

Abstract
Recent clinical data have implicated chronic adverse stress as a potential risk factor in the development of Alzheimer's disease (AD) and data also suggest that normal, physiological stress responses may be impaired in AD. It is possible that pathology associated with AD causes aberrant responses to chronic stress, due to potential alterations in the hypothalamic-pituitary-adrenal (HPA) axis. Recent work in rodent models of AD suggests that chronic adverse stress exacerbates the cognitive deficits and hippocampal pathology that are present in the AD brain. This review summarizes recent findings obtained in experimental AD models regarding the influence of chronic adverse stress on the underlying cellular and molecular disease processes including the potential role of glucocorticoids. Emerging findings suggest that both AD and chronic adverse stress affect hippocampal neural networks in a similar fashion. We describe alterations in hippocampal plasticity that occur in both chronic stress and AD including dendritic remodeling, neurogenesis and long-term potentiation. Finally, we outline potential roles for oxidative stress and neurotrophic factor signaling as key determinants of the impact of chronic stress on the plasticity of neural networks and AD pathogenesis.
Keywords: Alzheimer's disease, chronic stress, glucocorticoids, hippocampal plasticitiy
Introduction
A majority of AD cases are sporadic and do not have a genetic component (Mattson, 2004); environmental and social factors that can increase the risk of AD include incidence of head trauma, overeating, a sedentary lifestyle and severe adverse stress (Mejia et al., 2003). Chronic stresses such as loss of a spouse or sleep deprivation, may cause memory impairments and increase susceptibility to AD. The occurrence of a major stressful event seems to lower the age of onset of familial AD (Mejia et al., 2003). People vary greatly in their response to stressful events, and those most vulnerable to adverse outcomes exhibit a trait that can be described as negative affectivity, neuroticism or distress proneness (Watson and Clark, 1984; Lockenhoff et al., 2009; Weiss et al., 2009). Patients with a high level of distress proneness are 2.7 times more likely to develop AD than those not prone to distress, and the trait is also associated with a more rapid progression of the disease (Wilson et al., 2006, 2007).
It is widely accepted that in the normal central nervous system (CNS), stress induces activation of the hypothalamic-pituitary-adrenocorticol (HPA) axis which leads to increased release of steroid hormones known as glucocorticoids from the adrenal cortex (cortisol in humans, corticosterone in rodents) (Nelson, 1972; Munck and Guyre, 1986). Chronic stress and elevated glucocorticoids are associated with mood disorders such as depression (deKloet and Derijk, 2004; Sotiropoulos et al., 2008). Recent data shows efficacy for anti-depressant therapy in ameliorating deficits in spatial navigation and reducing the levels of Aβ in a transgenic model of AD, which supports a connection between depression and AD (Nelson et al., 2007). Patients with the AD type of dementia also exhibit an increase in circulating cortisol, which could indicate a dysregulation of the HPA-axis in AD, the potential for an altered stress-response in patients with AD, and a physiological connection between pathways in chronic stress, AD and depression (Davis et al., 1986; Umegaki et al., 2000; Rasmuson et al., 2001; Csernansky et al., 2006). A basal dysregulation of the HPA axis could contribute to abnormal cognitive and pathological responses to adverse environmental stress in AD patients. Even in neurologically normal subjects, excessive adverse stress can lead to cognitive impairment (Wolf, 2009), an effect that is mimicked in experimental rodent models of stress which demonstrate impairments in spatial memory, contextual memory and object recognition in response to psychosocial or environmental stress (Krugers et al., 1997; Li et al., 2008). Interestingly, in models of chronic adverse stress, the hippocampus, a region of the brain considered critical for learning and memory, appears to be extremely vulnerable, which is also the case in AD (Cequeira et al., 2007). Further, adverse stress drastically alters hippocampal neuronal circuitry, a phenomenon that manifests as remodeling of dendrties, decreases in neurogenesis and alterations in long-term potentiation (LTP) (Fuchs and Flugge, 1998; deKloet et al., 1999; McEwen, 2001; Fuchs et al., 2006). In AD, hippocampal neuronal networks are similarly altered, making the hippocampus particularly vulnerable to the effects of exposure to chronic stress.
Finally, it is noteworthy in the context of this review that not all stressful events are considered adverse. For example, exercise, dietary energy restriction and environmental enrichment can be considered mild stresses that have beneficial effects on cognitive function and may potentially increase an organism's resistance to further stress (Stranahan and Mattson, 2008; van Praag, 2009). Although the HPA axis is activated transiently during exercise and chronically during dietary energy restriction (Patel and Finch, 2002; Droste et al., 2009), these mild stressors confer numerous health benefits. Interestingly, beneficial and adverse stressors may differentially affect the ways in which neurons respond to elevated glucocorticoids. As evidence, adverse stressors (psychosocial stress and sleep deprivation, for example) decrease the expression of mineralocorticoid receptors (MR) whereas beneficial stressors such as dietary energy deprivation decrease the expression of glucocorticoid receptors (GR) in hippocampal neurons (Lee et al., 2000). For the purposes of this review, ‘stress’ should be considered ‘adverse stress’.
Chronic stress exacerbates cognitive deficits and amyloidogenesis in experimental models of AD
Despite the clinical connections between adverse stress, depression and the onset and progression of AD, few studies to date have utilized adverse behavioral stress paradigms in experimental models of AD (Table 1). Repeated adverse stress or chronic corticosterone administration are widely used to induce symptoms of clinical depression in laboratory rodents (Malberg and Duman 2003; David et al., 2009; Hajszan et al., 2009). Recent data obtained from a limited number of in vivo AD models demonstrate further declines in cognitive function after the application of adverse stress compared to the cognitive decline of AD alone (Dong et al., 2004; Jeong et al., 2006; Catania et al., 2009; Srivareerat et al., 2009). Chronic psychosocial stress induced by an intruder paradigm significantly exacerbated learning impairments in rats directly exposed to amyloid β-peptides (Aβs) Aβ1-40 and Aβ1-42, two cleavage products of the amyloid precursor protein (APP) that are believed to be neurotoxic in AD (Butterfield and Boyd-Kimball, 2004; Irie et al., 2007; Catania et al., 2009; Srivareerat et al., 2009). Similarly, in a transgenic model of AD, 6 months of intermittent immobilization stress worsened learning deficits as measured by both passive avoidance and olfaction tests (Jeong et al., 2006). Further, 6 months of social isolation in a transgenic model of AD accelerated the onset of impairments in contextual, but not cued memory in a fear conditioning test (Dong et al., 2004). Although these above-mentioned studies are the only to our knowledge (Table 1), to measure the cognitive effects of adverse stress in AD, many experimental models of stress demonstrate decreases in cognitive function following a wide range of stressors such as chronic stress paradigms consisting of unpredictable periods of individual housing, tilted cage, food and water deprivation, and wet cage, as well as prolonged subordination and sleep deprivation (Smith and Kelly, 1988; Krugers et al., 1997; Youngblood et al., 1997; Park et al., 2001; Graves et al., 2003; Yap et al., 2006; Li et al., 2008). Chronic administration of dexamethasone, a GR agonist, caused learning and memory impairments in a passive avoidance test for aged, but not young, mice suggesting an increased susceptibility to elevated glucocorticoids with age (Yao et al., 2007).
Table 1.
Summary of current research on the effects of chronic stress in various models of AD.
| Outcome | ||||
|---|---|---|---|---|
| AD model | Environmental Stress | Behavioral | Biochemical & Physiological | Reference |
| infusion of Aβ (rat) | • intruder psychosocial stress model (6 weeks) | • chronic stress worsens Aβ-induced deficits in spatial learning | • stress exacerbates impairment of early phase LTP in Aβ treated rats | Srivareerat et al., Biol Psychiat 2009 |
| • stress reduces basal levels of phosphorylated calcium/calmodulin dependent protein kinase II and increases basal calcineurin after Aβ infusion | ||||
| infusion of Aβ (rat) | • chronic unpredictable stress (4 weeks) • dex administration |
• chronic stress and dex both exacerbate Aβ-induced deficits in spatial learning | • stress and dex magnify increases in C99, BACE-1 and nicastrin induced by Aβ in the hippocampus and cortex | Catania et al., Mol Psychiat 2009 |
| • chronic stress and dex administration | • Aβ, stress and dex all induce anxiety but the effect is not compounded by their combination | |||
| APPV717I-CT100 (mouse) | • immobilization stress (6 or 8 months) | • stress accelerates learning and memory impairments in a passive avoidance paradigm and social transfer of food preference test | • stress increases extracellular amyloid plaques and intraneuronal Aβ deposition in the hippocampus | Jeong et al., FASEB 2006 |
| Tg 2576 (mouse) | • stress increases tau phosphorylation in the hippocampus and cortex | |||
| Tg 2576 (mouse) | • social isolation (6 months) | • social isolation accelerates the onset of memory impairments in contextual, but not cued, fear conditioning | • social isolation accelerates deposition of Aβ plaques in the hippocampus | Dong et al., Neuroscience 2004 |
| • social isolation exacerbates the decrease in hippocampal neurogenesis in AD | ||||
| Tg 2576 (mouse) | • social isolation (6 months) | • social isolation increases basal cort | Dong et al., Neuroscience 2008 | |
| • social isolation increases expression of GR in the cortex and CRF1 in the cortex and hippocampus | ||||
| • social isolation increases Aβ plaque deposition | ||||
| Tg 2576 (mouse) | • restraint stress (16 days) | • restraint increases plasma cort | Lee et al., J Neurochem 2009 | |
| • chronic restraint increases Aβ plaque deposition in the cortex, tau hyperphosphorylation and metabolic oxidative stress | ||||
| • chronic restraint downregulates expression of MMP-2 | ||||
| 3xTgAD (mouse) | • dex administration (7 days) | • basal plasma corticosterone is elevated in 3xTgAD mice | Green et al., J Neurosci 2006 | |
| • dex increases intraneuronal Aβ, total APP, C99 and BACE | ||||
| • dex accelerates accumulation of tau in dendrites | ||||
Abbreviations: AD (Alzheimer's Disease); dex (dexamethasone); LTP (long term potentiation); C99 (C-terminal fragment 99); BACE-1 (β-site APP-cleaving enzyme) cort (corticosterone); CRF1 (corticotropin releasing factor); MMP2 (matrix metalloproteinase 2); APP (amyloid precursor protein); GR (glucocorticoid receptor)
In addition to cognitive deficits measured after adverse stress in both AD and normal animals, several key pathological hallmarks of AD pathology are accelerated in response to adverse stress (Wisor et al., 2005; Green et al., 2006; Jeong et al., 2006; Catania et al., 2009; Lee et al., 2009) (Table 1). AD is characterized by extracellular deposits of Aβ which form ‘plaques’ and intracellular aggregates of the microtubule-associated protein tau which form neurofibrillary ‘tangles’ (Mattson, 2004). The number of Aβ immunoreactive plaques was increased in the cortex and hippocampus in a transgenic AD model after 6 months of social isolation (Dong et al., 2008). A similar increase in extracellular and neuronal Aβ as well as an increase in phosphorylated tau was noted in the hippocampus in a transgenic model of AD following long-term immobilization stress (Jeong et al., 2006). In fact, in a separate study with a similar transgenic mouse model of AD, only 16 days of immobilization stress accelerated Aβ plaque formation and tau phosphorylation implying that even short-term stress will alter amyloid processing in AD (Lee et al., 2009). Because adverse stress causes activation of the HPA axis and increases levels of circulating glucocorticoids, direct administration of glucocorticoids is often used to model chronic stress (Nelson, 1972; Munck and Guyre, 1986; Green et al., 2006; David et al., 2009). In a triple-transgenic model of AD, application of dexamethasone accelerated increases in intraneuronal Aβ40 and Aβ42 levels as well as increasing levels of tau (Green et al., 2006). In fact, corticosterone and dexamethasone increase levels of secreted Aβ40 and Aβ42 in vitro, implying that corticosterone can directly affect APP processing (Green et al., 2006). In the normal non-AD brain, exposure to chronic adverse stress modulates amyloid protein processing; 4 weeks of chronic unpredictable stress caused an increase in cleavage products of APP as well as an increase in amyloidogenic β-secretase cleavage of APP in the hippocampus, indicating a shift towards amyloidogenesis for the application of chronic stress (Sayer et al., 2008; Catania et al., 2009). Exposure to corticosterone or dexamethasone causes increased vulnerability to the neurotoxic effects of Aβ in vitro, potentially via increased intracellular calcium (Goodman et al., 1996; Yao et al., 2007). Collectively, this work strongly implicates adverse stress in exacerbating the cognitive and biochemical hallmarks of AD. Experimental results are beginning to show that the AD brain is particularly susceptible to challenges posed by adverse environmental stressors potentially due to dysregulation of the HPA axis and results seem to indicate that chronic stress and AD cause similar cognitive impairments and pathological hallmarks, specifically in the hippocampus (Table 1). Yet the pathways through which adverse stress promote AD pathogenesis are unknown.
