, Rosenfeld MG, Glass CK. Parallel SUMOylation-dependent pathways mediate gene- and signal-specific transrepression by LXRs and PPARgamma. Mol Cell. 2007;25:57–70. doi: 10.1016/j.molcel.2006.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green AR, Shuaib A. Therapeutic strategies for the treatment of stroke. Drug Discov Today. 2006;11:681–693. doi: 10.1016/j.drudis.2006.06.001. [DOI] [PubMed] [Google Scholar]
- Guerra-Crespo M, Gleason D, Sistos A, Toosky T, Solaroglu I, Zhang JH, Bryant PJ, Fallon JH. Transforming growth factor-alpha induces neurogenesis and behavioral improvement in a chronic stroke model. Neuroscience. 2009;160:470–483. doi: 10.1016/j.neuroscience.2009.02.029. [DOI] [PubMed] [Google Scholar]
- Hughes RN. The value of spontaneous alternation behavior (SAB) as a test of retention in pharmacological investigations of memory. Neurosci Biobehav Rev. 2004;28:497–505. doi: 10.1016/j.neubiorev.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Ji L, Nazarali AJ, Paterson PG. Protein-energy malnutrition increases activation of the transcription factor, nuclear factor kappaB, in the gerbil hippocampus following global ischemia. J Nutr Biochem. 2008;19:770–777. doi: 10.1016/j.jnutbio.2007.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph SB, Castrillo A, Laffitte BA, Mangelsdorf DJ, Tontonoz P. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat Med. 2003;9:213–219. doi: 10.1038/nm820. [DOI] [PubMed] [Google Scholar]
- Kainu T, Kononen J, Enmark E, Gustafsson JA, Pelto-Huikko M. Localization and ontogeny of the orphan receptor OR-1 in the rat brain. J Mol Neurosci. 1996;7:29–39. doi: 10.1007/BF02736846. [DOI] [PubMed] [Google Scholar]
- Karhunen H, Jolkkonen J, Sivenius J, Pitkanen A. Epileptogenesis after experimental focal cerebral ischemia. Neurochem Res. 2005;30:1529–1542. doi: 10.1007/s11064-005-8831-y. [DOI] [PubMed] [Google Scholar]
- Kusaka G, Ishikawa M, Nanda A, Granger DN, Zhang JH. Signaling pathways for early brain injury after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2004;24:916–925. doi: 10.1097/01.WCB.0000125886.48838.7E. [DOI] [PubMed] [Google Scholar]
- Magnoni S, Baker A, George SJ, Duncan WC, Kerr LE, McCulloch J, Horsburgh K. Differential alterations in the expression and activity of matrix metalloproteinases 2 and 9 after transient cerebral ischemia in mice. Neurobiol Dis. 2004;17:188–197. doi: 10.1016/j.nbd.2004.07.020. [DOI] [PubMed] [Google Scholar]
- Matchett GA, Calinisan JB, Matchett GC, Martin RD, Zhang JH. The effect of granulocyte-colony stimulating factor in global cerebral ischemia in rats. Brain Res. 2007;1136:200–207. doi: 10.1016/j.brainres.2006.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales JR, Ballesteros I, Deniz JM, Hurtado O, Vivancos J, Nombela F, Lizasoain I, Castrillo A, Moro MA. Activation of liver X receptors promotes neuroprotection and reduces brain inflammation in experimental stroke. Circulation. 2008;118:1450–1459. doi: 10.1161/CIRCULATIONAHA.108.782300. [DOI] [PubMed] [Google Scholar]
- Ninomiya Y, Yasuda T, Kawamoto M, Yuge O, Okazaki Y. Liver X receptor ligands inhibit the lipopolysaccharide-induced expression of microsomal prostaglandin E synthase-1 and diminish prostaglandin E2 production in murine peritoneal macrophages. J Steroid Biochem Mol Biol. 2007;103:44–50. doi: 10.1016/j.jsbmb.2006.07.009. [DOI] [PubMed] [Google Scholar]
- Ostrowski RP, Colohan AR, Zhang JH. Mechanisms of hyperbaric oxygen-induced neuroprotection in a rat model of subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2005;25:554–571. doi: 10.1038/sj.jcbfm.9600048. [DOI] [PubMed] [Google Scholar]
- Ostrowski RP, Graupner G, Titova E, Zhang J, Chiu J, Dach N, Corleone D, Tang J, Zhang JH. The hyperbaric oxygen preconditioning-induced brain protection is mediated by a reduction of early apoptosis after transient global cerebral ischemia. Neurobiol Dis. 2008;29:1–13. doi: 10.1016/j.nbd.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, Willson TM, Rosenfeld MG, Glass CK. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature. 2005;437:759–763. doi: 10.1038/nature03988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp E, Bottiger BW. Cerebral resuscitation: state of the art, experimental approaches and clinical perspectives. Neurol Clin. 2006;24:73–87. vi. doi: 10.1016/j.ncl.2005.10.008. [DOI] [PubMed] [Google Scholar]
- Pulsinelli WA, Brierley JB. A new model of bilateral hemispheric ischemia in the unanesthetized rat. Stroke. 1979;10:267–272. doi: 10.1161/01.str.10.3.267. [DOI] [PubMed] [Google Scholar]
