

Abstract
The electroporation mediated transdermal delivery (Protocol - 120V, 10ms, 30pulses at 1Hz with post pulse waiting period of 20min) of doxepin using pure drug solution (PDS) and doxepin-hydroxypropyl-β-cyclodextrin (HPCD) complex solution (CDS) was studied using porcine epidermis model. The stoichiometry of drug-HPCD inclusion complex was determined by differential scanning calorimetry (DSC). The amount of doxepin retained in the epidermis following electroporation did not differ significantly between PDS and CDS. When the drug loaded epidermis was subjected to “Release studies”, doxepin release attained a plateau within ~2.5 days in case of PDS, whereas in case of CDS, doxepin release was prolonged up to 5 days. Mechanistic studies across the nonbiological barriers demonstrated that the slow dissociation of complex was responsible for sustained release of drug from the epidermis. Pharmacodynamic studies were carried out by electroporation mediated delivery of CDS and PDS in hairless rats. The analgesic effect of doxepin was prolonged in case of CDS as compared to PDS.
Keywords: Doxepin, Hydroxypropyl-β-cyclodextrin, Hairless rats, Electroporation, Sustained release
1. Introduction
Postherpetic neuralgia (PHN) is the most common complication following an outbreak of herpes zoster. It is a chronic condition affecting nerve fibers in the affected dermatomic area. Burning sensation and continuous pain is generally associated with PHN, which may even last for months. Pain associated with PHN can be attributed to pathophysiological changes occurring due to both central and peripheral nervous systems [1, 2]. Commonly used agents for treatment of PHN include tricyclic antidepressants (TCAs), anticonvulsants and opiods. However, these can lead to unwanted anticholinergic, central nervous system and gastrointestinal effects.
Topical application to achieve regional delivery of drugs can aid in reducing the risk of systemic side effects as they elicit a localized effect avoiding high plasma concentrations [3–5]. Moreover, it is preferred due to the close proximity of the delivery site to the localization of nerve endings in the dermal region. Doxepin is a tricyclic antidepressant that is known to exhibit analgesic effect in chronic neuropathic pain when applied topically [6–9]. However, topical delivery through skin is limited due to poor permeability of drugs and relatively longer lag times [10]. Such limitations can be overcome by employing electrically mediated techniques such as iontophoresis and electroporation [11, 12].
The duration of action of topically delivered drugs is determined by the dermatokinetics of drugs. Generally, the drugs delivered across the skin are rapidly cleared from the dermal tissue extracellular fluid by the dermal circulation [13, 14]. Therefore, there is need for a sustained release transdermal delivery approach to deliver effective level of drugs to the affected region and to prolong the retention/activity of the drug as well. There are several sustained release formulations which could be administered invasively intradermally to prolong the drug activity at the site of administration [15]. However, combining the sustained release concept with a noninvasive drug delivery system would be relatively more patient compliant and safer than invasive methods. Previous studies by Murthy et al. showed that prolonged localization of drug in the skin can be achieved when complexed with hydroxypropyl-beta-cyclodextrin (HPCD) and delivered by electroporation [16].
Electroporation is a noninvasive method of drug administration into skin. Electroporation forms transient pores which allows high molecular weight substances and complexes to penetrate into the skin [17–20]. The efficiency of electroporation on drug delivery depends on variables like applied voltage, duration of application, rate and the number of pulses that are applied. The safety of skin electroporation was evaluated in animal models and human subjects by various researchers. Vanbever et al have reported that mild reversible skin reactions occurred following application of 15 pulses of 200ms pulse length at 250V in vivo in hairless rats [21]. Wong et al have reported that electroporation at 150V, 1ms and 60 pulses did not cause any pain in human subjects [22]. In all our studies including the present studies related to electroporation, an electrical protocol of 120V, 30 pulses each of 10ms duration was applied in rat model which is rather mild compared to other protocols. Therefore, the application of electrical pulses was well tolerated by the animals and there was no vocalization response as an indication of pain or discomfort. However, twitching of muscle was observed during pulse application [23, 24].
In the current project, an attempt was made to deliver doxepin-HPCD complex by transcutaneous electroporation to form a depot in the skin for sustained drug release. The prolonged analgesic activity would be useful in effective treatment of PHN.
2. Materials and methods
2.1. Chemicals
Doxepin Hydrochloride was purchased from Sigma-Aldrich Inc (St. Louis, MO). Phosphate buffered saline (PBS, pH 7.4) premixed powder was obtained from EMD Chemicals (Gibbstown, NJ). Hydroxypropyl-beta-cyclodextrin (HPCD), phenolphthalein and all the other chemicals used were obtained from Fischer Scientific (Fairway, NJ).
2.2. Porcine epidermis
Porcine belly skin was obtained from local abattoir. Porcine skin is considered to be a good model for human skin due to the similarities in structure of the stratum corneum [25–27]. Pieces of skin wrapped in aluminum foil were heated to 60°C for 2min and the epidermis was gently peeled off the skin. The fresh epidermis was placed on glass microscopic slides, kept dry at 4°C and was used within three days. Prior to use, the epidermis was hydrated with normal saline for an hour.
