

Abstract
Anti-inflammatory drugs prevent intestinal tumor formation, an activity related to their ability to inhibit inflammatory pathway signaling in the target tissue. We previously showed that treatment of Min/+ mice with the selective cyclooxygenase-2 (COX-2) inhibitor, celecoxib, induced rapid tumor regression, however, drug-resistant tumors appeared with long-term treatment. In this study, we investigated whole-tissue changes in inflammatory signaling by studying constituents of the tissue stroma and extracellular matrix (ECM). We found that celecoxib resistance was associated with changes in factors regulating autocrine TGFβ signaling. Chronic drug treatment expanded the population of bone marrow-derived CD34+ vimentin+ αSMA- myofibroblast precurors and αSMA+ vimentin+ F4/80- myofibroblasts in the lamina propria and submucosa, providing a source of increased TGFβ and COX-2 expression. Membrane constituents regulating TGFβ availability, including syndecan-1 and heparinase-1 (HPA-1) were also modified by chronic treatment in a manner promoting increased TGFβ signaling. Finally, long term celecoxib treatment induced tissue fibrosis, as indicated by increased expression of collagen, fibronectin, and laminin in the basement membrane. We conclude that chronic COX-2 inhibition alters TGFβ signaling in the intestinal mucosa, producing conditions consistent with chronic inflammation.
Introduction
Colorectal cancer (CRC) development is fostered by chronic inflammation, a condition associated with both sporadic tumor formation and inflammatory bowel disease (IBD). Consistent with this, non-steroidal anti-inflammatory drugs (NSAIDs) exhibit anti-tumor properties. In human clinical trials, these agents inhibited the formation of new colorectal adenomas, and also induced regression of already-established tumors (1, 2). The anti-tumor effect of NSAIDs is primarily achieved by inhibition of the cyclooxygenase-2 (COX-2) enzyme and its downstream product, prostaglandin E2 (PGE2), which is the primary mediator of inflammation in the colorectal mucosa. Recent human chemoprevention trials showed that the selective COX-2 inhibitor, celecoxib, reduced colorectal adenoma formation by as much as 68% in patients at high risk for CRC (3, 4). Unfortunately, treatment with this drug and others in its class was also associated with increased risk of serious cardiovascular events, revealing an uncharacterized role of COX-2 in maintaining normal cardiovascular function (3, 5, 6).
Previous work in our laboratory, using an animal model for CRC, showed that chronic administration of celecoxib was associated with resistance to its anti-tumor effect. In the Apc-deficient C57BL/6J-Min/+ (Min/+) mouse, short term dietary celecoxib treatment (3 weeks) inhibited adenoma formation, COX-2 expression, and PGE2 production, but long term treatment (4-5 months) induced resistant tumors, with the level of tumor formation similar to that of untreated mice (7). Both the tumors and non-tumor intestinal mucosa of chronically treated mice demonstrated recurrence of high levels of PGE2 and COX-2 expression (7). In this tissue, however, we found minimal changes in the expression of PGE2 receptors, lipoxygenases, or the multi-drug resistance transporter, MDR1 (7). Understanding the cellular and molecular basis for this treatment resistance is important to improving application of NSAIDs for chemoprevention.
In the setting of chronic inflammation, the intestinal stroma plays an active role in colorectal tumorigenesis, engaging in dynamic crosstalk with epithelial cells. In the normal intestine, COX-2 expression is restricted to the stromal compartment, with expression by fibroblasts, endothelial cells or macrophages (8). Myofibroblasts reside subjacent to the basement membrane and interact with enterocytes to regulate epithelial cell restitution and barrier function. These stromal cells also contribute to fibrosis and intestinal tumor progression (9). Myofibroblasts participate in innate immune responses via signaling from surface pattern recognition receptors (TLRs) that bind microbial products (10). As a result of inflammatory conditions, myofibroblasts increase in number, and can be expected to produce greater amounts of PGE2 in this setting. Myofibroblasts, therefore, may be critical in driving the occurrence and progression of precancerous lesions (11).