Regulation of glucocorticoids and the HPA axis
Increased levels of glucocorticoids, associated with chronic stress and activation of the HPA axis, are linked with impaired memory function and endogenous cortisol levels are elevated in patients with AD leading to the hypothesis that AD causes dysregulation of the HPA axis leading to increased cortisol release and impairment of memory (Davis et al., 1986; Umegaki et al., 2000; McEwen, 2001 Rasmuson et al., 2001; Csernansky et al., 2006). Further support for this hypothesis comes from studies showing that the basal and stress-induced corticosterone levels are elevated in several transgenic rodent models of AD (Pedersen et al., 1999; Touma et al., 2004; Green et al., 2006; Pedersen et al., 2006; Dong et al., 2008). Interestingly, accumulations in Aβ in the hippocampus precede elevations in basal levels of plasma corticosterone in a triple-transgenic model of AD, implying that the observed alteration in the HPA axis in AD may be due to AD neuropathology (Green et al., 2006).
The hippocampus directly regulates the HPA axis, as demonstrated by studies that show that hippocampal CA3 lesions cause increases in corticosterone levels (Herman et al., 1989; Jacobson and Sapolsky, 1991; Roozendaal et al., 2001) (Fig. 1A). It has therefore been hypothesized that in AD, an increase in Aβ plaques in the hippocampus causes disinhibition of the HPA axis which then leads to an increase in basal glucocorticoid levels (Kulstad et al., 2005; Breyhan et al., 2009; Nuntagij et al., 2009). As mentioned above, increases in glucocorticoid levels may promote accelerated Aβ production and a decrease in Aβ degradation in AD (Green et al., 2006). The early progression of AD pathology could be leading to the initiation of a powerful positive-feedback loop that exacerbates AD pathology via dis-inhibition of the HPA axis and production of glucocorticoids (Fig. 1A). Indeed, elevated basal levels of corticosterone are further increased in AD animal models with the application of adverse stress implying an elevated stress response in AD which may lead to exacerbated AD pathology and, potentially, a worsening of cognitive deficits (Green et al., 2006; Jeong et al., 2006; Dong et al., 2008; Lee et al., 2009). A one-time administration of metyrapone, a corticosterone synthesis inhibitor, to un-stressed mice in a transgenic model of AD attenuated losses in spatial working and reference memory implying that production of corticosterone leads to these cognitive deficits (Pedersen et al., 2006). Further, in a transgenic model of AD, chronic restraint stress caused an increase in Aβ plaques, yet this effect was reversed by pharmacologically blocking the restraint-induced surge in plasma corticosterone, which could indicate that elevated corticosterone directly mediates accelerated plaque formation after chronic stress in AD (Lee et al., 2009).
Figure 1.


Schematic showing the components of the HPA axis (A). Activation of the HPA axis results in release of glucocorticoids (corticosterone in rodents, cortisol in humans). The CA3 region of the hippocampus inhibits activation of the HPA axis. Schematic of the hippocampus showing areas CA1, CA3 and the dentate gyrus (B). Specific sites of hippocampal plasticity as well as control of the HPA axis and the site of glucocorticoid receptors (GR) are noted.
Glucocorticoids exert their biological effects via two receptors, the high affinity MR, and the GR which has roughly one-tenth of the affinity of MR receptors (Reul and de Kloet, 1985; de Kloet et al., 1999). MRs are predominantly expressed in the hippocampus and GRs, while expressed throughout the brain, are most densely present in the CA1 region of the hippocampus (Nishi et al., 2007; Patel et al., 2008; Romeo et al., 2008). In the resting state, when glucocorticoid levels are low, high affinity MR receptors are occupied, whereas GR are relatively unoccupied. When glucocorticoid levels rise, such as in cases of chronic adverse stress, GR receptors begin to become activated, suggesting that the adverse affects of glucocorticoids may be mediated mainly through GR rather than MR (Conrad et al., 1999; Pavlides et al., 2002; deKloet and Kerijk, 2004). As we will discuss later, the relative activation of these receptors may help explain how glucocorticoids regulate long-term potentiation (LTP), the increase in synaptic strength that is thought to underlie learning and memory function (Martin and Clark, 2007; Raymond, 2007). Both clinical and experimental studies of AD show that neurons in the CA1 region of the hippocampus, which express very high levels of GR receptors, are particularly prone to dysfunction and degeneration (Fig. 1B) (Bobinksi et al., 1998; West et al., 1994, 2000; Casas et al., 2004). It is possible that in AD, increased levels of circulating glucocorticoids are related to a loss of GR-expressing neurons. In the normal hippocampus, the expression of GRs is usually decreased in response to adverse stress, perhaps as an attempt to limit glucocorticoid-mediated damage (Sapolsky et al., 1986; McEwen, 2006). However, in a transgenic model of AD, chronic isolation stress caused an increase in GR expression in the hippocampus (Dong et al., 2008). The increase in GR expression in the hippocampus after the application of stress in AD could indicate an aberrant response of dying or degenerating CA1 neurons and could explain the altered reaction to adverse stress in AD.
AD, stress and hippocampal plasticity
Both adverse stress and AD cause alterations in hippocampal plasticity, including shortening and remodeling of dendrites, altered patterns of neurogenesis and impairments in LTP and learning and memory. Although no study to date has established changes in hippocampal plasticity in AD after chronic stress, it is noteworthy that the changes in plasticity in AD and adverse stress paradigms share several common features.
Dendritic remodeling
Cognitive deficits resulting from chronic adverse stress and hyperactivation of the HPA axis, are likely mediated through alterations in hippocampal networks. The hippocampus plays a critical role in learning and memory processes, and alterations in hippocampal neuronal network function and plasticity are detected after corticosterone administration and chronic adverse stress and in models of AD (Jacobsen et al., 2006; Shankar et al., 2008; Aisa et al., 2009). Dendritic remodeling, in the form of reduced/retracted dendtritic spines, occurs in the CA3 region of the hippocampus after chronic psychosocial stress, chronic mild unpredictable stress or corticosterone administration (Fig. 1B) (Magariños, 1996; Sousa, 2000). Shortening of dendrites appears to also be connected to an overall decrease in the number of synapses in the hippocampus after chronic stress (Magariños et al., 1998; Sousa et al., 2000). This affect is noted in vivo after a wide range of chronic stress paradigms as well as after corticosterone administration (Woolley et al., 1990; Watanabe et al., 1992; Magariños et al, 1998). A similar reduction in dendritic spine density is observed in AD (Jacobsen, 2006). Clinical postmortem studies of AD determined that the overall number of synapses is decreased in both the dentate gyrus and the CA1 region, even in cases of mild AD (Selkoe, 2002; Scheff et al., 2006, 2007).
Several studies show that deposits of Aβ can lead to alterations in dendritic spines and a decrease in the number of synapses in AD. Oligomers of Aβ isolated from AD brains attach selectively to dendrites in vitro implying that Aβ directly influences alterations in dendritic morphology in AD (Gong, 2003; Lacor, 2004). Application of Aβ oligomers to normal organotypic hippocampal slice cultures induces reductions in spine density (Selkoe, 2008). Notably, electrophysiologic studies demonstrate that decreased spine density is manifest as a loss of excitatory synapses (Selkoe, 2008). Synaptic degeneration in the hippocampus in a transgenic model of AD was ameliorated by lowering of Aβ levels using Aβ immunotheraphy, further implicating Aβ in mediating synaptic alterations in AD (Buttini, 2005). It is also possible that basal increases in glucocorticoids in AD are responsible for dendritic alterations in the hippocampus, particularly considering that corticosterone administration alone is sufficient to induce a decrease in dendritic spines and the number of synapses in the hippocampus (Magariños et al., 1998).
Neurogenesis
Although it was previously thought that the birth of new neurons in the brain did not occur beyond development, it is now widely accepted that adult neurogenesis occurs in both the subventricular zone/olfactory bulb and the hippocampus. In the subgranular zone of the hippocampal dentate gyrus the newly generated cells become incorporated into the granule cell layer and become morphologically and biochemically neuronal in nature (Fig. 1B) (Cameron et al., 1993; Eriksson et al., 1998; van Praag et al., 2002). The production of new cells is believed to be one mechanism through which the hippocampus exerts plasticity and alters neural networks in response to external stimuli. While the exact mechanisms through which memory function and neurogenesis are linked are not yet known, inhibition of neurogenesis causes impairments in spatial and contextual memory, which could indicate a direct role for adult neurogenesis in maintaining normal memory function (Fig. 2) (Snyder et al., 2005; Saxe et al., 2006; Imayoshi et al., 2009). Neurogenesis is regulated by many environmental factors, including both adverse and beneficial stresses. Chronic stresses such as exposure to predator odor, psychosocial stress and restraint, as well as direct corticosterone administration all cause decreases in adult neurogenesis in the hippocampus (Gould et al., 1997; Czeh et al., 2003; Malberg and Duman, 2003; David et al., 2009). Further, the effect of chronic stress on newly generated neurons in the dentate gyrus, as measured by BrdU, is age-dependent with older animals showing greater vulnerability to stress (Simon et al., 2005). Chronic adverse stress may also lead to increased apoptosis of newly generated neurons in the hippocampus (Lucassen et al., 2006). Both clinical and experimental studies show that overall hippocampal volume is decreased following chronic stress, in depression and in AD, which could indicate that a decrease in neurogenesis and an increase in cell loss in the hippocampus contributes to the cognitive deficits noted in these three neurological conditions (Sheline et al., 1996, 2003; Donohue et al., 2006; Breyhan et al., 2009; Henneman et al., 2009).
Figure 2.