2.3. General experimental setup
A vertical Franz-type diffusion apparatus (Logan instruments, Somerset, NJ) was used for all resistance and transport measurements across the porcine epidermis. The temperature of the chamber was regulated at 37±1°C by water circulation. A piece of porcine epidermis was placed between two compartments of the diffusion apparatus, one serving as the donor and other as the receiver compartment. The active diffusion area of the epidermis was 0.64 cm2. The receiver compartment (5ml) was filled with PBS (pH 7.4) and 0.5ml of the permeant solution was placed in the donor compartment. Ag/AgCl electrode wires of 0.5mm diameter (Alfa Aesar, Ward Hill, MA) made in the form of concentric rings were placed 2mm away from the porcine epidermis in both the donor and the receiver compartments. Electrical pulses were applied using an ECM 830 Electro Square Porator (BTX Harvard apparatus, Holliston, MA).
2.4. Epidermal electrical resistance
The AC electrical resistance of the epidermis was measured by placing a load resistor RL (100 kΩ) in series with the epidermis. The voltage drop across the whole circuit (VO) and across the epidermis (VS) was measured using an electrical set up consisting of a wave form generator and a digital multimeter (Agilent Technologies, Santa Clara, CA). For measuring resistance, voltage of 100 mv was applied at 10 Hz and the skin resistance in kΩ was approximated [28, 29]. The piece of skin, which had a resistance greater than 20 kΩ.cm2 only, was used for the experiment.
2.5. Preparation and characterization of drug-HPCD complex
Drug-HPCD complex was prepared by dissolving doxepin and HPCD (1:1, 2:1, 5:1 and 10:1 molar ratios) in methanol by common solvent method [30]. The residue obtained after evaporation of solvent obtained was dried and characterized by using DSC. Doxepin, HPCD and different molar ratios of doxepin-HPCD complex preparations were subjected to DSC between 50 and 250°C using Perkin Elmer Pyris 1 DSC. Samples (2mg) were placed in aluminum pan, covered and crimped and heated at 10°C/min with an empty pan as reference.
2.6. In vitro transport across the porcine epidermis
Electroporation mediated transport of doxepin was carried out across the porcine epidermis using pure drug solution (PDS) and doxepin-HPCD complex solution (CDS) prepared in PBS. The donor compartments were filled with 0.5ml of PDS or CDS (equivalent to 10mg/ml of doxepin) and receiver compartments were filled with 5ml of PBS. Thirty electrical pulses (at 1Hz) of 10ms duration at 120V were applied during transport studies. The donor solution was retained for 20min after cessation of electrical pulses and the total amount of doxepin and HPCD (in CDS only) transported across epidermis was measured in the receiver compartment buffer. The amount of doxepin and HPCD retained in the epidermis was also measured after extraction as described in section 2.9. In control experiments (the epidermis was not electroporated), passive transport of doxepin from PDS-control and CDS-control in 20 minutes was determined.
2.6.1. In vitro drug release from reservoir/depot in the porcine epidermis
In separate set of studies, the drug loaded epidermis from PDS and CDS, was subjected to release studies. The drug loaded epidermis was mounted on a Franz diffusion cell. PBS buffer (pH 7.4) was placed in the receiver compartment and the donor compartment was left empty and open. Doxepin retained (drug reservoir/depot) in the epidermis was allowed to release into the receiver compartment buffer and the amount of doxepin in the receiver compartment at different time intervals (until cumulative drug concentration attained a plateau) was analyzed in the samples.
2.7. Drug permeation studies across nonbiological membranes
Passive permeation studies were carried out across the nonbiological membranes to investigate the mechanism responsible for release of doxepin from the epidermis. The permeation experimental set up was similar to that discussed in section 2.3, except that the porcine epidermis was replaced with a nonbiological membrane in this case. The passive permeation studies were carried out using PDS and CDS in the donor compartment.
2.7.1. Permeation across dialysis membrane
The permeation of doxepin from PDS of different concentrations 2.5, 5, 7.5 and 10mg/ml and CDS containing 10mg/ml of doxepin were carried out across dialysis membrane of 1000 dalton molecular weight cut off. The samples were withdrawn at different time intervals.
2.7.2. Permeation across 0.2μm polycarbonate membrane
The permeation of doxepin from PDS (10mg/ml) and CDS (containing 10mg/ml of doxepin) was carried out across 0.2μm membrane with samples being withdrawn at regular time intervals.
2.8. In Vivo studies
All the in vivo experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Mississippi (Protocol # 09-002). Male hairless rats weighing ~250–300g were purchased from Taconic (Hudson, NY) and maintained on a 12-h light-dark cycle in an animal facility with unlimited access to food and water. All the studies were carried out without anaesthetizing the animals.
2.8.1. Pharmacodynamic studies
The rats were divided into 6 groups (n=3). Custom made applicators were used to apply different preparations (volume of 100 μl). The patch system was applied to one side of lumbar region of the rats. In group-1 rats, PBS was loaded on the patch and no pulses were applied which served as no-pulse vehicle control. Group-2 was same as group-1, but electroporation was carried out, which served as pulse-control. Patch systems loaded with 100μl of PDS (5%) and CDS (containing 5% of drug) were placed and electroporation was carried out in rats of group-3 and group-4 respectively. The electrical pulses were applied by connecting the electrodes on the patch applicator to the BTX 830 Squareporator. The electrical protocol applied was same as that applied during in vitro transport studies. In group-5 and group-6 rats, patch systems loaded with 100μl of PDS (5%) and CDS (containing 5% of drug) were placed and were removed after 20 min. In case of group-5 and 6 electrical pulses were not applied. The patch systems from all the rats were removed 20 min after the application and the surface was washed to remove the adhering drug on the surface. The test area was checked for pain avoiding response using a blunt needle and this was compared with the contralateral control area. The pain avoiding response was indicated by withdrawal and vocalization of the animal. Three sets of six pinpricks were applied (at 2, 4, 6 h and every 12 h thereafter) at the test and control area and the number of pinpricks to which rat failed to respond were recorded on a scale of 0–6 [31, 32].