PGE2 works in concert with ubiquitously expressed Transforming Growth Factor β (TGFβ during normal wound healing, but antagonizes the growth inhibiting function of this cytokine during inflammation-associated tumorigenesis (12, 13). TGFβ acts as a tumor suppressor and promoter depending on the cellular context (14). TGFβ is secreted as part of a large complex that maintains a reservoir of latent ligand in the extracellular matrix (ECM) and requires specific processing for activation (15). Targeted knockout mice showed that loss of TGFβ signaling in the intestine by epithelial, mesenchymal, or immune cells stimulated polyp formation, suggesting that balanced signaling by or between these cell types in the intestine promotes proper growth-regulating intracellular communication (16, 17). Further supporting the crucial role of TGFβ in intestinal homeostasis, patients with a germline mutation inactivating SMAD4, a downstream effector, develop multiple intestinal polyps and have an increased risk of CRC, a syndrome known as Familial Juvenile Polyposis (18). Finally, later loss of TGFβ signaling by inactivation of its primary receptor (TβRII) may be a factor in progression of adenomas to invasive CRC in certain settings; however, TGFβ may enhance tumorigenesis (21) TGFβ expression is typically increased in the setting of chronic inflammation and tumor progression. TGFβ signaling is required for the differentiation of precursor cells into myofibroblasts, and it also engenders their acquisition of muscle-like contractility and ECM-remodeling capabilities (18). Importantly, TGFβ signaling in myofibroblasts promotes intestinal fibrosis in the setting of IBD (19, 20). Together, these findings suggest that chronic intestinal inflammation changes the roles of TGFβ signaling and myofibroblasts in tumor promoting ways.
Chronic intestinal inflammation also results in dramatic changes in the ECM. Heparan sulfate proteogylans (HSPGs) are complex polysaccharides attached to cell surface membranes that regulate ECM homeostasis (22). The transmembrane HSPG, syndecan-1, is expressed at the basolateral membranes of normal enterocytes, and acts as a co-receptor by binding cytokines and growth factors at the cell surface. Syndecan-1 regulates the downstream signaling of both Wnt and TGFβ ligands, which are critical positive and negative regulators of intestinal cell growth and tumorigenesis (23-25). In the intestine, the intact syndecan-1 ectodomain modulates innate immunity, and maintains barrier function (26). Cleavage of the HSPG extracellular moiety (ectodomain) or the heparan sulfate side chain by metalloproteinases, reactive oxygen species, or heparanase-1 (HPA-1), significantly alters the activity of syndecan-1 ligands (27-29). In patients with IBD, mucosal HPA-1 levels are increased, suggesting that cleavage of the syndecan-1 ectodomain is pro-inflammatory (30).
The Min/+ tumor model allows us to observe how epithelial-stromal interactions change during tumorigenesis. We hypothesized that celecoxib resistance arises from time-dependent adaptations in enterocytes, stromal cells, and the ECM that act cooperatively to promote PGE2 production.
Materials and Methods
Materials
5-week old female C57BL/6J-Min/+ and Apc+/+ (WT) mice were purchased from The Jackson Laboratory (Bar Harbor, ME). AIN-76A diet with/without celecoxib (1,500 ppm) was prepared by Research Diets (New Brunswick, NJ). Heparinase (from Flavobacterium heparinum) was from Seikagaku Corp. (Tokyo, Japan), recombinant TGFβ1 (rTGFβ1) was from R&D Systems (Minneapolis, MI), and PGE2 was from Cayman Chemicals (Ann Arbor, MI). The Trichrome stain (Masson) kit, HT15, was from Sigma (Saint Louis, MO). Antibodies used are listed in Supplementary Data Table 1. All other reagents were the same as previously reported (7, 27).