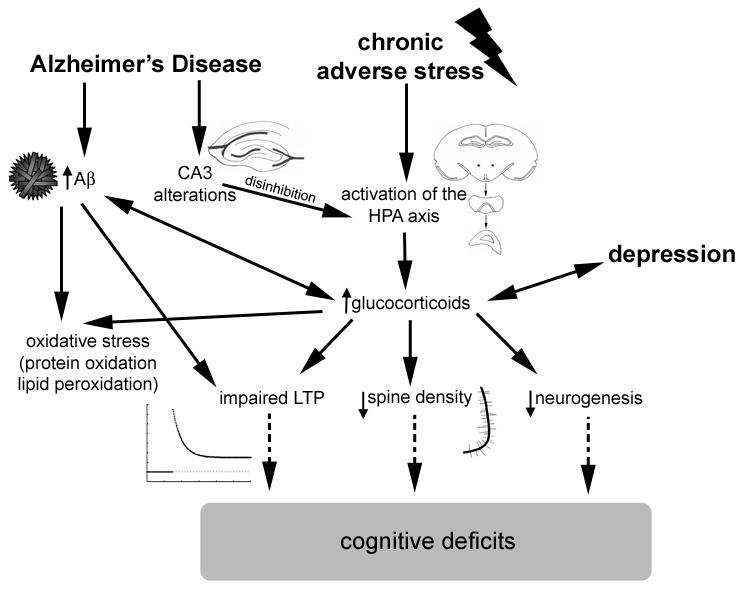
Pathways through which AD and chronic adverse stress potentially lead to cognitive impairments. Chronic stress and AD share similar pathways via increased release of glucocorticoids. Dashed arrows indicate that impairments in LTP, alterations in dendritic morphology and decreases in neurogenesis are all thought to lead to cognitive impairments although the exact mechanisms are not entirely known.
Neurogenesis can be enhanced by stressors known to improve overall health including regular exercise (van Praag, 2009), dietary energy restriction (Lee et al., 2002a, 2002b) and environmental/social enrichment (Kempermann et al., 2002). The proliferation of the neural progenitor cells may be increased and/or the survival of newly generated neurons (many of which normally undergo apoptosis) may be enhanced by beneficial types of stress.
While the loss of hippocampal CA1 neurons is accepted as pathological characteristic of AD, the influence of the AD process on hippocampal neurogenesis is currently under debate. A clinical post-mortem study of AD patients indicated an increased expression of some markers of immature neurons in the hippocampus, such as doublecourtin and TUC-4, which could indicate increased neurogenesis in AD. However, several experimental studies in transgenic mouse models have detected a decrease in neurogenesis using markers of dividing cells as well as markers of immature neurons (Haughey et al., 2002; Jin et al., 2004; Zhang et al., 2007; Rodriguez et al., 2008). It is likely that confounding factors in the clinical setting, such as a history of head trauma or other brain damage, could be causing the appearance of mitotic cells in AD. Further, it is possible that cell loss in the CA1 region coupled with a decrease in neurogenesis in the dentate gyrus causes the reduction in the overall volume of the hippocampus that is noted in AD, resulting in an artifactual appearance of increased density of proliferative cells (Fig. 1B). Potentially, basal levels of neurogenesis remain normal in AD, but survival and differentiation of newly generated neurons is impaired. Coupling AD and chronic stress, which is proven to reduce neurogenesis, would then certainly cause a reduction in the birth of new neurons in the hippocampus. Although no study to date has evaluated neurogenesis in the hippocampus after chronic stress in AD animal models, it is likely that stress exacerbates any reductions in neurogenesis in AD, since both AD and stress alone cause decreases in neurogenesis in the hippocampus.
Long-term potentiation
Long-term potentiation (LTP), a persistent increase in synaptic strength, is considered a likely candidate to be the neuronal mechanism that underlies learning and memory and is consequently of considerable interest in the study of AD as well as stress-induced impairments in memory function. Conversely, long-term depression is the weakening of synapses likely due to persistent weak synaptic stimulation (Sheng and Kim, 2002). Adverse stress reduces LTP in the hippocampus, a phenomenon that was initially demonstrated in models of inescapable foot shock and has since been replicated for exposure to predators, psychosocial stress, chronic mild unpredictable stress and sleep deprivation (Shors et al., 1989; Holderbach et al., 2007; Aleisa et al., 2006; Tadavarty et al., 2009). This effect is likely mediated through release of glucocorticoids; both in vitro and in vivo studies show that LTP is impaired in hippocampal neurons after exposure to high doses of corticosterone (Zhou et al., 2000, Alfarez et al., 2002; Joels and Krugers, 2007). Alterations in hippocampal plasticity from chronic stress appear to be longer-lasting than from acute stress paradigms; hippocampal slices taken from rats after 21 days of chronic mild stress did not respond to synaptic stimulation by corticosterone implying that the induction of LTP was severely impaired (Alfarez et al., 2003). The relationship between corticosterone levels and LTP follows a well-characterized ‘inverted U-shape’ pattern; extremely low levels of corticosterone will reduce LTP, intermediate levels of glucocorticoids are required for normal LTP, and higher levels that occur in stress paradigms impair LTP (Diamond et al., 1992). It is noteworthy that this inverted U-shape is similar to the relationship between chronic stress and cognitive function, with a lack of stimulation having an adverse affect on cognitive function, small stimulations such as exercise and environmental enrichment having a positive effect on memory function, and adverse stressful stimulations such as psychosocial stresses or chronic restraint having an adverse effect on cognitive function (Smith and Kelly, 1988; Youngblood et al., 1997; Park et al., 2001; Graves et al., 2003; van Praag et al., 2005; Yap et al., 2006; Ibi et al., 2008; Li et al., 2008).
In some transgenic models of AD, impairment of LTP begins early in the progression of the disease, before significant accumulation of insoluble plaque depositions (Jacobsen et al., 2006; Liu et al., 2008). Evidence implicates soluble pre-plaque Aβ oligomers in these alterations in LTP; LTP deficits are induced in rats as well as in hippocampal slices after exposure to Aβ and these deficits are alleviated by Aβ immunotherapy or pretreatment of Aβ solutions with proteolytic agents that hydrolyze Aβ (Walsh et al., 2002; Shankar et al., 2008). In a rat model of AD, both chronic infusion of Aβ and chronic psychosocial stress caused impairments in the early phase of LTP in vivo, and the combination of the two worsened these impairments and also impaired activation of calcium-calmodulin kinase II (CaMKII) after in vivo LTP induction (Srivareerat, 2009). Although infusion of Aβ does not represent the most relevant model of AD, the latter findings do provide compelling evidence that Aβ-induced alterations in LTP can be exacerbated by chronic mild stress, which could explain the worsening of cognitive symptoms in this and other AD models when chronic stress is also present.
Cellular and Molecular Mechanisms of Stress-Induced Neuronal Dysfunction and Degeneration
Adverse stressors have been shown to increase oxidative and metabolic stress in neurons on the one hand, while impairing adaptive cellular stress response pathways, on the other hand. A better understanding of the molecular alterations responsible for the neuronal degeneration-promoting and repair-inhibiting effects of adverse stress is leading to novel therapeutic interventions designed to preserve and restore functional neuronal circuits.
Oxidative stress
In addition to similar alterations in hippocampal plasticity, chronic stress and AD are both associated with oxidative stress. In AD, increased oxidative stress is closely linked to Aβ accumulation and neurofibrillary tangle pathology (Butterfield et al., 2001) (Fig. 2). Aβ induces the formation of reactive oxygen species which can then cause lipid peroxidation and protein oxidation (Hensley et al., 2004; Goodman and Mattson, 1994; Yatin et al., 1999; Butterfield et al., 2001; Matsuoka et al., 2001). When Aβ aggregates at the synapse, it induces lipid peroxidation and oxidation of membrane proteins (Keller et al., 1997; Mark et al., 1997). Data indicate the presence of oxidative stress in the brain in AD, including increases in reactive oxygen species, lipid peroxidation products and protein oxidation (Butterfield et al., 2001). Chronic restraint stress, sleep deprivation and social isolation cause increases in markers of oxidative stress in the brain, including lipid peroxidation, protein carbonyls and nitrite levels (Pajovic et al., 2006; Atif et al., 2008; Singh and Kumar, 2008; Zafir and Banu, 2009). Corticosterone may promote oxidative stress, since exogenous administration of corticosterone causes an increase in oxidative stress in the brain (Zafir and Banu, 2009).
Chronic restraint induced a significant increase in lipid peroxidation in the hippocampus in both wild-type mice and in a transgenic mouse model of AD. However, increases in lipid peroxidation were three-fold greater in AD mice subjected to chronic restraint stress compared to unstressed controls (Lee et al., 2009) implying that the AD hippocampus is vulnerable to oxidative stress induced by adverse stress. The increases in lipid peroxidation products after restraint stress were reduced by a pharmacologic block of corticosterone release, implying that this effect is mediated by corticosterone (Lee et al., 2009). It is possible that early increases in Aβ in AD, as well as elevated corticosterone levels cause oxidative stress in the brain. The addition of chronic stress further increases corticosterone levels and exacerbates oxidative damage to neurons (Fig. 2).
Metabolic Stress
Brain imaging studies in which cerebral blood flow or glucose utilization were evaluated demonstrated that cellular energy metabolism is decreased in patients with mild cognitive impairment, and to an even greater extent in AD patients, in brain regions involved in cognition including the hippocampus and associated structures (Friedland et al., 1989; Johnson et al., 2005; Perneczky et al., 2007). While cell loss likely contributes to the deficits in regional brain energy metabolism detected by brain imaging methods, increasing evidence suggests that the ability of neurons to generate energy (ATP and NAD+) efficiently is impaired in neurons well prior to cell death and the onset of cognitive symptoms. In particular, mitochondrial function is compromised (see Mattson et al., 2008 for review). For example, the activities of the α-ketoglutarate dehydrogenase complex, the pyruvate dehydrogenase complex and isocitrate dehydrogenase are decreased in brain regions most affected in AD (Bubber et al., 2005). In addition, oxidative damage to mitochondrial DNA is greater in brain tissue samples from patients with mild cognitive impairment and AD compared to samples from age-matched control subjects (Shao et al., 2008). Other studies have documented mitochondrial deficits in peripheral cells from patients with mild cognitive impairment or AD, including platelets (Trimmer et al., 2004; Valla et al., 2006). Finally, studies of cell culture and animal models have documented multiple adverse effects of genetic factors and stressors relevant to aging and AD (oxidative stress, Aβ and others) on neuronal energy metabolism (Mark et al., 1997; Begley et al., 1999; Hauptmann et al., 2008; Reddy and Beal, 2008).
Adverse stressors may endanger neurons, in part, by impairing energy metabolism in neurons themselves, as well in peripheral cells. Levels of glucocorticoids achieved during chronic stress can inhibit glucose transport in neurons (Horner et al., 1990), a process which may contribute to energy deficits that increase the vulnerability of neurons to aging and AD. Indeed, the damaging effects of levels of glucocorticoids that occur during exposures to adverse stress can be prevented by administering “brain fuels” including ketone bodies (Sapolsky, 1986). Interestingly, low doses of glucocorticoids enhance, whereas high doses impair, mitochondrial function in neural cells (Du et al., 2009). Increasing evidence suggests that impaired peripheral energy (glucose) metabolism (insulin resistance and diabetes) is a risk factor for cognitive impairment (Stranahan et al., 2008a; Craft, 2009), and adverse stress can impair peripheral glucose metabolism by reducing the insulin sensitivity of muscle, liver and other cells (Fu et al., 2009). Treatments that improve glucose metabolism, including dietary energy restriction (Bruce-Keller et al., 1999; Stranahan et al., 2009), exercise (Radak et al., 2001; Stranahan et al., 2008b, 2009) and insulin administration (Bohringer et al., 2008) can enhance cognitive function in animal models. Analysis of the effects of exercise on hippocampal gene expression suggest that this anti-diabetic factor upregulates genes associated with synaptic plasticity, mitochondrial function, energy metabolism, and insulin, MAP kinase and Wnt signaling; genes associated with oxidative stress and infl ammation were downregulated by exercise (Stranahan et al., 2008b). It has been known for several decades that the hypothalamic/sympathetic neural network that regulates energy balance responds to stress and glucocorticoids on the one hand, and that energy intake and output influence HPA stress responses, on the other hand (Dallman, 1995). More recent findings, such as those described above, reveal a much larger neural network (that includes the hippocampus and associated structures) which integrates cognitive responses to stress that may be adaptive or maladaptive depending upon the nature, frequency and intensity of the stressor.