2.8.2. Pharmacokinetic studies
Pharmacokinetic studies were carried out in two groups of rats (n=3). In both the groups, the time course of drug in the plasma was determined following administration of PDS or CDS. In case of PDS group, custom made patch system loaded with 100μl of doxepin solution (5%) was applied and electroporation was carried out and the patch system was removed 20 minutes after application of electrical pulses. In case of CDS group 100μl of doxepin-HPCD complex solution (containing doxepin equivalent to 5%) was administered using custom made patch system and was removed 20 minutes after application of electrical pulses. The electrical protocol followed was similar to that applied in in vitro transport studies. Blood (50μl) was withdrawn every 8 h from the tail vein of rats and placed in heparinized tubes, diluted with 200μl of PBS and centrifuged at 2000g. The plasma proteins were precipitated with acetonitrile and the supernatant was analyzed for drug content by HPLC [33].
2.9. Extraction of doxepin and HPCD from porcine epidermis
The active diffusional area of the porcine epidermis was cut using a biopsy punch after the transport studies. The epidermis was washed with deionized water to remove any adhering drug and HPCD. Then the epidermis was dried and cut into small pieces and added to 20ml of deionized water in a glass vial and kept on a shaker bath for 48h. The samples were then centrifuged and filtered and analyzed for the amount of doxepin and HPCD retained in the porcine epidermis. Appropriate dilutions were made wherever necessary and the extraction procedure was standardized.
2.10. Analytical method
Doxepin was analyzed by HPLC using Symmetry® C18 column (4.6 × 150mm) with UV detection at 210nm. Mobile phase consisted of a mixture of phosphate buffer (0.005mol/l, pH 6), methanol, acetonitrile (19:32:49, v/v/v) and the flow rate was 1ml/min [34]. The sensitivity of the method was 10ng/ml and linearity was between 10–1000ng/ml (R2 = 0.99). To the plasma samples, equal volume of acetonitrile was added to precipitate proteins and then centrifuged at 2000g for 10 min at room temperature and the supernatant was analyzed for drug content. HPCD was estimated by UV spectrometric method at 550nm using phenolphthalein in sodium carbonate solution [35].
2.11. Statistical analysis
Statistical analysis was carried out for comparing the amount of drug permeated across and retained in the epidermis in case of PDS vs. CDS in vitro, plasma elimination rate constants in vivo using Unpaired t- test (GraphPad Prism 5, GraphPad software, Inc., CA, USA). P<0.05 was considered as the level of significance.
3. Results and discussion
3.1. Doxepin –HPCD complex
Inclusion complex of doxepin with HPCD was prepared by solvent evaporation technique at different molar rations of doxepin and HPCD. The dried mixture was subjected to differential scanning calorimetry (DSC) to confirm the stoichiometry. The DSC thermographs of the samples are shown in Fig. 1. In general, incorporation of a drug molecule in cyclodextrin cavity leads to disappearance of melting point of drug molecule [36–38]. An endothermic peak at 189°C representing the melting point of doxepin was observed for doxepin powder. The thermograph of doxepin-HPCD complex obtained by solvent evaporation exhibited no endothermic melt transition when doxepin and HPCD are incorporated in 1:1 molar ratio indicating complete molecular inclusion of doxepin into the HPCD cavity. Endothermic melting peak of doxepin was observed in case of 10:1, 5:1 and 2:1 molar ratios of doxepin: HPCD and the peak height increased with the increase in the amount of doxepin incorporated. This indicates that at ratios higher than 1:1, the entire doxepin was not utilized in complexation and the excess free drug is responsible for the endothermic peaks observed in DSC. Disappearance of the endothermic peak in Fig. 1E suggests that the stoichiometry of doxepin and HPCD complex was most likely 1:1. Recently, Cruz and coworkers have also confirmed that doxepin forms inclusion complex with cyclodextrin with 1:1 stoichiometry using NMR characterization [39].
Fig. 1.
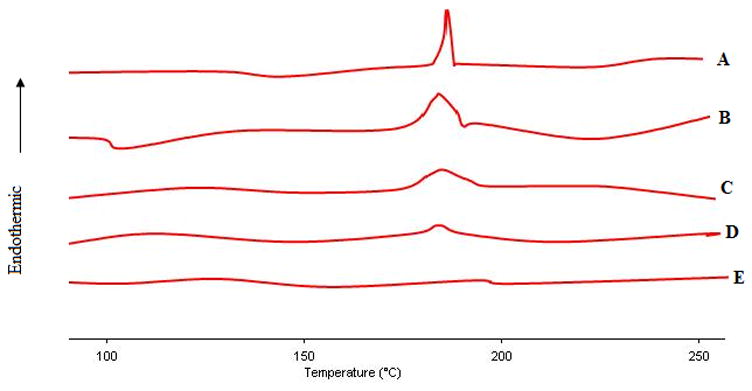
DSC thermograms of pure doxepin (A) and doxepin-HPCD complex of different molar ratios of doxepin: HPCD (B- 10:1; C- 5:1; D-2:1; E-1:1).