Immunohistochemical (IHC) and histochemical staining analyses
Formalin-fixed paraffin-embedded 4 μm sections of ileum from Min/+ and WT mice were used for IHC by standard techniques (7), except that in certain cases (syndecan-1, TGFβ, F4/80, CD34, fibronectin, and collagen IV) the blocking step was omitted and all solutions were prepared in Antibody Diluent (Invitrogen, Carlsbad, CA). Masson's trichrome staining to detect connective tissue used a kit by the vendor's protocol. For Laminin-5 γ2 IHC, deparaffinized tissue sections were reacted with Proteinase XXIV (0.125 units/50 μl) for 15 min prior to reaction with D4B5 antibody (27). All experiments were repeated using tissues from at least 3 different mice of each treatment group.
Ex vivo treatments of WT small intestine
Freshly harvested ileum of 4-month old female WT mice was opened longitudinally, rinsed with PBS, and placed in tissue culture medium supplemented with 0.1% FBS and containing drug or vehicle only. Tissues were next incubated at 37°C in a humidified 5% CO2 incubator for 30 min, as detailed (7). Specimens were then preserved by formalin-fixation and paraffin-embedding prior to further analysis.
Immunoblotting (IB)
IBs used total cell lysates of enterocytes scraped from the small intestinal lumen of Min/+ mice, as described (31).
Results
Chronic treatment of Min/+ mice with celecoxib increased TGBβ signaling in the intestinal stroma
Female Min/+ mice were treated for 4 months with celecoxib (1,500 ppm) incorporated into AIN-76A diet, starting at 8 weeks of age. Tumor counts in the small and large intestine confirmed that short term celecoxib treatment significantly reduced the number of lesions; however, re-growth of resistant tumors occurred during long term celecoxib exposure (Supplementary Data, Fig. S1). Western blotting showed that celecoxib treatment, both short and long term, increased the overall level of TGFβ1, 2, 3 (henceforth referred to as TGFβ) expression in Min/+ ileum (Fig. 1). Serial sections of ileum were used to localize TGFβ expression, comparing the epithelial to the stromal compartments (Fig. 1A). In untreated Min/+ ileum, TGFβ was expressed in the membrane of enterocytes, and in a moderate number of stromal cells. Short term treatment produced little change in the stromal compartment, but was associated with increased TGFβ expression in the enterocyte cell membranes. A striking change in TGFβ expression was observed in the long term treated intestine, with a marked increase in the percentage of stromal cells expressing TGFβ accompanied by an overall decrease in enterocyte membrane expression. To learn whether the stromal cells expressing TGFβ engaged in canonical TGFβ signaling, we examined the expression and location of its transcriptional effector, Smad4, in treated tissues (Fig. 1B). Smad4 nuclear expression in the crypts of normal Min/+ enterocytes was reduced in ileum of long term-treated mice suggesting that in this compartment, signaling by the TGFβ family of ligands (TGFβ, BMPs, activins, etc) was inhibited. Moreover, TGFβ signaling in the stroma was significantly altered by the duration of celecoxib treatment. Few stromal cells were positively stained for Smad4 after short term celecoxib treatment, however most were positive after long term drug exposure.
Figure 1. Long term celecoxib treatment increases TGFβ signaling in the intestinal stroma.