Neurotrophic Factors
Studies show that the normal nervous system responds to a moderate level of stress by enhancing its ability to withstand more severe stress, a phenomenon that is termed ‘neurohormesis’ (Mattson, 2008). An important class of adaptive stress response proteins upregulated by beneficial stressors is the neurotrophic factors. For example, exercise and cognitive stimulation activate an adaptive stress response pathway in neurons involving the transcription factor CREB (cyclic AMP response element-binding protein) which induces the expression of brain-derived neurotrophic factor (BDNF) (see Mattson et al., 2004 for review). Similarly, the expression of fibroblast growth factor 2 (FGF2) is increased in the hippocampus in response to exercise and cognitive stimulation (Gomez-Pinilla et al., 1997). In contrast, to upregulation in response to beneficial stressors, neurotrophic factor expression is suppressed under conditions of chronic adverse stress. BDNF is normally produced in the entorhinal cortex where it is anterogradely transported to the hippocampus, however in AD, BDNF levels are reduced in both the entorhinal cortex and hippocampus, suggesting that production and transport of the neurotrophin is decreased in AD (Connor et al., 1997; Hock et al., 2000). Reductions in BDNF in AD may have wide-reaching effects since this neurotrophin is involved in a wide range of processes that govern neural networks. Moreover, BDNF protects neurons against damage caused by oxidative, metabolic and excitotoxic stress (Mattson, 2004a; Marini, 2007; Nagahara, 2009).
Adverse stress and aging have been consistently shown to reduce the expression of neurotrophic factors. For example, BDNF levels in the hippocampus are decreased in animals subjected to chronic immobilization stress (Smith et al., 1995). Interestingly, chronic social stress during adolescence in mice results in reduced hippocampal BDNF levels and cognitive impairment when the animals are older (Sterlemann et al., 2009) suggesting that adverse stress during early life can endanger the brain during aging. Acute stress enhances, whereas chronic stress inhibits, cocaine-induced bFGF production in a brain region-specific manner (Fumagalli et al., 2008). It is possible that changes in basal BDNF expression that occur in the hippocampus in normal aging and AD render brain cells vulnerable to the adverse effects of chronic stress on neuroprotective signaling pathways. Specifically, BDNF mediates the formation of memories via long-term potentiation (LTP). Animals subjected to exercise demonstrate increased neurogenesis and acquisition and memory retention, effects that are mediated by increases in BDNF in response to exercise (van Praag et al., 2005; Gomez-Pinilla et al., 2008; Stranahan et al., 2009). In contrast, animals subjected to chronic adverse stress exhibit reduces levels of BDNF in hippocampal neurons (Li et al., 2008), suggesting an adverse effect chronic stress on the ability of neurons to protect themselves against injury and disease. Although no research to date has quantified how BDNF regulation in AD affects the response to chronic stress, it is likely that administration of BDNF would improve the stress response, considering that recent work has shown that overexpression of BDNF increases cognitive performance in mice and administration of BDNF reverses synapse loss and restores learning and memory in rodent and primate models of AD (Nakajo et al., 2008; Nagahara et al. 2009).
Finally, it should be noted that although recent data have begun to identify the mechanisms underlying the vulnerability of the hippocampus to chronic stress and AD, it is likely that the type and intensity of the stressful stimulus greatly affects cognitive and pathological outcomes. At least one recent study found no affect of chronic social isolation on Aβ deposits and only a limited affect of social isolation on cognitive measures (Pietropaolo et al., 2009). In a study using chronic restraint stress, levels of Aβ were increased and this effect was blocked by corticotrophic hormone receptor antagonists implying that regulation of the HPA axis was involved (Lee et al., 2009). However in a study of sleep deprivation-induced memory impairment in normal mice, the deficits were not abolished by a corticosterone synthesis inhibitor indicating that restraint stress and sleep deprivation potentially induce varying pathological outcomes even if cognitive deficits are similar (Tiba et al., 2008).
Implications for Therapeutic Interventions to Preserve and Restore Cognitive Function
Several approaches to preserving cognitive function during aging and preventing or delaying the onset of AD are suggested by the findings described above. Lifestyles that incorporate mild intermittent beneficial stressors (exercise, dietary energy restriction and cognitive challenges) and minimize adverse chronic stressors (psychosocial stress, sleep deprivation and the like) provide the first line of defense. Improving peripheral and brain cell energy metabolism through dietary supplementation (with creatine, for example) or drugs (exendin-4, for example) is another approach that may be used in individuals at risk (Sullivan et al., 2000; Pan et al., 2007; Li et al., 2009; Martin et al., 2009). Suppressing glucocorticoid production pharmacologically would be expected to counteract the adverse effects of chronic stress on neural networks in the brain (Smith-Swintosky et al., 1996), although such drugs may compromise the beneficial effects of glucocorticoids in acute stress conditions. Upregulation of the expression of neurotrophic factors using drugs that increase serotonergic and/or noradrenergic signaling is another approach for combating the potentially damaging effects of adverse stress on the integrity and plasticity of nerve cell circuits involved in learning and memory (Mowla et al., 2007; Nelson et al., 2007). Further interrogation of the signaling pathways involved in adaptive and maladaptive responses of neural circuits to stress will likely lead to additional preventative and therapeutic approaches for optimizing cognitive performance throughout the lifespan.
Acknowledgments
This research was supported entirely by the Intramural Research Program of the NIH, National Institute on Aging
References
- Aisa B, Elizalde N, Tordera R, Lasheras B, Del Río J, Ramírez MJ. Effects of neonatal stress on markers of synaptic plasticity in the hippocampus: Implications for spatial memory. Hippocampus. 2009 doi: 10.1002/hipo.20586. epub. [DOI] [PubMed] [Google Scholar]
- Aleisa AM, Alzoubi KH, Gerges NZ, Alkadhi KA. Chronic psychosocial stress-induced impairment of hippocampal LTP: possible role of BDNF. Neurobiol Dis. 2006;22:453–462. doi: 10.1016/j.nbd.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Alfarez DN, Joëls M, Krugers HJ. Chronic unpredictable stress impairs long-term potentiation in rat hippocampal CA1 area and dentate gyrus in vitro. Eur J Neurosci. 2003;17:1928–1934. doi: 10.1046/j.1460-9568.2003.02622.x. [DOI] [PubMed] [Google Scholar]
- Alfarez DN, Weigert O, Joëls M, Krugers HJ. Corticosterone and stress reduce synaptic potentiation in mouse hippocampal slices with mild stimulation. Neurosci. 2002;115:1119–1126. doi: 10.1016/s0306-4522(02)00483-9. [DOI] [PubMed] [Google Scholar]
- Atif F, Yousuf S, Agrawal SK. Restraint stress-induced oxidative damage and its amelioration with selenium. Eur J Pharmacol. 2008;600:59–63. doi: 10.1016/j.ejphar.2008.09.029. [DOI] [PubMed] [Google Scholar]
- Begley JG, Duan W, Chan S, Duff K, Mattson MP. Altered calcium homeostasis and mitochondrial dysfunction in cortical synaptic compartments of presenilin-1 mutant mice. J Neurochem. 1999;72:1030–1039. doi: 10.1046/j.1471-4159.1999.0721030.x. [DOI] [PubMed] [Google Scholar]
- Bobinski M, de Leon MJ, Tarnawski M, Wegiel J, Reisberg B, Miller DC, Wisniewski HM. Neuronal and volume loss in CA1 of the hippocampal formation uniquely predicts duration and severity of Alzheimer disease. Brain Res. 1998;805:267–269. doi: 10.1016/s0006-8993(98)00759-8. [DOI] [PubMed] [Google Scholar]
- Bohringer A, Schwabe L, Richter S, Schachinger H. Intranasal insulin attenuates the hypothalamic-pituitary-adrenal axis response to psychosocial stress. Psychoneuroendocrinology. 2008;33:1394–1400. doi: 10.1016/j.psyneuen.2008.08.002. [DOI] [PubMed] [Google Scholar]
- Breyhan H, Wirths O, Duan K, Marcello A, Rettig J, Bayer TA. APP/PS1KI bigenic mice develop early synaptic deficits and hippocampus atrophy. Acta Neuropathol. 2009;117:677–685. doi: 10.1007/s00401-009-0539-7. [DOI] [PubMed] [Google Scholar]
- Bruce-Keller AJ, Umberger G, McFall R, Mattson MP. Food restriction reduces brain damage and improves behavioral outcome following excitotoxic and metabolic insults. Ann Neurol. 1999;45:8–15. [PubMed] [Google Scholar]
- Bubber P, Haroutunian V, Fisch G, Blass JP, Gibson GE. Mitochondrial abnormalities in Alzheimer brain: mechanistic implications. Ann Neurol. 2005;57:695–703. doi: 10.1002/ana.20474. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Boyd-Kimball D. Amyloid beta-peptide(1-42) contributes to the oxidative stress and neurodegeneration found in Alzheimer disease brain. Brain Pathol. 2004;14:426–432. doi: 10.1111/j.1750-3639.2004.tb00087.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield DA, Drake J, Pocernich C, Castegna A. Evidence of oxidative damage in Alzheimer's disease brain: central role for amyloid beta-peptide. Trends Mol Med. 2001;7:548–554. doi: 10.1016/s1471-4914(01)02173-6. [DOI] [PubMed] [Google Scholar]
- Buttini M, Masliah E, Barbour R, Grajeda H, Motter R, Johnson-Wood K, Khan K, Seubert P, Freedman S, Schenk D, Games D. Beta-amyloid immunotherapy prevents synaptic degeneration in a mouse model of Alzheimer's disease. J Neurosci. 2005;25:9096–9101. doi: 10.1523/JNEUROSCI.1697-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron HA, Woolley CS, McEwen BS, Gould E. Differentiation of newly born neurons and glia in the dentate gyrus of the adult rat. Neuroscience. 1993;56:337–344. doi: 10.1016/0306-4522(93)90335-d. [DOI] [PubMed] [Google Scholar]