3.2. In vitro transport across porcine epidermis
The total amount of drug transported from PDS-control (i.e., passive permeation across porcine epidermis in 20 min duration) was found to be 0.16±0.03μg/cm2. As compared to control, the amount of drug transported by electroporation in case of PDS was ~100 fold higher (16.77±2.52μg/cm2). On the other hand, the amount of drug transported by passive permeation within 20 minutes from CDS-control was less than detectable levels and in case of electroporation it was found to be 4.82±1.08μg/cm2 from CDS.
The amount of drug retained in the epidermis (after 20min) by electroporation in case of PDS was 289.77±45.38μg/cm2, whereas in case of PDS-control, it was significantly less (1.32±0.48μg/cm2). In case of CDS, the amount of drug retained in epidermis by electroporation was 249.33±46.77μg/cm2 whereas in case of CDS-control it was 0.40±0.11μg/cm2. From the results it was evident that electroporation enhances the drug transport across the epidermis and drug deposition in the epidermis as well.
The amount of drug transported across the epidermis by electroporation in case of PDS was higher than CDS (P<0.01). However, the amount of drug retained in the epidermis did not differ significantly between PDS and CDS. These are represented in Fig. 2A, 2B.
Fig. 2.
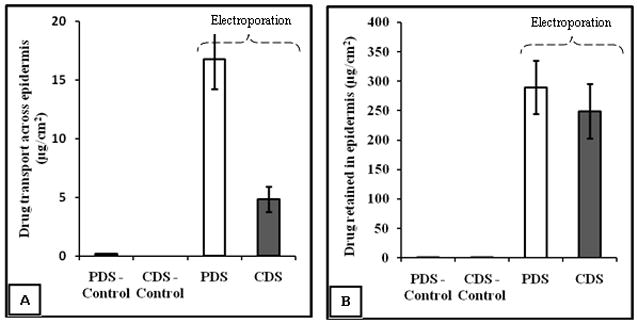
Amount of doxepin transported (A) and retained (B) in the epidermis in case of passive and electroporation mediated delivery. The amount of drug transported and retained in epidermis in case of PDS-control was 0.16±0.03μg/cm2 and 1.32±0.48μg/cm2 respectively. The amount of drug transported across epidermis in case of CDS-control was below detectable levels and the amount of drug retained in the epidermis was 0.40±0.11μg/cm2. The data points represent an average of n=5±S.D.
The amount of HPCD transported across and retained in the epidermis was measured in case of CDS. There was no detectable level of HPCD either in epidermis or in the receiver compartment fluid in case of CDS-control, whereas significant amount of HPCD was delivered by electroporation. The amount of HPCD retained in the epidermis (782.77±46.81μg/cm2) was relatively higher than that transported (271.44±24.02μg/cm2) across the epidermis following electroporation. This was in agreement with the previous observations by Murthy and coworkers [16].
3.3. In vitro drug release from the skin reservoir/depot formed in the porcine epidermis
The amount of drug retained in the porcine epidermis due to electroporation in PDS and CDS did not differ significantly. The release profile of the drug retained in the epidermis is shown in Fig. 3. In case of epidermis from PDS, the drug release profile showed a clear burst release of about ~50% within the first 24h. Further, the drug release attained a plateau in about ~2.5 days. Whereas in case of epidermis from CDS, the drug release was steady and sustained up to 5 days. This shows that, when delivered in the form of complex, the drug is likely to retain in the skin relatively longer than pure drug and the drug is released slowly from the depot formed in the skin. When the drug concentration in the donor in PDS was increased (20 mg/ml) by two fold, the drug load in the epidermis increased to ~420μg/cm2. In this case also a burst release of ~50% was observed and the drug release attained a plateau in 3 days. This suggests that increasing the drug concentration in donor would increase the drug load in the epidermis. However, this approach would not lead to sustained release of drug unlike in the case of CDS. The likely reasons for sustained release of drug from epidermis loaded with CDS are discussed in subsequent sections.
Fig. 3.
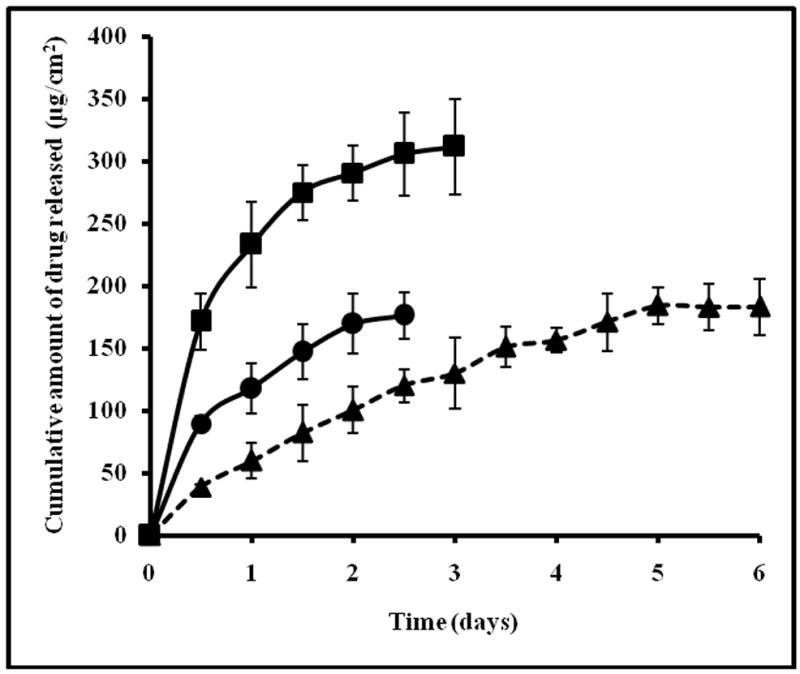
Cumulative amount of drug released from skin reservoir formed in the porcine epidermis from PDS-10mg/ml (●), PDS-20mg/ml (■) and CDS (▲). The data represents an average of n=5±S.D.