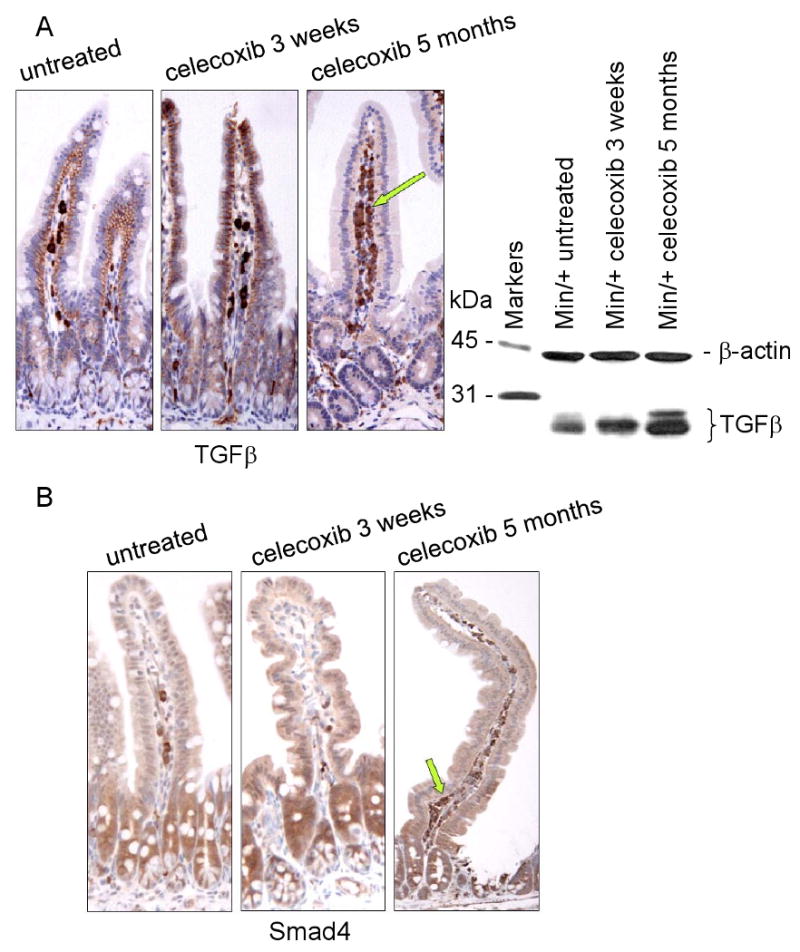
(A) Representative photomicrographs of IHC for TGFβ using sectioned ileum from untreated Min/+ mice, or those treated with celecoxib short (3 weeks) or long term (5 months). Arrow points to positively stained stromal cells in the lamina propria. IB analysis of total cell lysates from control and treated mice measured the expression of TGFβ using 1D11 antibody. The loading control shown below was probed for β-actin using AC-40 antibody.
(B) IHC for Smad-4 correlated the presence or absence of membrane-localized TGFβ on enterocytes with low or high TGFβ downstream signaling in the stroma, respectively. Arrows highlight the relative abundance of stained stromal cells. The antibody dilutions used for all IHC performed in this study are provided in Supplementary Table 1. The original magnification on all IHC images was 20×, unless otherwise indicated. All IHC was performed on the complete treatment set in parallel and all experiments used tissues from at least 3 different mice of the same treatment group.
Long term celecoxib treatment was associated with syndecan-1 ectodomain shedding from enterocytes and release of ECM-sequestered TGFβ
TGFβ ligands associate with HSPGs in the ECM adjacent to the basal membrane of enterocytes, and cellular concentrations of HSPGs regulate ligand availability by sequestering these soluble mediators (15, 25). To compare the location and levels of syndecan-1 and TGFβ in ileum of untreated Min/+ mice from short and long term-treated mice, we performed parallel IHC and IB analyses (Fig. 2A). The sections stained for syndecan-1 were those immediately adjacent to those stained for TGFβ (Fig. 1A), allowing us to assess the co-localization of these two proteins. As expected, syndecan-1 expression was observed in the basolateral membranes of enterocytes in untreated Min/+ small bowel, and within the crypt-villus unit, syndecan-1 expression appeared invariant (Fig. 2A). Several stromal cells in the lamina propria also expressed syndecan-1. In a pattern the same as that found for TGFβ expression, syndecan-1 levels were increased in enterocyte membranes of Min/+ mice treated short term with celecoxib, but were strikingly reduced with long term treatment. Since the epitope of clone 291-2 antibody is specific for the extracellular domain of syndecan-1, this loss from enterocyte membranes indicates surface shedding, a process associated with tumor promotion (29). The number of stromal cells in the lamina propria expressing syndecan-1 was also modulated by the duration of celecoxib treatment, with low expression in short term-treated ileum and high expression with long term treatment. IB of total cell lysates to detect steady-state syndecan-1 levels showed that short term treatment modestly increased syndecan-1 expression, but long term treatment decreased this protein relative to the untreated control (Fig. 2A). If the syndecan-1 ectodomain is required to maintain latent TGFβ in the proper context for autocrine signaling in enterocytes, especially those in the proliferative compartment of crypts, then shedding of these HSPG-bearing portions after long term treatment is expected to inhibit the negative growth control function of this cytokine.