- Casas C, Sergeant N, Itier JM, Blanchard V, Wirths O, van der Kolk N, Vingtdeux V, van de Steeg E, Ret G, Canton T, Drobecq H, Clark A, Bonici B, Delacourte A, Benavides J, Schmitz C, Tremp G, Bayer TA, Benoit P, Pradier L. Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated Abeta42 accumulation in a novel Alzheimer transgenic model. Am J Pathol. 2004;165:1289–1300. doi: 10.1016/s0002-9440(10)63388-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catania C, Sotiropoulos I, Silva R, Onofri C, Breen KC, Sousa N, Almeida OFX. The amyloidogenic potential and behavioral correlates of stress. Mol Psychiat. 2009;14:95–105. doi: 10.1038/sj.mp.4002101. [DOI] [PubMed] [Google Scholar]
- Cerqueira JJ, Mailliet F, Almeida OF, Jay TM, Sousa N. The prefrontal cortex as a key target of the maladaptive response to stress. J Neurosci. 2007;27:2781–7. doi: 10.1523/JNEUROSCI.4372-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor B, Young D, Yan Q, Faull RL, Synek B, Dragunow M. Brain-derived neurotrophic factor is reduced in Alzheimer's disease. Brain Res Mol Brain Res. 1997;49:71–81. doi: 10.1016/s0169-328x(97)00125-3. [DOI] [PubMed] [Google Scholar]
- Conrad CD, Lupien SJ, McEwen BS. Support for a bimodal role for type II adrenal steroid receptors in spatial memory. Neurobiol Learn Mem. 1999;72:39–46. doi: 10.1006/nlme.1998.3898. [DOI] [PubMed] [Google Scholar]
- Craft S. The role of metabolic disorders in Alzheimer disease and vascular dementia: two roads converged. Arch Neurol. 2009;66:300–305. doi: 10.1001/archneurol.2009.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csernansky JG, Dong H, Fagan AM, Wang L, Xiong C, Holtzman DM, Morris JC. Plasma cortisol and progression of dementia in subjects with Alzheimer-type dementia. Am J Psychiatry. 2006;163:2164–2169. doi: 10.1176/appi.ajp.163.12.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czéh B, Welt T, Fischer AK, Erhardt A, Schmitt W, Müller MB, Toschi N, Fuchs E, Keck ME. Chronic psychosocial stress and concomitant repetitive transcranial magnetic stimulation: effects on stress hormone levels and adult hippocampal neurogenesis. Biol Psychiatry. 2003;52:1057–1065. doi: 10.1016/s0006-3223(02)01457-9. [DOI] [PubMed] [Google Scholar]
- Dallman MF, Akana SF, Strack AM, Hanson ES, Sebastian RJ. The neural network that regulates energy balance is responsive to glucocorticoids and insulin and also regulates HPA axis responsivity at a site proximal to CRF neurons. Ann N Y Acad Sci. 1995;771:730–742. doi: 10.1111/j.1749-6632.1995.tb44724.x. [DOI] [PubMed] [Google Scholar]
- David DJ, Samuels BA, Rainer Q, Wang JW, Marsteller D, Mendez I, Drew M, Craig DA, Guiard BP, Guilloux JP, Artymyshyn RP, Gardier AM, Gerald C, Antonijevic IA, Leonardo ED, Hen R. Neurogenesis-dependent and -independent effects of fluoxetine in an animal model of anxiety/depression. Neuron. 2009;62:479–493. doi: 10.1016/j.neuron.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis KL, Davis BM, Greenwald BS, Mohs RC, Mathe AA, Johns CA, Horvath TB. Cortisol and Alzheimer's disease, I: Basal studies. Am J Psychiat. 1986;143:300–305. doi: 10.1176/ajp.143.3.300. [DOI] [PubMed] [Google Scholar]
- de Kloet ER, Derijk R. Signaling pathways in brain involved in predisposition and pathogenesis of stress-related disease: genetic and kinetic factors affecting the MR/GR balance. Ann N Y Acad Sci. 2004;1032:14–34. doi: 10.1196/annals.1314.003. [DOI] [PubMed] [Google Scholar]
- de Kloet ER, Oitzl MS, Joëls M. Stress and cognition: are corticosteroids good or bad guys? Trends Neurosci. 1999;22:422–426. doi: 10.1016/s0166-2236(99)01438-1. [DOI] [PubMed] [Google Scholar]
- Diamond DM, Bennett MC, Fleshner M, Rose GM. Inverted-U relationsip between the level of peripheral corticosterone and the magnitude of hippocampal primed burst potentiation. Hippocampus. 1992;2:421–430. doi: 10.1002/hipo.450020409. [DOI] [PubMed] [Google Scholar]
- Dong H, Goico B, Martin M, Csernansky CA, Bertchume A, Csernansky JG. Modulation of hippocampal cell proliferation, memory, and amyloid plaque deposition in APPsw (Tg2576) mutant mice by isolation stress. Neuroscience. 2004;127:601–609. doi: 10.1016/j.neuroscience.2004.05.040. [DOI] [PubMed] [Google Scholar]
- Dong H, Yuede CM, Yoo HS, Martin MV, Deal C, Mace AG, Csernansky JG. Corticosterone and related receptor expression are associated with increased β-amyloid plaques in isolated Tg2576 mice. Neuroscience. 2008;155:154–163. doi: 10.1016/j.neuroscience.2008.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donohue HS, Gabbott PL, Davies HA, Rodríguez JJ, Cordero MI, Sandi C, Medvedev NI, Popov VI, Colyer FM, Peddie CJ, Stewart MG. Chronic restraint stress induces changes in synapse morphology in stratum lacunosum-moleculare CA1 rat hippocampus: a stereological and three-dimensional ultrastructural study. Neuroscience. 2006;140:597–606. doi: 10.1016/j.neuroscience.2006.02.072. [DOI] [PubMed] [Google Scholar]
- Droste SK, Collins A, Lightman SL, Linthorst AC, Reul JM. Distinct, time-dependent effects of voluntary exercise on circadian and ultradian rhythms and stress responses of free corticosterone in the rat hippocampus. Endocrinology. 2009 doi: 10.1210/en.2009-0402. Epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Wang Y, Hunter R, Wei Y, Blumenthal R, Falke C, Khairova R, Zhou R, Yuan P, Machado-Vieira R, McEwen BS, Manji HK. Dynamic regulation of mitochondrial function by glucocorticoids. Proc Natl Acad Sci U S A. 2009;106:3543–3548. doi: 10.1073/pnas.0812671106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson PS, Perfilieva E, Björk-Eriksson T, Alborn AM, Nordborg C, Peterson DA, Gage FH. Neurogenesis in the adult human hippocampus. Nat Med. 1998;4:1313–1317. doi: 10.1038/3305. [DOI] [PubMed] [Google Scholar]
- Friedland RP, Jagust WJ, Huesman RH, Koss E, Knittel B, Mathis CA, Ober BA, Mazoyer BM, Budinger TF. Regional cerebral glucose transport and utilization in Alzheimer's disease. Neurology. 1989;39:1427–1434. doi: 10.1212/wnl.39.11.1427. [DOI] [PubMed] [Google Scholar]
- Fu JH, Xie SR, Kong SJ, Wang Y, Wei W, Shan Y, Luo YM. The Combination of a High-fat Diet and Chronic Stress Aggravates Insulin Resistance in Wistar Male Rats. Exp Clin Endocrinol Diabetes. 2009 doi: 10.1055/s-0028-1119406. Epub. [DOI] [PubMed] [Google Scholar]
- Fuchs E, Flugge G. Stress, glucocorticoids and structural plasticitiy of the hippocampus. Neurosci Behav Rev. 1998;23:295–300. doi: 10.1016/s0149-7634(98)00031-1. [DOI] [PubMed] [Google Scholar]
- Fuchs E, Flugge G, Czeh B. Remodeling of neural networks by stress. Frontiers Bioscience. 2006;11:2746–2758. doi: 10.2741/2004. [DOI] [PubMed] [Google Scholar]
- Fumagalli F, Di Pasquale L, Caffino L, Racagni G, Riva MA. Stress and cocaine interact to modulate basic fibroblast growth factor (FGF-2) expression in rat brain. Psychopharmacology (Berl) 2008;196:357–364. doi: 10.1007/s00213-007-0966-x. [DOI] [PubMed] [Google Scholar]
- Gómez-Pinilla F, Dao L, So V. Physical exercise induces FGF-2 and its mRNA in the hippocampus. Brain Res. 1997;764:1–8. doi: 10.1016/s0006-8993(97)00375-2. [DOI] [PubMed] [Google Scholar]
- Gomez-Pinilla F, Vaynman S, Ying Z. Brain-derived neurotrophic factor functions as a metabotrophin to mediate the effects of exercise on cognition. Eur J Neurosci. 2008;28:2278–2287. doi: 10.1111/j.1460-9568.2008.06524.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Y, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, Krafft GA, Klein WL. Alzheimer's disease-affected brain: presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. PNAS. 2003;100:10417–10422. doi: 10.1073/pnas.1834302100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman Y, Bruce AJ, Cheng B, Mattson MP. Estrogens attenuate and corticosterone exacerbates excitotoxicity, oxidative injury, and amyloid beta-peptide toxicity in hippocampal neurons. J Neurochem. 1996;66:1836–1844. doi: 10.1046/j.1471-4159.1996.66051836.x. [DOI] [PubMed] [Google Scholar]
- Goodman Y, Mattson MP. Secreted forms of beta-amyloid precursor protein protect hippocampal neurons against amyloid beta-peptide-induced oxidative injury. Exp Neurol. 1994;128:1–12. doi: 10.1006/exnr.1994.1107. [DOI] [PubMed] [Google Scholar]
- Gould E, McEwen BS, Tanapat P, Galea LA, Fuchs E. Neurogenesis in the dentate gyrus of the adult tree shrew is regulated by psychosocial stress and NMDA receptor activation. J Neurosci. 1997;17:2492–2498. doi: 10.1523/JNEUROSCI.17-07-02492.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves LA, Heller EA, Pack AI, Abel T. Sleep deprivation selectively impairs memory consolidation for contextual fear conditioning. Learn Mem. 2003;10:168–176. doi: 10.1101/lm.48803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green KN, Billings LM, Roozendaal B, McGaugh JL, LaFerla FM. Glucocorticoids increase amyloid-β and tau pathology in a mouse model of alzheimer's disease. J Neurosci. 2006;26:9047–9056. doi: 10.1523/JNEUROSCI.2797-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajszan T, Dow A, Warner-Schmidt JL, Szigeti-Buck K, Sallam NL, Parducz A, Leranth C, Duman RS. Remodeling of hippocampal spine synapses in the rat learned helplessness model of depression. Biol Psychiatry. 2009;65:392–400. doi: 10.1016/j.biopsych.2008.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haughey NJ, Nath A, Chan SL, Borchard AC, Rao MS, Mattson MP. Disruption of neurogenesis by amyloid beta-peptide, and perturbed neural progenitor cell homeostasis, in models of Alzheimer's disease. J Neurochem. 2002;83:1509–1524. doi: 10.1046/j.1471-4159.2002.01267.x. [DOI] [PubMed] [Google Scholar]
- Hauptmann S, Scherping I, Dröse S, Brandt U, Schulz KL, Jendrach M, Leuner K, Eckert A, Müller WE. Mitochondrial dysfunction: An early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol Aging. 2008 doi: 10.1016/j.neurobiolaging.2007.12.005. Epub. [DOI] [PubMed] [Google Scholar]
- Henneman WJ, Sluimer JD, Barnes J, van der Flier WM, Sluimer IC, Fox NC, Scheltens P, Vrenken H, Barkhof F. Hippocampal atrophy rates in Alzheimer disease: added value over whole brain volume measures. Neurology. 2009;72:999–1007. doi: 10.1212/01.wnl.0000344568.09360.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensley K, Carney JM, Mattson MP, Aksenova M, Harris M, Wu JF, Floyd RA, Butterfield DA. A model for beta-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to Alzheimer disease. Proc Natl Acad Sci U S A. 1994;91:3270–3274. doi: 10.1073/pnas.91.8.3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman JP, Schäfer MK, Young EA, Thompson R, Douglass J, Akil H, Watson SJ. Evidence for hippocampal regulation of neuroendocrine neurons of the hypothalamo-pituitary-adrenocortical axis. J Neurosci. 1989;9:3072–3082. doi: 10.1523/JNEUROSCI.09-09-03072.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hock C, Heese K, Hulette C, Rosenberg C, Otten U. Region-specific neurotrophin imbalances in Alzheimer disease: decreased levels of brain-derived neurotrophic factor and increased levels of nerve growth factor in hippocampus and cortical areas. Arch Neurol. 2000;57:846–851. doi: 10.1001/archneur.57.6.846. [DOI] [PubMed] [Google Scholar]