3.4. What controls the release of drug from the porcine epidermis?
Apparently, the two mechanisms that could be responsible for slow drug release from the porcine epidermis are slow dissociation of free drug from the complex and slow diffusion of the complex owing to its relatively higher molecular weight as compared to the drug. To investigate the relative contribution of each of these mechanisms further, permeation studies were carried out across the dialysis membrane of 1000Da cut-off and 0.2μm polycarbonate membrane.
The 0.2 micron pore size in the polycarbonate membrane allows both the complex and the free drug to diffuse through. As these pores act as continuous channels between the donor and the receiver compartment, the diffusion rate of substrates across the membrane is nothing but a function of the diffusion coefficient of the substrate in water. In Fig. 4, the slopes of the graph plotted with the amount of doxepin permeated from PDS and CDS across the membrane against time were found to be 1.04±0.02 and 0.92±0.05 respectively, indicating that the increase in molecular weight due to complexation with HPCD did not lead to a drastic decrease in the diffusion coefficient of drug.
Fig. 4.
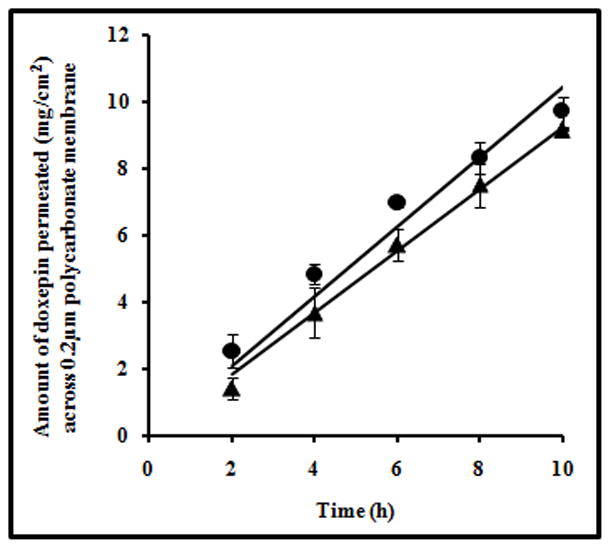
Plot showing the amount of drug permeated at different time points from PDS (●) and CDS (▲) across 0.2μm pore size polycarbonate membrane. The data points are average of n=3±S.D.
The diffusion flux of doxepin from CDS was determined across the dialysis membrane as well. The doxepin-HPCD complex solution present in the donor compartment exists in equilibrium with the free doxepin and free HPCD. Only the free drug which is dissociated from complex is able to diffuse across 1000Da molecular weight cut-off dialysis membrane. Doxepin-HPCD complex (~1850Da) and HPCD (~1500Da) will be retained owing to their larger molecular size compared to the size of the pores in dialysis membrane.

Therefore, in case of CDS, the amount of free drug that exists in equilibrium with the complex determines the flux of doxepin across the membrane. The flux of doxepin across the dialysis membrane from CDS was found to be ~82μg/cm2/h.
An indirect method was followed to approximate the fraction of free drug present in CDS. First, in a separate set of experiment, the diffusion flux of drug across the dialysis membrane from different concentrations of PDS was determined. A clear linear relationship was observed between the drug concentration in the donor and the diffusion flux of drug across the membrane (Fig. 5, R2 =0.96). Now that the diffusion flux of doxepin from CDS was known to be ~82μg/cm2/h, the corresponding free drug concentration in the donor (which is in equilibrium with complex) could be approximated by interpolation in Fig. 5 (point A with point B). This corresponds to ~3.2mg/ml of free doxepin which is only about 1/3rd of the total drug in the CDS (10mg/ml) placed in the donor compartment. This study demonstrates that the complex dissociates slowly to liberate the free drug.
Fig. 5.
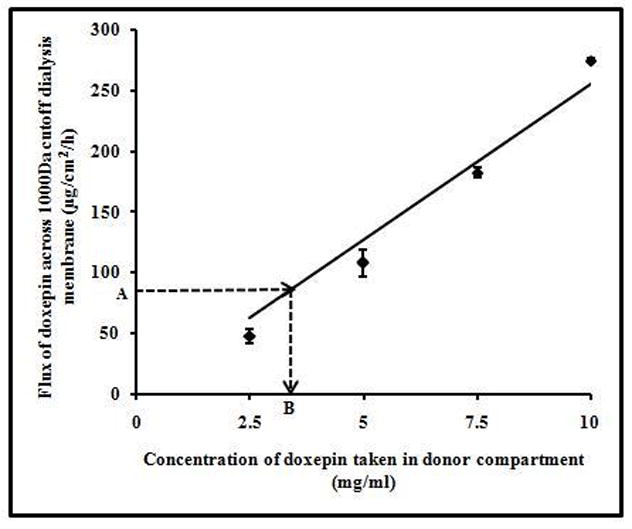
Flux of doxepin from different concentrations of PDS across 1000Da molecular weight cut off dialysis membrane. There was a linear relationship (y=25.48x; R2=0.96) between the donor concentrations of PDS and doxepin flux. Point A (~82μg/cm2/h) is the flux of doxepin from CDS across dialysis membrane. Point B (~3.2mg/ml) indicates the amount of free doxepin present in CDS in the donor. The data points are average of n=3±S.D.