- Rehni AK, Bhateja P, Singh N. Diethyl dithiocarbamic acid, a possible nuclear factor kappa B inhibitor, attenuates ischemic postconditioning-induced attenuation of cerebral ischemia-reperfusion injury in mice. Can J Physiol Pharmacol. 2009;87:63–68. doi: 10.1139/Y08-100. [DOI] [PubMed] [Google Scholar]
- Repa JJ, Mangelsdorf DJ. The role of orphan nuclear receptors in the regulation of cholesterol homeostasis. Annu Rev Cell Dev Biol. 2000;16:459–481. doi: 10.1146/annurev.cellbio.16.1.459. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Kitagawa K, Yamagata K, Takemiya T, Tanaka S, Omura-Matsuoka E, Sugiura S, Matsumoto M, Hori M. Amelioration of hippocampal neuronal damage after transient forebrain ischemia in cyclooxygenase-2-deficient mice. J Cereb Blood Flow Metab. 2004;24:107–113. doi: 10.1097/01.WCB.0000100065.36077.4A. [DOI] [PubMed] [Google Scholar]
- Schultz JR, Tu H, Luk A, Repa JJ, Medina JC, Li L, Schwendner S, Wang S, Thoolen M, Mangelsdorf DJ, Lustig KD, Shan B. Role of LXRs in control of lipogenesis. Genes Dev. 2000;14:2831–2838. doi: 10.1101/gad.850400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamura N, Matchett G, Solaroglu I, Tsubokawa T, Ohkuma H, Zhang J. Inhibition of integrin alphavbeta3 reduces blood-brain barrier breakdown in focal ischemia in rats. J Neurosci Res. 2006;84:1837–1847. doi: 10.1002/jnr.21073. [DOI] [PubMed] [Google Scholar]
- Sironi L, Mitro N, Cimino M, Gelosa P, Guerrini U, Tremoli E, Saez E. Treatment with LXR agonists after focal cerebral ischemia prevents brain damage. FEBS Lett. 2008;582:3396–3400. doi: 10.1016/j.febslet.2008.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titova E, Ostrowski RP, Kevil CG, Tong W, Rojas H, Sowers LC, Zhang JH, Tang J. Reduced brain injury in CD18-deficient mice after experimental intracerebral hemorrhage. J Neurosci Res. 2008;86:3240–3245. doi: 10.1002/jnr.21762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno T, Sawa Y, Kitagawa-Sakakida S, Nishimura M, Morishita R, Kaneda Y, Kohmura E, Yoshimine T, Matsuda H. Nuclear factor-kappa B decoy attenuates neuronal damage after global brain ischemia: a future strategy for brain protection during circulatory arrest. J Thorac Cardiovasc Surg. 2001;122:720–727. doi: 10.1067/mtc.2001.115917. [DOI] [PubMed] [Google Scholar]
- Wang YY, Dahle MK, Steffensen KR, Reinholt FP, Collins JL, Thiemermann C, Aasen AO, Gustafsson JA, Wang JE. Liver X receptor agonist GW3965 dose-dependently regulates LPSmediated liver injury and modulates post-transcriptional TNFalpha production and p38 MAPK activation in liver macrophages. Shock. 2009 doi: 10.1097/SHK.0b013e3181a47f85. [DOI] [PubMed] [Google Scholar]
- Webster CM, Kelly S, Koike MA, Chock VY, Giffard RG, Yenari MA. Inflammation and NFkappaB activation is decreased by hypothermia following global cerebral ischemia. Neurobiol Dis. 2009;33:301–312. doi: 10.1016/j.nbd.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitney KD, Watson MA, Collins JL, Benson WG, Stone TM, Numerick MJ, Tippin TK, Wilson JG, Winegar DA, Kliewer SA. Regulation of cholesterol homeostasis by the liver X receptors in the central nervous system. Mol Endocrinol. 2002;16:1378–1385. doi: 10.1210/mend.16.6.0835. [DOI] [PubMed] [Google Scholar]
- Xiang Z, Thomas S, Pasinetti G. Increased neuronal injury in transgenic mice with neuronal overexpression of human cyclooxygenase-2 is reversed by hypothermia and rofecoxib treatment. Curr Neurovasc Res. 2007;4:274–279. doi: 10.2174/156720207782446342. [DOI] [PubMed] [Google Scholar]
- Yamaguchi M, Calvert JW, Kusaka G, Zhang JH. One-stage anterior approach for four-vessel occlusion in rat. Stroke. 2005;36:2212–2214. doi: 10.1161/01.STR.0000182238.08510.c5. [DOI] [PubMed] [Google Scholar]
- Yatsushige H, Ostrowski RP, Tsubokawa T, Colohan A, Zhang JH. Role of c-Jun N-terminal kinase in early brain injury after subarachnoid hemorrhage. J Neurosci Res. 2007;85:1436–1448. doi: 10.1002/jnr.21281. [DOI] [PubMed] [Google Scholar]
- Zhang N, Ahsan MH, Zhu L, Sambucetti LC, Purchio AF, West DB. Regulation of IkappaBalpha expression involves both NF-kappaB and the MAP kinase signaling pathways. J Inflamm (Lond) 2005;2:10. doi: 10.1186/1476-9255-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Saito K, Murakami Y, Asano M, Iwakura Y, Seishima M. Early increase in mRNA levels of pro-inflammatory cytokines and their interactions in the mouse hippocampus after transient global ischemia. Neurosci Lett. 2006;393:122–126. doi: 10.1016/j.neulet.2005.08.072. [DOI] [PubMed] [Google Scholar]