- Holderbach R, Clark K, Moreau JL, Bischofberger J, Normann C. Enhanced long-term synaptic depression in an animal model of depression. Biol Psychiatry. 2007;62:92–100. doi: 10.1016/j.biopsych.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Horner HC, Packan DR, Sapolsky RM. Glucocorticoids inhibit glucose transport in cultured hippocampal neurons and glia. Neuroendocrinology. 1990;52:57–64. doi: 10.1159/000125539. [DOI] [PubMed] [Google Scholar]
- Ibi D, Takuma K, Koike H, Mizoguchi H, Tsuritani K, Kuwahara Y, Kamei H, Nagai T, Yoneda Y, Nabeshima T, Yamada K. Social isolation rearing-induced impairment of the hippocampal neurogenesis is associated with deficits in spatial memory and emotion-related behaviors in juvenile mice. J Neurochem. 2008;105:921–932. doi: 10.1111/j.1471-4159.2007.05207.x. [DOI] [PubMed] [Google Scholar]
- Imayoshi I, Sakamoto M, Ohtsuka T, Kageyama R. Continuous neurogenesis in the adult brain. Dev Growth Differ. 2009;51:379–386. doi: 10.1111/j.1440-169X.2009.01094.x. [DOI] [PubMed] [Google Scholar]
- Irie K, Murakami K, Masuda Y, Morimoto A, Ohigashi H, Hara H, Ohashi R, Takegoshi K, Fukuda H, Nagao M, Shimizu T, Shirasawa T. The toxic conformation of the 42-residue amyloid beta peptide and its relevance to oxidative stress in Alzheimer's disease. Mini Rev Med Chem. 2007;7:1001–1008. doi: 10.2174/138955707782110187. [DOI] [PubMed] [Google Scholar]
- Jacobsen JS, Wu CC, Redwine JM, Comery TA, Arias R, Bowlby M, Martone R, Morrison JH, Pangalos MN, Reinhart PH, Bloom FE. Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer's disease. PNAS. 2006;103:5161–5166. doi: 10.1073/pnas.0600948103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson L, Sapolsky R. The role of the hippocampus in feedback regulation of the hypothalamic-pituitary-adrenocortical axis. Endocr Rev. 1991;12:118–134. doi: 10.1210/edrv-12-2-118. [DOI] [PubMed] [Google Scholar]
- Jeong YH, Park CH, Yoo J, Shin KJ, Ahn SM, Kim HS, Lee SH, Emson PC, Suh YH. Chronic stress accelerates learning and memory impairments and increases amyloid deposition in APPV7171-CT100 transgenic mice, an Alzheimer's disease model. FASEB. 2006;20:729–731. doi: 10.1096/fj.05-4265fje. [DOI] [PubMed] [Google Scholar]
- Jin K, Peel AL, Mao XO, Xie L, Cottrell BA, Henshall DC, Greenberg DA. Increased hippocampal neurogenesis in Alzheimer's disease. PNAS. 2004;101:343–347. doi: 10.1073/pnas.2634794100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joëls M, Krugers HJ. LTP after stress: up or down? Neural Plasticity. 2007;2007:93202. doi: 10.1155/2007/93202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson NA, Jahng GH, Weiner MW, Miller BL, Chui HC, Jagust WJ, Gorno-Tempini ML, Schuff N. Pattern of cerebral hypoperfusion in Alzheimer disease and mild cognitive impairment measured with arterial spin-labeling MR imaging: initial experience. Radiology. 2005;234:851–859. doi: 10.1148/radiol.2343040197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller JN, Mark RJ, Bruce AJ, Blanc E, Rothstein JD, Uchida K, Waeg G, Mattson MP. 4-Hydroxynonenal, an aldehydic product of membrane lipid peroxidation, impairs glutamate transport and mitochondrial function in synaptosomes. Neuroscience. 1997;80:685–696. doi: 10.1016/s0306-4522(97)00065-1. [DOI] [PubMed] [Google Scholar]
- Kempermann G, Gast D, Gage FH. Neuroplasticity in old age: sustained fivefold induction of hippocampal neurogenesis by long-term environmental enrichment. Ann Neurol. 2002;52:135–143. doi: 10.1002/ana.10262. [DOI] [PubMed] [Google Scholar]
- Krugers HJ, Douma BR, Andringa G, Bohus B, Korf J, Luiten PG. Exposure to chronic psychosocial stress and corticosterone in the rat: effects on spatial discrimination learning and hippocampal protein kinase Cgamma immunoreactivity. Hippocampus. 1997;7:427–436. doi: 10.1002/(SICI)1098-1063(1997)7:4<427::AID-HIPO8>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Kulstad JJ, McMillan PJ, Leverenz JB, Cook DG, Green PS, Peskind ER, Wilkinson CW, Farris W, Mehta PD, Craft S. Effects of chronic glucocorticoid administration on insulin-degrading enzyme and amyloid-beta peptide in the aged macaque. J Neuropathol Exp Neurol. 2005;64:139–146. doi: 10.1093/jnen/64.2.139. [DOI] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, Krafft GA, Klein WL. Synaptic targeting by Alzheimer's-related amyloid beta oligomers. J Neurosci. 2004;24:10191–10200. doi: 10.1523/JNEUROSCI.3432-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Duan W, Mattson MP. Evidence that brain-derived neurotrophic factor is required for basal neurogenesis and mediates, in part, the enhancement of neurogenesis by dietary restriction in the hippocampus of adult mice. J Neurochem. 2002b;82:1367–1375. doi: 10.1046/j.1471-4159.2002.01085.x. [DOI] [PubMed] [Google Scholar]
- Lee J, Herman JP, Mattson MP. Dietary restriction selectively decreases glucocorticoid receptor expression in the hippocampus and cerebral cortex of rats. Exp Neurol. 2000;166:435–441. doi: 10.1006/exnr.2000.7512. [DOI] [PubMed] [Google Scholar]
- Lee J, Seroogy KB, Mattson MP. Dietary restriction enhances neurotrophin expression and neurogenesis in the hippocampus of adult mice. J Neurochem. 2002a;80:539–547. doi: 10.1046/j.0022-3042.2001.00747.x. [DOI] [PubMed] [Google Scholar]
- Lee KW, Kim JB, Seo JS, Kim TK, Im JY, Baek IS, Kim SH, Lee JK, Han PL. Behavioral stress accelerates plaque pathogenesis in the brain of Tg2576 mice via generation of metabolic oxidative stress. J Neurochem. 2009;108:165–175. doi: 10.1111/j.1471-4159.2008.05769.x. [DOI] [PubMed] [Google Scholar]
- Li S, Wang C, Wang W, Dong H, Hou P, Tang Y. Chronic mild stress impairs cognition in mice: from brain homeostasis to behavior. Life Sci. 2008;82:934–942. doi: 10.1016/j.lfs.2008.02.010. [DOI] [PubMed] [Google Scholar]
- Li Y, Perry T, Kindy MS, Harvey BK, Tweedie D, Holloway HW, Powers K, Shen H, Egan JM, Sambamurti K, Brossi A, Lahiri DK, Mattson MP, Hoffer BJ, Wang Y, Greig NH. GLP-1 receptor stimulation preserves primary cortical and dopaminergic neurons in cellular and rodent models of stroke and Parkinsonism. Proc Natl Acad Sci U S A. 2009;106:1285–1290. doi: 10.1073/pnas.0806720106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Orozco IJ, Planel E, Wen Y, Bretteville A, Krishnamurthy P, Wang L, Herman M, Figueroa H, Yu WH, Arancio O, Duff K. A transgenic rat that develops Alzheimer's disease-like amyloid pathology, deficits in synaptic plasticity and cognitive impairment. Neurobiol Dis. 2008;31:46–57. doi: 10.1016/j.nbd.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löckenhoff CE, Terracciano A, Patriciu NS, Eaton WW, Costa PT., Jr Self-reported extremely adverse life events and longitudinal changes in five-factor model personality traits in an urban sample. J Trauma Stress. 2009;22:53–59. doi: 10.1002/jts.20385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucassen PJ, Heine VM, Muller MB, van der Beek EM, Wiegant VM, De Kloet ER, Joels M, Fuchs E, Swaab DF, Czeh B. Stress, depression and hippocampal apoptosis. CNS Neurol Disord Drug Targets. 2006;5:531–546. doi: 10.2174/187152706778559273. [DOI] [PubMed] [Google Scholar]
- Magariños AM, McEwen BS, Flügge G, Fuchs E. Chronic psychosocial stress causes apical dendritic atrophy of hippocampal CA3 pyramidal neurons in subordinate tree shrews. J Neurosci. 1996;16:3534–3540. doi: 10.1523/JNEUROSCI.16-10-03534.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magariños AM, Orchinik M, McEwen BS. Morphological changes in the hippocampal CA3 region induced by non-invasive glucocorticoid administration: a paradox. Brain Res. 1998;809:314–318. doi: 10.1016/s0006-8993(98)00882-8. [DOI] [PubMed] [Google Scholar]
- Malberg JE, Duman RS. Cell proliferation in adult hippocampus is decreased by inescapable stress: reversal by fluoxetine treatment. Neuropsychopharmacology. 2003;28:1562–1571. doi: 10.1038/sj.npp.1300234. [DOI] [PubMed] [Google Scholar]
- Marini AM, Jiang X, Wu X, Pan H, Guo Z, Mattson MP, Blondeau N, Novelli A, Lipsky RH. Preconditioning and neurotrophins: a model for brain adaptation to seizures, ischemia and other stressful stimuli. Amino Acids. 2007;32:299–304. doi: 10.1007/s00726-006-0414-y. [DOI] [PubMed] [Google Scholar]
- Mark RJ, Pang Z, Geddes JW, Uchida K, Mattson MP. Amyloid beta-peptide impairs glucose transport in hippocampal and cortical neurons: involvement of membrane lipid peroxidation. J Neurosci. 1997;17:1046–1054. doi: 10.1523/JNEUROSCI.17-03-01046.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin B, Golden E, Carlson OD, Pistell P, Zhou J, Kim W, Frank BP, Thomas S, Chadwick WA, Greig NH, Bates GP, Sathasivam K, Bernier M, Maudsley S, Mattson MP, Egan JM. Exendin-4 improves glycemic control, ameliorates brain and pancreatic pathologies, and extends survival in a mouse model of Huntington's disease. Diabetes. 2009;58:318–328. doi: 10.2337/db08-0799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SJ, Clark RE. The rodent hippocampus and spatial memory: from synapses to systems. Cell Mol Life Sci. 2007;64:401–431. doi: 10.1007/s00018-007-6336-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka Y, Picciano M, La Francois J, Duff K. Fibrillar beta-amyloid evokes oxidative damage in a transgenic mouse model of Alzheimer's disease. Neuroscience. 2001;104:609–613. doi: 10.1016/s0306-4522(01)00115-4. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Pathways towards and away from Alzheimer's disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Awareness of hormesis will enhance future research in basic and applied neuroscience. Crit Rev Toxicol. 2008;38:633–639. doi: 10.1080/10408440802026406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Duan W, Wan R, Guo Z. Prophylactic activation of neuroprotective stress response pathways by dietary and behavioral manipulations. NeuroRx. 2004;1:111–116. doi: 10.1602/neurorx.1.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Gleichmann M, Cheng A. Mitochondria in neuroplasticity and neurological disorders. Neuron. 2008;60:748–766. doi: 10.1016/j.neuron.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen BS. Plasticity of the hippocampus: adaptation to chronic stress and allostatic load. Ann N Y Acad Sci. 2001;933:265–277. doi: 10.1111/j.1749-6632.2001.tb05830.x. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Protective and damaging effects of stress mediators: central role of the brain. Dialogues Clin Neurosci. 2006;8:367–381. doi: 10.31887/DCNS.2006.8.4/bmcewen. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejia S, Giraldo M, Pineda D, Ardila A, Lopera F. Nongenetic factors as modifiers of the age of onset of familial alzheimer's disease. Int Psychogeriatr. 2003;15:337–349. doi: 10.1017/s1041610203009591. [DOI] [PubMed] [Google Scholar]