Figure 2. Regulation of TGFβ availability by altered membrane expression of syndecan-1.
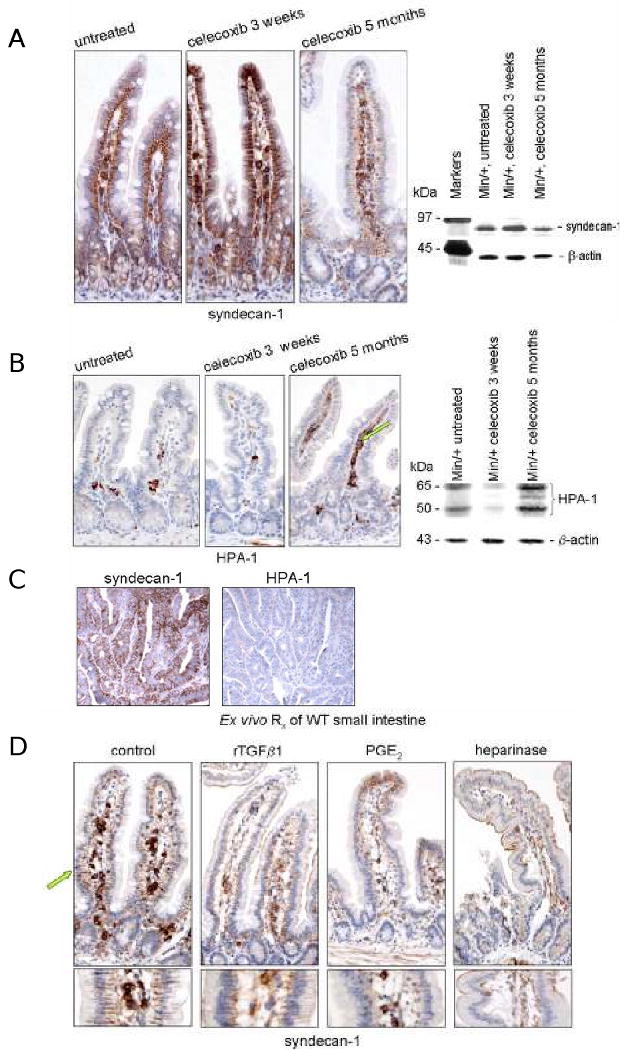
(A) Representative photomicrographs of IHC for syndecan-1 using sectioned ileum from untreated and treated Min/+ mice. IB analysis of total cell lysates from control and treated mice assessed the steady-state expression of syndecan-1 using 291-2 antibody. (B) Representative photomicrographs of IHC for HPA-1 of sectioned ileum from untreated and treated Min/+ mice. IB analysis for HPA-1 expression used HP3/17 antibody, a probe that detects two isoforms of HPA-1 in intestinal lysates (20). (C) Serial sections of an adenoma from an untreated Min/+ mouse immunostained for syndecan-1 and HPA-1. (D) Representative photomicrographs of sections of ex vivo-treated wild type ileum immunostained for syndecan-1. Tissues were incubated in medium with or without a drug treatment (PGE2, rTGFβ1, or heparinase) prior to formalin-fixing and paraffin-embedding. Treatment conditions were those specified in the Materials and Methods (42).
HPA-1 cleaves intact transmembrane syndecan-1, releasing HSPG-bound growth factors. As a result, the expression of HPA-1 in stromal cells is expected to inversely correlate with enterocyte membrane syndecan-1 expression. This relationship was confirmed in treated Min/+ tissues by IB and IHC analyses (Fig. 2B). In all specimens, HPA-1 expression was limited predominantly to stromal cells in the lamina propria and submucosa. The number of these HPA-1 positive stromal cells was strongly increased after long-term treatment of Min/+ mice with celecoxib. As shown in an adenoma from an untreated Min/+ mouse (Fig 2C), the expression of syndecan-1 and HPA-1 was inversely related in all tumors. This representative image also illustrates that these untreated tumors contained few, if any, HPA-1- or syndecan-1-positive infiltrating stromal cells.