From all the above studies, it can be inferred that the complex is retained in the epidermis longer than the pure drug alone. The drug from the depot formed in the epidermis is released relatively slowly due to slow dissociation of the complex.
3.5. Pharmacodynamic studies
Prolonged retention of the drug in the affected region is likely to elicit longer pharmacological activity. To assess this, pharmacodynamic studies were carried out in rats to evaluate the analgesic activity of doxepin based on the pin prick scores. The number of pinpricks to which rat failed to respond were recorded on a scale of 0–6. There was no analgesic effect in case of no-pulse control group and pulse control group. Analgesic effect was seen only for 2 h in case of rats to which PDS and CDS were administered without electroporation. The group of rats to which CDS was administered by electroporation showed analgesic effect for up to 60 h when compared to group of rats that were administered with PDS by electroporation, which showed analgesic effect for only 24 h. These are shown in Fig. 6. Prolonged analgesic effect in case of CDS as compared to PDS could be attributed to the prolonged retention and sustained release of drug in the skin.
Fig. 6.
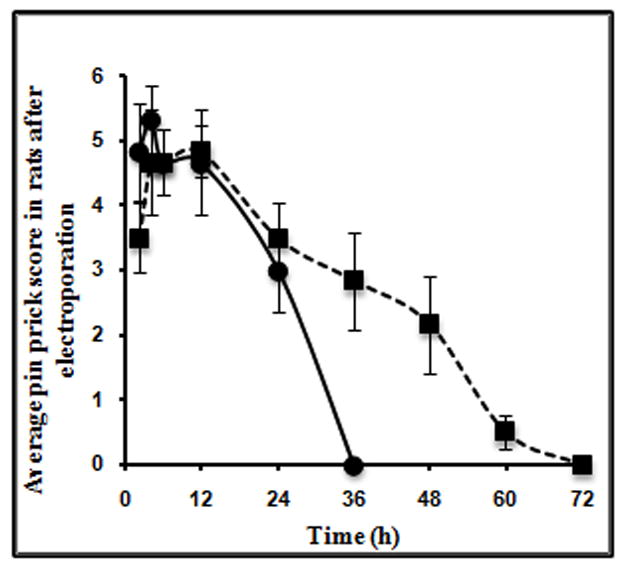
Average pin prick score at different time points in vivo in hairless rats (n=3) after administration of PDS (●) and CDS (■) by electroporation.
3.6. Pharmacokinetic studies
The plasma elimination rate constant (Kel) of drugs is generally less when administered in the form of sustained release dosage form. In the present case, we presumed that, if CDS is leading to formation of a reservoir in the skin and the drug is released relatively slowly, then the plasma elimination rate constant of doxepin in case of CDS should be lesser than that of PDS. Pharmacokinetic studies were carried out in rats. The drug was administered transcutaneously by electroporation using PDS and CDS. Detectable amount of drug was present in plasma for up to 16 h in case of PDS group whereas, the profile was extended up to 24 h in case of CDS group (Fig. 7).
Fig. 7.
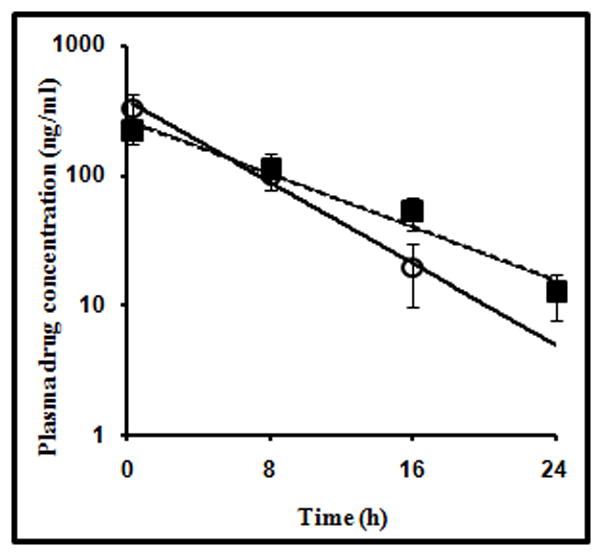
Plasma concentration-time profile of doxepin in hairless rats (n=3) after administration of pure drug solution (PDS group) (○) and doxepin-HPCD complex solution (CDS group) (■) by electroporation.
The plasma elimination rate constant (Kel) was found to be 0.11±0.006 h−1 when CDS was administered and 0.17±0.010 h−1 with PDS. There was statistically significant difference (p<0.01) between the elimination rate constants. These results further confirmed that the drug retained in the cutaneous tissue is released slowly when it is delivered as a complex of HPCD.
4. Conclusion
Electroporation resulted in retention of significant amount of drug in epidermis when doxepin was delivered from PDS and CDS. The drug was released rapidly from epidermis in case of PDS. Sustained drug release was observed from epidermis in case of CDS. Prolonged analgesic activity was observed in case of CDS group of rats as compared to PDS group due to longer retention and sustained release of drug in the former. Transcutaneous delivery of doxepin-HPCD complex by electroporation could be developed as a potential noninvasive sustained release approach for treatment of chronic pain condition like PHN.