- Mowla A, Mosavinasab M, Pani A. Does fluoxetine have any effect on the cognition of patients with mild cognitive impairment? A double-blind, placebo-controlled, clinical trial. J Clin Psychopharmacol. 2007;27:67–70. doi: 10.1097/JCP.0b013e31802e0002. [DOI] [PubMed] [Google Scholar]
- Munck A, Guyre PM. Glucocorticoid physiology, pharmacology and stress. Adv Exp Med Biol. 1986;196:81–96. doi: 10.1007/978-1-4684-5101-6_6. [DOI] [PubMed] [Google Scholar]
- Nagahara AH, Merrill DA, Coppola G, Tsukada S, Schroder BE, Shaked GM, Wang L, Blesch A, Kim A, Conner JM, Rockenstein E, Chao MV, Koo EH, Geschwind D, Masliah E, Chiba AA, Tuszynski MH. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer's disease. Nat Med. 2009;15:331–337. doi: 10.1038/nm.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajo Y, Miyamoto S, Nakano Y, Xue JH, Hori T, Yanamoto H. Genetic increase in brain-derived neurotrophic factor levels enhances learning and memory. Brain Res. 2008;1241:103–109. doi: 10.1016/j.brainres.2008.08.080. [DOI] [PubMed] [Google Scholar]
- Nelson DH. Regulation of glucocorticoid release. Am J Med. 1972;53:590–594. doi: 10.1016/0002-9343(72)90155-6. [DOI] [PubMed] [Google Scholar]
- Nelson RL, Guo Z, Halagappa VM, Pearson M, Gray AJ, Matsuoka Y, Brown M, Martin B, Iyun T, Maudsley S, Clark RF, Mattson MP. Prophylactic treatment with paroxetine ameliorates behavioral deficits and retards the development of amyloid and tau pathologies in 3xTgAD mice. Exp Neurol. 2007;205:166–176. doi: 10.1016/j.expneurol.2007.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi M, Usuku T, Itose M, Fujikawa K, Hosokawa K, Matsuda KI, Kawata M. Direct visualization of glucocorticoid receptor positive cells in the hippocampal regions using green fluorescent protein transgenic mice. Neuroscience. 2007;146:1555–1560. doi: 10.1016/j.neuroscience.2007.03.020. [DOI] [PubMed] [Google Scholar]
- Nuntagij P, Oddo S, LaFerla FM, Kotchabhakdi N, Ottersen OP, Torp R. Amyloid deposits show complexity and intimate spatial relationship with dendrosomatic plasma membranes: an electron microscopic 3D reconstruction analysis in 3xTg-AD mice and aged canines. J Alzheimers Dis. 2009;16:315–323. doi: 10.3233/JAD-2009-0962. [DOI] [PubMed] [Google Scholar]
- Pajović SB, Pejić S, Stojiljković V, Gavrilović L, Dronjak S, Kanazir DT. Alterations in hippocampal antioxidant enzyme activities and sympatho-adrenomedullary system of rats in response to different stress models. Physiol Res. 2006;55:453–460. doi: 10.33549/physiolres.930807. [DOI] [PubMed] [Google Scholar]
- Pan JW, Takahashi K. Cerebral energetic effects of creatine supplementation in humans. Am J Physiol Regul Integr Comp Physiol. 2007;292:R1745–1750. doi: 10.1152/ajpregu.00717.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CR, Campbell AM, Diamond DM. Chronic psychosocial stress impairs learning and memory and increases sensitivity to yohimbine in adult rats. Biol Psychiatry. 2001;50:994–1004. doi: 10.1016/s0006-3223(01)01255-0. [DOI] [PubMed] [Google Scholar]
- Patel PD, Katz M, Karssen AM, Lyons DM. Stress-induced changes in corticosteroid receptor expression in primate hippocampus and prefrontal cortex. Psychoneuroendocrinology. 2008;33:360–367. doi: 10.1016/j.psyneuen.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel NV, Finch CE. The glucocorticoid paradox of caloric restriction in slowing brain aging. Neurobiol Aging. 2002;23:707–717. doi: 10.1016/s0197-4580(02)00017-9. [DOI] [PubMed] [Google Scholar]
- Pavlides C, Nivón LG, McEwen BS. Effects of chronic stress on hippocampal long-term potentiation. Hippocampus. 2002;12:245–257. doi: 10.1002/hipo.1116. [DOI] [PubMed] [Google Scholar]
- Pedersen WA, Culmsee C, Ziegler D, Herman JP, Mattson MP. Aberrant stress response associated with severe hypoglycemia in a transgenic mouse model of Alzheimer's disease. J Mol Neurosci. 1999;13:159–165. doi: 10.1385/JMN:13:1-2:159. [DOI] [PubMed] [Google Scholar]
- Pedersen WA, McMillan PJ, Kulstad JJ, Leverenz JB, Craft S, Haynatzki GR. Rosiglitazone attenuates learning and memory deficits in Tg2576 Alzheimer mice. Exp Neurol. 2006;199:265–273. doi: 10.1016/j.expneurol.2006.01.018. [DOI] [PubMed] [Google Scholar]
- Perneczky R, Hartmann J, Grimmer T, Drzezga A, Kurz A. Cerebral metabolic correlates of the clinical dementia rating scale in mild cognitive impairment. J Geriatr Psychiatry Neurol. 2007;20:84–88. doi: 10.1177/0891988706297093. [DOI] [PubMed] [Google Scholar]
- Pietropaolo S, Sun Y, Li R, Brana C, Feldon J, Yee BK. Limited impact of social isolation on Alzheimer-like symptoms in a triple transgenic mouse model. Behav Neurosci. 2009;123:181–195. doi: 10.1037/a0013607. [DOI] [PubMed] [Google Scholar]
- Radák Z, Kaneko T, Tahara S, Nakamoto H, Pucsok J, Sasvári M, Nyakas C, Goto S. Regular exercise improves cognitive function and decreases oxidative damage in rat brain. Neurochem Int. 2001;38:17–23. doi: 10.1016/s0197-0186(00)00063-2. [DOI] [PubMed] [Google Scholar]
- Rasmuson S, Andrew R, Näsman B, Seckl JR, Walker BR, Olsson T. Increased glucocorticoid production and altered cortisol metabolism in women with mild to moderate Alzheimer's disease. Biol Psychiat. 2001;49:547–552. doi: 10.1016/s0006-3223(00)01015-5. [DOI] [PubMed] [Google Scholar]
- Raymond CR. LTP forms 1, 2 and 3: different mechanisms for the “long” in long-term potentiation. Trends Neurosci. 2007;30:167–175. doi: 10.1016/j.tins.2007.01.007. [DOI] [PubMed] [Google Scholar]
- Reddy PH, Beal MF. Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer's disease. Trends Mol Med. 2008;14:45–53. doi: 10.1016/j.molmed.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reul JM, de Kloet ER. Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology. 1985;117:2505–2511. doi: 10.1210/endo-117-6-2505. [DOI] [PubMed] [Google Scholar]
- Rodríguez JJ, Jones VC, Tabuchi M, Allan SM, Knight EM, LaFerla FM, Oddo S, Verkhratsky A. Impaired adult neurogenesis in the dentate gyrus of a triple transgenic mouse model of Alzheimer's disease. PLoS One. 2008 Aug 13;3(8):e2935. doi: 10.1371/journal.pone.0002935. 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romeo RD, Ali FS, Karatsoreos IN, Bellani R, Chhua N, Vernov M, McEwen BS. Glucocorticoid receptor mRNA expression in the hippocampal formation of male rats before and after pubertal development in response to acute or repeated stress. Neuroendocrinology. 2008;87:160–167. doi: 10.1159/000109710. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, Phillips RG, Power AE, Brooke SM, Sapolsky RM, McGaugh JL. Memory retrival impairment induced by hippocampal CA3 lesions is blocked by adrenocortical suppression. Nat Neurosci. 2001;4:1169–1171. doi: 10.1038/nn766. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM. Glucocorticoid toxicity in the hippocampus: reversal by supplementation with brain fuels. J Neurosci. 1986;6:2240–2244. doi: 10.1523/JNEUROSCI.06-08-02240.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapolsky RM, Krey LC, McEwen BS. The neuroendocrinology of stress and aging: the glucocorticoid cascade hypothesis. Endocr Rev. 1986;7:284–301. doi: 10.1210/edrv-7-3-284. [DOI] [PubMed] [Google Scholar]
- Saxe MD, Battaglia F, Wang JW, Malleret G, David DJ, Monckton JE, Garcia AD, Sofroniew MV, Kandel ER, Santarelli L, Hen R, Drew MR. Ablation of hippocampal neurogenesis impairs contextual fear conditioning and synaptic plasticity in the dentate gyrus. PNAS. 2006;103:17501–17506. doi: 10.1073/pnas.0607207103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayer R, Robertson D, Balfour DJ, Breen KC, Stewart CA. The effect of stress on the expression of the amyloid precursor protein in rat brain. Neurosci Lett. 2008;431:197–200. doi: 10.1016/j.neulet.2007.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Schmitt FA, DeKosky ST, Mufson EJ. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology. 2007;68:1501–1508. doi: 10.1212/01.wnl.0000260698.46517.8f. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Schmitt FA, Mufson EJ. Hippocampal synaptic loss in early Alzheimer's disease and mild cognitive impairment. Neurobiol Aging. 2006;27:1372–1384. doi: 10.1016/j.neurobiolaging.2005.09.012. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav Brain Res. 2008;192:106–113. doi: 10.1016/j.bbr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao C, Xiong S, Li GM, Gu L, Mao G, Markesbery WR, Lovell MA. Altered 8-oxoguanine glycosylase in mild cognitive impairment and late-stage Alzheimer's disease brain. Free Radic Biol Med. 2008;45:813–819. doi: 10.1016/j.freeradbiomed.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheline YI, Gado MH, Kraemer HC. Untreated depression and hippocampal volume loss. Am J Psychiatry. 2003;160:1516–1518. doi: 10.1176/appi.ajp.160.8.1516. [DOI] [PubMed] [Google Scholar]
- Sheline YI, Wang PW, Gado MH, Csernansky JG, Vannier MW. Hippocampal atrophy in recurrent major depression. PNAS. 1996;93:3908–3913. doi: 10.1073/pnas.93.9.3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Kim MJ. Postsynaptic signaling and plasticity mechanisms. Science. 2002;298:776–780. doi: 10.1126/science.1075333. [DOI] [PubMed] [Google Scholar]
- Shors TJ, Seib TB, Levine S, Thompson RF. Inescapable versus escapable foot shock modulates long-term potentiation in the rat hippocampus. Science. 1989;244:224–226. doi: 10.1126/science.2704997. [DOI] [PubMed] [Google Scholar]
- Simon M, Czéh B, Fuchs E. Age-dependent susceptibility of adult hippocampal cell proliferation to chronic psychosocial stress. Brain Res. 2005;1049:244–248. doi: 10.1016/j.brainres.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Singh A, Kumar A. Protective effect of alprazolam against sleep deprivation-induced behavior alterations and oxidative damage in mice. Neurosci Res. 2008;60:372–379. doi: 10.1016/j.neures.2007.12.003. [DOI] [PubMed] [Google Scholar]