To characterize the effects of inflammatory mediators on the membrane-localized expression of syndecan-1 in normal mucosa, we performed separate 30 min ex vivo treatments of WT small intestine with PGE2 (200 nM), bacterial heparinase (0.5 mU/ml), or rTGFβ1 (5 ng/ml) (Fig. 2D). Tissues were processed for syndecan-1 IHC immediately after treatment. In this experiment, the negative control tissue (incubated in medium without drug) retained the syndecan-1 ectodomain at the basolateral membranes of enterocytes. However, syndecan-1 was completely lost from these sites after separate treatments with PGE2 and heparinase, and diminished after treatment with rTGFβ1. We assume that our treatment conditions simulated physiological responses since near-normal tissue morphology was preserved in all of the specimens. These results show that the appearance of Min/+ enterocytes following chronic treatment with celecoxib was induced in normal untreated tissue in response to PGE2, TGFβ1, and heparinase.
Long term celecoxib treatment increased the number of myofibroblasts in the intestinal stroma
Next we identified the cell responsible for expression of syndecan-1, TGFβ, and HPA-1 in the stroma of Min/+ ileum. Serial sections were stained to identify myofibroblasts, which co-express vimentin and α-smooth muscle actin (αSMA) (9). We then counted and analyzed the number of double positive cells in the lamina propria as a function of treatment time (Fig. 3A). Short term celecoxib treatment significantly decreased the number of myofibroblasts in Min/+ mucosa, but long term treatment produced the opposite effect. Celecoxib-resistant tumors serially stained for αSMA, vimentin, Smad4, and HPA-1 showed clustered infiltrates of myofibroblasts, which were absent in untreated tumors (Fig. 3B). We also determined the relationship between stromal Cox-2 expression and treatment. Consistent with previous studies (7), stromal cells with the characteristic morphology of myofibroblasts showed high expression of Cox-2 in long term treated Min/+ ileum but not in this tissue from mice treated for 3 weeks (Fig. 3C). Finally, IB analysis for TGFβ in lysates of pool small bowel tumors from treated and untreated Min/+ mice showed that expression of this cytokine increased after long term celecoxib treatment (Supplementary Data Fig. S2).
Figure 3. Chronic celecoxib treatment increased the number of myofibroblasts in Min/+ intestine.
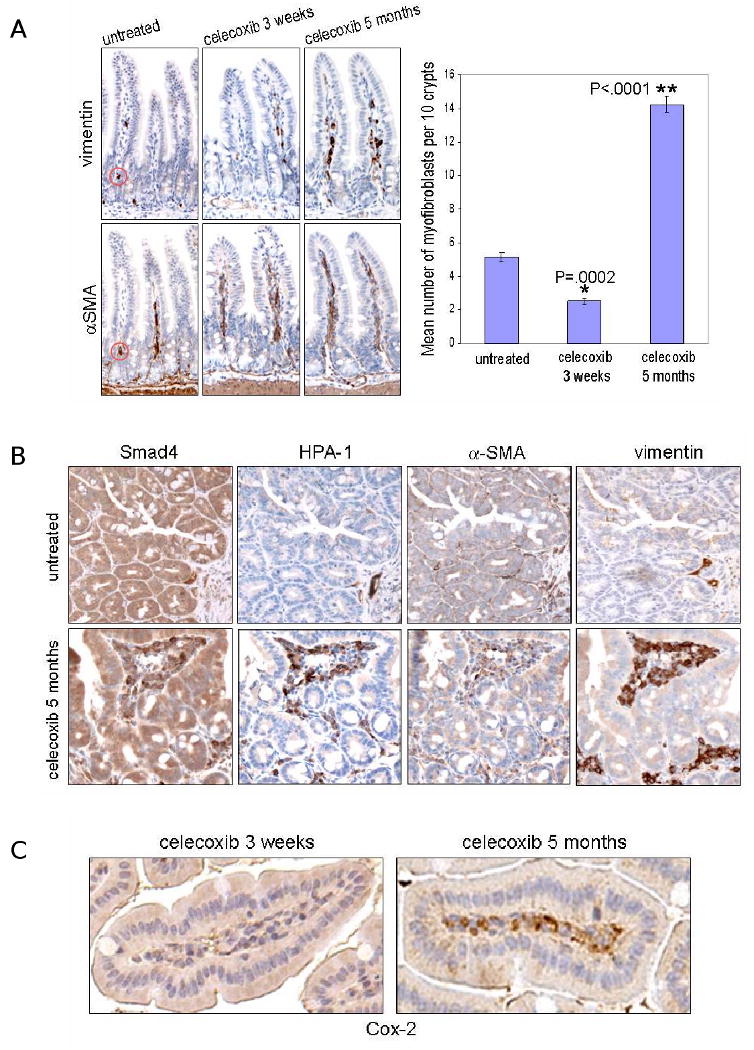