Acknowledgments
The project described was supported by Grant Number 5P20RR021929 from the National Center for Research Resources. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Niv D, Tseikhin AM. Postherpetic neuralgia: The never ending challenge. Pain Pract. 2005;5:327–340. doi: 10.1111/j.1533-2500.2005.00035.x. [DOI] [PubMed] [Google Scholar]
- 2.Argoff CE, Katz N, Backonja M. Treatment of postherpetic neuralgia: a review of therapeutic options. J Pain Symptom Manage. 2004;28:396–411. doi: 10.1016/j.jpainsymman.2004.01.014. [DOI] [PubMed] [Google Scholar]
- 3.Sawynok J, Esser MJ, Reid AR. Antidepressants as analgesics: an overview of central and peripheral mechanisms of action. J Psychiatry Neurosci. 2001;26:21–29. [PMC free article] [PubMed] [Google Scholar]
- 4.Sudoh Y, Cahoon EE, Gerner P, Wang GK. Tricyclic antidepressants as long acting local anesthetics. Pain. 2003;103:49–55. doi: 10.1016/s0304-3959(02)00375-5. [DOI] [PubMed] [Google Scholar]
- 5.Davies PS, Galer BS. Review of lidocaine patch 5% studies in the treatment of postherpetic neuralgia. Drugs. 2004;64:937–947. doi: 10.2165/00003495-200464090-00002. [DOI] [PubMed] [Google Scholar]
- 6.McCleane G. Topical application of doxepin hydrochloride, capsaicin and a combination of both produces analgesia in chronic human neuropathic pain: a randomized, double blind, placebo-controlled study. Br J Clin Pharmacol. 2000;49:574–579. doi: 10.1046/j.1365-2125.2000.00200.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McCleane G. Topical doxepin hydrochloride reduces neuropathic pain: a randomized, double-blind, placebo controlled study. The Pain Clinic. 2000;2:47–50. doi: 10.1046/j.1365-2125.2000.00200.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sawynok J. Topical and peripherally acting analhesics. Pharmacol Rev. 2003;55:1–20. doi: 10.1124/pr.55.1.1. [DOI] [PubMed] [Google Scholar]
- 9.Sawynok J. Recent findings surrounding topical antidepressants as analgesics and review of existing and emerging topical analgesics. Advanced Studies in Medicine. 2003;3:S635–S641. [Google Scholar]
- 10.Vanbever R, Preat V. In vivo efficacy and safety of skin electroporation. Adv Drug Deliv Rev. 1999;35:77–88. doi: 10.1016/s0169-409x(98)00064-7. [DOI] [PubMed] [Google Scholar]
- 11.Kalia YN, Naik A, Garrison J, Guy RH. Iontophoretic drug delivery. Adv Drug Deliv Rev. 2004;56:619–658. doi: 10.1016/j.addr.2003.10.026. [DOI] [PubMed] [Google Scholar]
- 12.Prausnitz MR, Bose VG, Langer R, Weaver JC. electroporation of mammalian skin: a mechanism to enhance transdermal drug delivery. Proc Natl Acad Sci. 1993;90:10504–10508. doi: 10.1073/pnas.90.22.10504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cross SE, Roberts MS. Targeting local tissues by transdermal application: understanding drug physicochemical properties that best exploit protein binding and blood flow effects. Drug Dev Res. 1999;46:309–315. [Google Scholar]
- 14.Cevc G, Mazgareanu S, Rother M. Preclinical characterization of NSAIDs in ultradeformable carriers or conventional topical gels. Int J Pharm. 2008;360:29–39. doi: 10.1016/j.ijpharm.2008.01.051. [DOI] [PubMed] [Google Scholar]
- 15.Magerl W, Fuchs PN, Meyer RA, Treede RD. Roles of capsaicin-insensitive nociceptors in cutaneous pain and secondary hyperalgesia. Brain. 2001;124:1754–1764. doi: 10.1093/brain/124.9.1754. [DOI] [PubMed] [Google Scholar]
- 16.Murthy SN, Zhao YL, Sen A, Hui SW. Cyclodextrin enhanced transdermal delivery of piroxicam and carboxyfluorescein by electroporation. J Control Release. 2004;99:393–402. doi: 10.1016/j.jconrel.2004.07.026. [DOI] [PubMed] [Google Scholar]
- 17.Murthy SN, Zhao YL, Marlan K, Hui SW, Kazim AL, Sen A. Lipid and electroosmosis enhanced transdermal delivery of insulin by electroporation. J Pharm Sci. 2006;95:2041–2050. doi: 10.1002/jps.20682. [DOI] [PubMed] [Google Scholar]
- 18.Murthy SN, Sen A, Hui SW. Surfactant-enhanced transdermal delivery by electroporation. J Control Release. 2004;98:307–315. doi: 10.1016/j.jconrel.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 19.Vanbever R, Langers G, Montmayeur S, Preat V. Transdermal delivery of fentanyl: rapid onset of analgesia using skin electroporation. J Control Release. 1998;50:225–235. doi: 10.1016/s0168-3659(97)00147-8. [DOI] [PubMed] [Google Scholar]
- 20.Denet AR, Vanbever R, Preat V. Skin electroporation for transdermal and topical delivery. Adv Drug Deliv Rev. 2004;56:659–674. doi: 10.1016/j.addr.2003.10.027. [DOI] [PubMed] [Google Scholar]