- Smith C, Kelly G. Paradoxical sleep deprivation applied two days after end of training retards learning. Physiol Behav. 1988;43:213–216. doi: 10.1016/0031-9384(88)90240-5. [DOI] [PubMed] [Google Scholar]
- Smith MA, Makino S, Kvetnansky R, Post RM. Stress and glucocorticoids affect the expression of brain-derived neurotrophic factor and neurotrophin-3 mRNAs in the hippocampus. J Neurosci. 1995;15:1768–1777. doi: 10.1523/JNEUROSCI.15-03-01768.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith-Swintosky VL, Pettigrew LC, Sapolsky RM, Phares C, Craddock SD, Brooke SM, Mattson MP. Metyrapone, an inhibitor of glucocorticoid production, reduces brain injury induced by focal and global ischemia and seizures. J Cereb Blood Flow Metab. 1996;16:585–598. doi: 10.1097/00004647-199607000-00008. [DOI] [PubMed] [Google Scholar]
- Snyder JS, Hong NS, McDonald RJ, Wojtowicz JM. A role for adult neurogenesis in spatial long-term memory. Neuroscience. 2005;130:843–852. doi: 10.1016/j.neuroscience.2004.10.009. [DOI] [PubMed] [Google Scholar]
- Sotiropoulos I, Cerqueira JJ, Catania C, Takashima A, Sousa N, Almeida OFX. Stress and glucocorticoid footprints in the brain- the path from depression to Alzheimer's disease. Neurosci Biobehav Rev. 2008;32:1161–1173. doi: 10.1016/j.neubiorev.2008.05.007. [DOI] [PubMed] [Google Scholar]
- Sousa N, Lukoyanov NV, Madeira MD, Almeida OF, Paula-Barbosa MM. Reorganization of the morphology of hippocampal neurites and synapses after stress-induced damage correlates with behavioral improvement. Neuroscience. 2000;97:253–266. doi: 10.1016/s0306-4522(00)00050-6. [DOI] [PubMed] [Google Scholar]
- Srivareerat M, Tran TT, Alzoubi KH, Alkadhi KA. Chronic psychosocial stress exacerbates impairment of cognition and long-term potentiation in β-amyloid rat model of alzheimer's disease. Bio Psychiat. 2009;65:918–926. doi: 10.1016/j.biopsych.2008.08.021. [DOI] [PubMed] [Google Scholar]
- Sterlemann V, Rammes G, Wolf M, Liebl C, Ganea K, Müller MB, Schmidt MV. Chronic social stress during adolescence induces cognitive impairment in aged mice. Hippocampus. 2009 doi: 10.1002/hipo.20655. Epub. [DOI] [PubMed] [Google Scholar]
- Stranahan AM, Arumugam TV, Cutler RG, Lee K, Egan JM, Mattson MP. Diabetes impairs hippocampal function through glucocorticoid-mediated effects on new and mature neurons. Nat Neurosci. 2008a;11:309–317. doi: 10.1038/nn2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranahan AM, Lee K, Becker KG, Zhang Y, Maudsley S, Martin B, Cutler RG, Mattson MP. Hippocampal gene expression patterns underlying the enhancement of memory by running in aged mice. Neurobiol Aging. 2008b doi: 10.1016/j.neurobiolaging.2008.10.016. Epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranahan AM, Lee K, Martin B, Maudsley S, Golden E, Cutler RG, Mattson MP. Voluntary exercise and caloric restriction enhance hippocampal dendritic spine density and BDNF levels in diabetic mice. Hippocampus. 2009 doi: 10.1002/hipo.20577. 2009 epub March 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranahan AM, Mattson MP. Impact of energy intake and expenditure on neuronal plasticity. Neuromolecular Med. 2008;10:209–218. doi: 10.1007/s12017-008-8043-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PG, Geiger JD, Mattson MP, Scheff SW. Dietary supplement creatine protects against traumatic brain injury. Ann Neurol. 2000;48:723–729. [PubMed] [Google Scholar]
- Tadavarty R, Kaan TK, Sastry BR. Long-term depression of excitatory synaptic transmission in rat hippocampal CA1 neurons following sleep-deprivation. Exp Neurol. 2009;216:239–242. doi: 10.1016/j.expneurol.2008.11.012. [DOI] [PubMed] [Google Scholar]
- Teng E, Lu PH, Cummings JL. Neuropsychiatric symptoms are associated with progression from mild cognitive impairment to Alzheimer's disease. Dement Geriatr Cogn Disord. 2007;24:253–259. doi: 10.1159/000107100. [DOI] [PubMed] [Google Scholar]
- Tiba PA, Oliveira MG, Rossi VC, Tufik S, Suchecki D. Glucocorticoids are not responsible for paradoxical sleep deprivation-induced memory impairments. Sleep. 2008;31:505–15. doi: 10.1093/sleep/31.4.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touma C, Ambrée O, Görtz N, Keyvani K, Lewejohann L, Palme R, Paulus W, Schwarze-Eicker K, Sachser N. Age- and sex-dependent development of adrenocortical hyperactivity in a transgenic mouse model of Alzheimer's disease. Neurobiol Aging. 2004;25:893–904. doi: 10.1016/j.neurobiolaging.2003.09.004. [DOI] [PubMed] [Google Scholar]
- Trimmer PA, Keeney PM, Borland MK, Simon FA, Almeida J, Swerdlow RH, Parks JP, Parker WD, Jr, Bennett JP., Jr Mitochondrial abnormalities in cybrid cell models of sporadic Alzheimer's disease worsen with passage in culture. Neurobiol Dis. 2004;15:29–39. doi: 10.1016/j.nbd.2003.09.011. [DOI] [PubMed] [Google Scholar]
- Umegaki H, Ikari H, Nakahata H, Endo H, Suzuki Y, Ogawa O, Nakamura A, Yamamoto T, Iguchi A. Plasma cortisol levels in elderly female subjects with Alzheimer's disease: a cross-sectional and longitudinal study. Brain Res. 2000;881:241–243. doi: 10.1016/s0006-8993(00)02847-x. [DOI] [PubMed] [Google Scholar]
- Valla J, Schneider L, Niedzielko T, Coon KD, Caselli R, Sabbagh MN, Ahern GL, Baxter L, Alexander G, Walker DG, Reiman EM. Impaired platelet mitochondrial activity in Alzheimer's disease and mild cognitive impairment. Mitochondrion. 2006;6:323–330. doi: 10.1016/j.mito.2006.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Praag H. Exercise and the brain: something to chew on. Trends Neurosci. 2009;32:283–290. doi: 10.1016/j.tins.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Praag H, Schinder AF, Christie BR, Toni N, Palmer TD, Gage FH. Functional neurogenesis in the adult hippocampus. Nature. 2002;415:1030–1034. doi: 10.1038/4151030a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Praag H, Shubert T, Zhao C, Gage FH. Exercise enhances learning and hippocampal neurogenesis in aged mice. J Neurosci. 2005;25:8680–8685. doi: 10.1523/JNEUROSCI.1731-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Gould E, Cameron HA, Daniels DC, McEwen BS. Phenytoin prevents stress- and corticosterone-induced atrophy of CA3 pyramidal neurons. Hippocampus. 1992;2:431–436. doi: 10.1002/hipo.450020410. [DOI] [PubMed] [Google Scholar]
- Watson D, Clark LA. Negative affectivity: the disposition to experience aversive emotional states. Psychol Bull. 1984;96:465–490. [PubMed] [Google Scholar]
- Weiss A, Sutin AR, Duberstein PR, Friedman B, Bagby RM, Costa PT., Jr The personality domains and styles of the five-factor model are related to incident depression in Medicare recipients aged 65 to 100. Am J Geriatr Psychiatry. 2009;17:591–601. doi: 10.1097/jgp.0b013e31819d859d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MJ, Coleman PD, Flood DG, Troncoso JC. Differences in the pattern of hippocampal neuronal loss in normal ageing and Alzheimer's disease. Lancet. 1994;344:769–772. doi: 10.1016/s0140-6736(94)92338-8. [DOI] [PubMed] [Google Scholar]
- West MJ, Kawas CH, Martin LJ, Troncoso JC. The CA1 region of the human hippocampus is a hot spot in Alzheimer's disease. Ann N Y Acad Sci. 2000;908:255–259. doi: 10.1111/j.1749-6632.2000.tb06652.x. [DOI] [PubMed] [Google Scholar]
- Wilson RS, Arnold SE, Schneider JA, Kelly JF, Tang Y, Bennett DA. Chronic psychological distress and risk of Alzheimer's disease in old age. Neuroepidemiology. 2006;27:143–153. doi: 10.1159/000095761. [DOI] [PubMed] [Google Scholar]
- Wilson RS, Arnold SE, Schneider JA, Li Y, Bennett DA. Chronic distress, age-related neuropathology, and late-life dementia. Psychosom Med. 2007;69:47–53. doi: 10.1097/01.psy.0000250264.25017.21. [DOI] [PubMed] [Google Scholar]
- Wisor JP, Edgar DM, Yesavage J, Ryan HS, McCormick CM, Lapustea N, Murphy GM., Jr Sleep and circadian abnormalities in a transgenic mouse model of Alzheimer's disease: a role for cholinergic transmission. Neuroscience. 2005;131:375–385. doi: 10.1016/j.neuroscience.2004.11.018. [DOI] [PubMed] [Google Scholar]
- Wolf OT. Stress and memory in humans: Twelve years of progress. Brain Res. 2009 doi: 10.1016/j.brainres.2009.04.013. Epub. [DOI] [PubMed] [Google Scholar]
- Woolley CS, Gould E, McEwen BS. Exposure to excess glucocorticoids alters dendritic morphology of adult hippocampal pyramidal neurons. Brain Res. 1990;531:225–231. doi: 10.1016/0006-8993(90)90778-a. [DOI] [PubMed] [Google Scholar]
- Yao YY, Liu DM, Xu DF, Li WP. Memory and learning impairment induced by dexamethasone in senescent but not young mice. Eur J Pharmacol. 2007;574:20–28. doi: 10.1016/j.ejphar.2007.07.021. [DOI] [PubMed] [Google Scholar]
- Yap JJ, Takase LF, Kochman LJ, Fornal CA, Miczek KA, Jacobs BL. Repeated brief social defeat episodes in mice: effects on cell proliferation in the dentate gyrus. Behav Brain Res. 2006;172:344–350. doi: 10.1016/j.bbr.2006.05.027. [DOI] [PubMed] [Google Scholar]
- Yatin SM, Aksenov M, Butterfield DA. The antioxidant vitamin E modulates amyloid beta-peptide-induced creatine kinase activity inhibition and increased protein oxidation: implications for the free radical hypothesis of Alzheimer's disease. Neurochem Res. 1999;24:427–435. doi: 10.1023/a:1020997903147. [DOI] [PubMed] [Google Scholar]
- Youngblood BD, Zhou J, Smagin GN, Ryan DH, Harris RB. Sleep deprivation by the “flower pot” technique and spatial reference memory. Physiol Behav. 1997;61:249–256. doi: 10.1016/s0031-9384(96)00363-0. [DOI] [PubMed] [Google Scholar]
- Zafir A, Banu N. Modulation of in vivo oxidative status by exogenous corticosterone and restraint stress in rats. Stress. 2009;12:167–177. doi: 10.1080/10253890802234168. [DOI] [PubMed] [Google Scholar]
- Zhang C, McNeil E, Dressler L, Siman R. Long-lasting impairment in hippocampal neurogenesis associated with amyloid deposition in a knock-in mouse model of familial Alzheimer's disease. Exp Neurol. 2007;204:77–87. doi: 10.1016/j.expneurol.2006.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Zhang F, Zhang Y. Corticosterone inhibits generation of long-term potentiation in rat hippocampal slice: involvement of brain-derived neurotrophic factor. Brain Res. 2000;885:182–191. doi: 10.1016/s0006-8993(00)02934-6. [DOI] [PubMed] [Google Scholar]