(A) Stromal myofibroblasts identified in representative photomicrographs of IHC for vimentin and αSMA of serially sectioned ileum from untreated and treated Min/+ mice. Doubly-positive cells were identified as indicated (red circles). The graph depicts the mean number of myofibroblasts per 10 crypts for the different celecoxib treatment times. N = 260 crypts from Min/+ ileum per treatment group; error bars represent SEM. (B) Adenomas from long term-treated Min/+ mice showed infiltrated clusters of HPA-1- and Smad4-expressing myofibroblasts, while these cells were largely absent in untreated tumors; 40× magnification. (C) Representative photomicrographs of IHC for Cox-2 used anti-murine Cox-2 antibody on sections of long or short term-treated Min/+ ileum.
Myofibroblast precursors, or fibrocytes, originate from CD14+ CD16- bone marrow-derived monocytes, and home to sites of inflammation and wound healing (9). Fibrocytes are distinguished from myofibroblasts in that they are CD34+, vimentin+, but lack expression of αSMA. We performed IHC for CD34 on untreated and treated Min/+ ileum and confirmed that CD34-positve staining in the submucosa and surrounding crypts was increased in Min/+ treated long term with celecoxib relative to untreated and short term-treated mice. This difference is best visualized upon viewing tissue sections at lower magnification (Fig. 4A). We then immunostained serial sections to detect vimentin+ CD34+ cells. Double positive cells were very rare in untreated and short term-treated Min/+ ileum (data not shown), but were readily visible in the submucosa of Min/+ treated long term (Fig. 4B). Consistent with recruitment of precursor cells from the circulation, Ki-67 and vimentin immunostaining of serial sections showed minimal stromal cell proliferation in the mucosa of any of the treatment groups (data not shown). Bone marrow-derived myeloid precursors also home to inflamed tissue and differentiate into macrophages. Using antibody for the mature macrophage marker, F4/80, IHC of the entire treatment set of tissues showed that these cells were not abundant in the Min/+ mucosa and did not significantly change with the duration of celecoxib treatment. Representative F4/80 immunostaining is shown (Supplementary Data Fig. S3). Taken together, these data suggest that chronic celecoxib exposure recruited bone marrow-derived precursors to the ileum and that a fibrocyte lineage subsequently expanded the resident myofibroblast population size.
Figure 4. Increased vimentin+ CD34+ stromal cells suggests recruitment of bone marrow-derived myofibroblast precursors to the intestinal submucosa of Min/+ during long term celecoxib treatment.