- 21.Vanbever R, Fouchard D, Jaduol A, De Morre N, Preat V, Marty JP. In vivo non-invasive evaluation of hairless rat skin after high-voltage pulse exposure. Skin Pharmacol Appl Skin Physiol. 1998;11:23–24. doi: 10.1159/000029805. [DOI] [PubMed] [Google Scholar]
- 22.Wong TW, Chen CH, Huang CC, Lin CD, Hui SW. Painless electroporation with a needle free-microelectrode array to enhance transdermal drug delivery. J Control Release. 2006;110:557–565. doi: 10.1016/j.jconrel.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 23.Sammeta SM, Vaka SRK, Murthy SN. Transcutaneous sampling of ciprofloxacin and 8-methoxypsoralen by electroporation (ETS) technique. Int J Pharm. 2009;369:24–29. doi: 10.1016/j.ijpharm.2008.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sammeta SM, Vaka SRK, Murthy SN. Dermal levels of antibiotic (cephalexin) determined by electroporation and transcutaneous sampling (ETS) technique. J Pharm Sci. 2009;98:2677–2685. doi: 10.1002/jps.21642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pliquett U, Gallo S, Hui SW, Gusbeth C, Neumann E. Local and transient structural changes in stratum corneum at high electric fields: contribution of joule heating. Bioelectrochemistry. 2005;67:37–46. doi: 10.1016/j.bioelechem.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 26.Ferry LL, Argentieri G, Lochner DH. The comparative histology of porcine and guinea pig skin with respect to iontophoretic drug delivery. Pharma Acta Helv. 1995;70:43–56. doi: 10.1016/0031-6865(94)00050-6. [DOI] [PubMed] [Google Scholar]
- 27.Schurer NY, Elias PM. The biochemistry and function of stratum corneum lipids. Adv Lipid Res. 1991;24:27–56. doi: 10.1016/b978-0-12-024924-4.50006-7. [DOI] [PubMed] [Google Scholar]
- 28.Murthy SN, Sen A, Hui SW. Surfactant-enhanced transdermal drug delivery by electroporation. J Control Release. 2004;98:307–315. doi: 10.1016/j.jconrel.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 29.Srinivasa Murthy S, Siva Ram Kiran V, Mathur SK, Murthy SN. Noninvasive transcutaneous sampling of glucose by electroporation. J Diabetes Sci Technol. 2008;2:250–254. doi: 10.1177/193229680800200213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Badaway SIF, Ghorab MM, Adeyeye CM. Characterization and bioavailability of danazol-hydroxypropyl β-cyclodextrin coprecipitates. Int J Pharm. 1996;128:45–54. [Google Scholar]
- 31.Gerner P, Srinivasa V, Zizza AM, Zhuang ZY, Luo SH, Zurakowski D, Eppan S, Wang GK. Doxepin by topical application and intrathecal route in rats. Anesth Analg. 2006;102:283–287. doi: 10.1213/01.ane.0000183639.37428.4d. [DOI] [PubMed] [Google Scholar]
- 32.Haderer A, Gerner P, Kao G, Srinivasa V, Wang GK. Cutaneous analgesia after transdermal application of amitriptyline versus lidocaine in rats. Anesth Analg. 2003;96:1707–1710. doi: 10.1213/01.ANE.0000060456.91215.90. [DOI] [PubMed] [Google Scholar]
- 33.Heinig K, Henion J. Fast liquid chromatographic-mass spectrometric determination of pharmaceutical compounds. J Chromatogr B Biomed Sci Appl. 1999;732:445–458. doi: 10.1016/s0378-4347(99)00313-8. [DOI] [PubMed] [Google Scholar]
- 34.Meyer-Barner M, Meineke I, Schreeb KH. Pharmacokinetics of doxepin and desmethyldoxepin: an evaluation with the population approach. Eur J Clin Pharmacol. 2002;58:253–257. doi: 10.1007/s00228-002-0448-3. [DOI] [PubMed] [Google Scholar]
- 35.Goel A, Nene SN. Modifications in the phenolphthalein method for spectrophotometric estimation of beta cyclodextrin. Starch. 1995;47:399–400. [Google Scholar]
- 36.Yang J, Wiley CJ, Godwin DA, Felton LA. Influence of hydroxypropyl-β-cyclodextrin on transdermal penetration and photostability of avobenzene. Eur J Pharm Biopharm. 2008;69:605–612. doi: 10.1016/j.ejpb.2007.12.015. [DOI] [PubMed] [Google Scholar]
- 37.Dias MMR, Raghavan SL, Pellett MA, Hadgraft J. The effect of β-cyclodextrins on the permeation of diclofenac from supersaturated solutions. Int J Pharm. 2003;263:173–181. doi: 10.1016/s0378-5173(03)00366-1. [DOI] [PubMed] [Google Scholar]
- 38.Doliwa A, Santoyo S, Ygartua P. Influence of piroxicam:hydroxypropyl-beta-cyclodextrin complexation on the in vitro permeation and skin retention of piroxicam. Skin Pharmacol Appl Skin Physiol. 2001;14:97–107. doi: 10.1159/000056339. [DOI] [PubMed] [Google Scholar]
- 39.Cruz JR, Becker BA, Morris KF, Larive CK. NMR characterization of the host-guest inclusion complex between β-cyclodextrin and doxepin. Magn Reson Chem. 2008;46:838–845. doi: 10.1002/mrc.2267. [DOI] [PubMed] [Google Scholar]