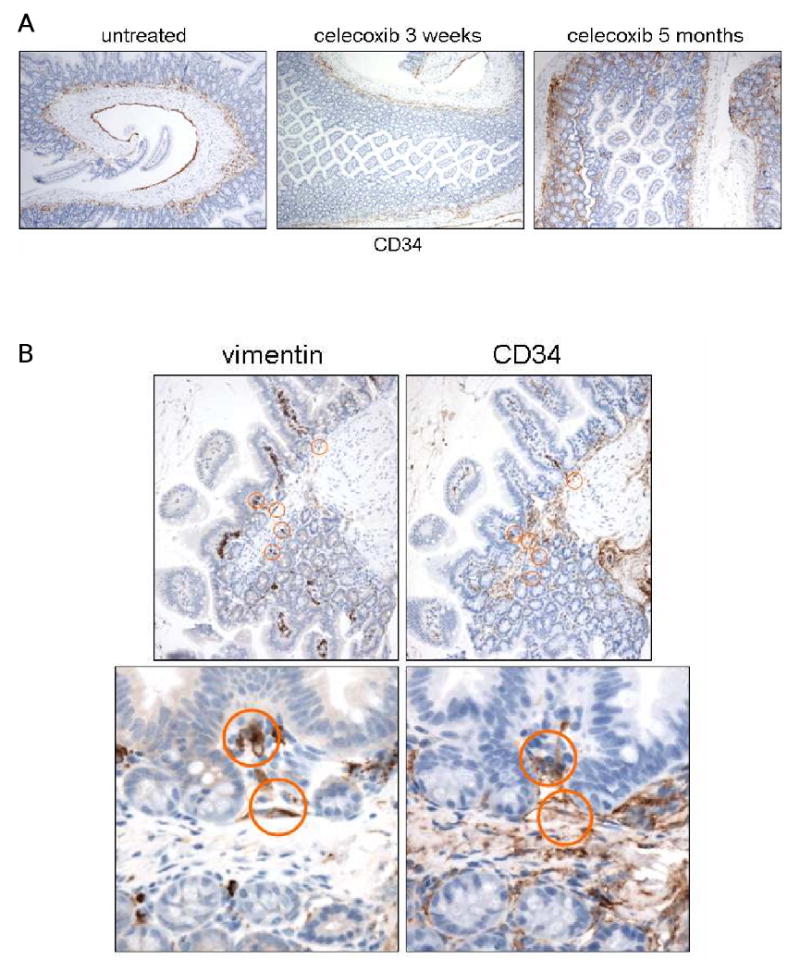
(A) Representative photomicrographs of IHC for CD34 of sectioned ileum from untreated and treated Min/+ mice; 10× magnification. (B) Representative photomicrographs of IHC for vimentin and CD34 of serially-sectioned ileum from long term-treated Min/+ mice at 20× and 40× magnifications show double positive stromal cells (circled) in the submucosa.
Long term celecoxib treatment induced intestinal fibrosis
In response to activation of TGFβ signaling, both fibrocytes and myofibroblasts express proteins that alter ECM composition (32). Downstream TGFβ transcriptional targets include various collagens, fibronectin, and laminins. These observations indicate that the presence of increased numbers of myofibroblasts and their elevated TGFβ signaling in the intestine of long term-treated Min/+ mice would be associated with increased ECM deposition. In our experiments, Masson's trichrome staining of long term-treated Min/+ ileum showed increased connective tissue (blue staining) in both the submucosa and the basement membrane subjacent to villus enterocytes (Fig. 5A). The opposite effect was observed in short term-treated Min/+ mice. Cross-sections obtained at the crypt level showed that collagen surrounding the crypts was present in long term-treated Min/+ mice but not in the short term-treated animals (Fig. 5A, Right). Because collagen deposition can be enhanced by aging, and the lifespan of Min/+ mice treated long term with celecoxib was extended by 2 months relative to untreated animals, we performed Masson's staining on additional controls to determine if collagen deposition was due to aging or drug resistance (Supplementary Data Fig. S4A). Increased collagen was not found in the submucosa or basement membranes of age- and gender-matched Min/+ mice fed a normal chow diet. Moreover, a similar negative result was also observed in 8-month old Apc1638/+ mice that, like Min/+, bear a germline Apc mutation but have an attenuated polyposis/tumor phenotype and a lifespan of ∼1 year. Finally, to confirm that collagen deposition was increased in long term-treated Min/+ ileum, we performed IHC for collagen IV, the predominant type of collagen in the intestinal submucosa, and obtained results consistent with those of the Masson stain (Supplementary Data Fig. S4B).
Figure 5. Long term celecoxib treatment induced increased collagen, fibronectin, and laminin-5 expression in Min/+ intestinal stroma.
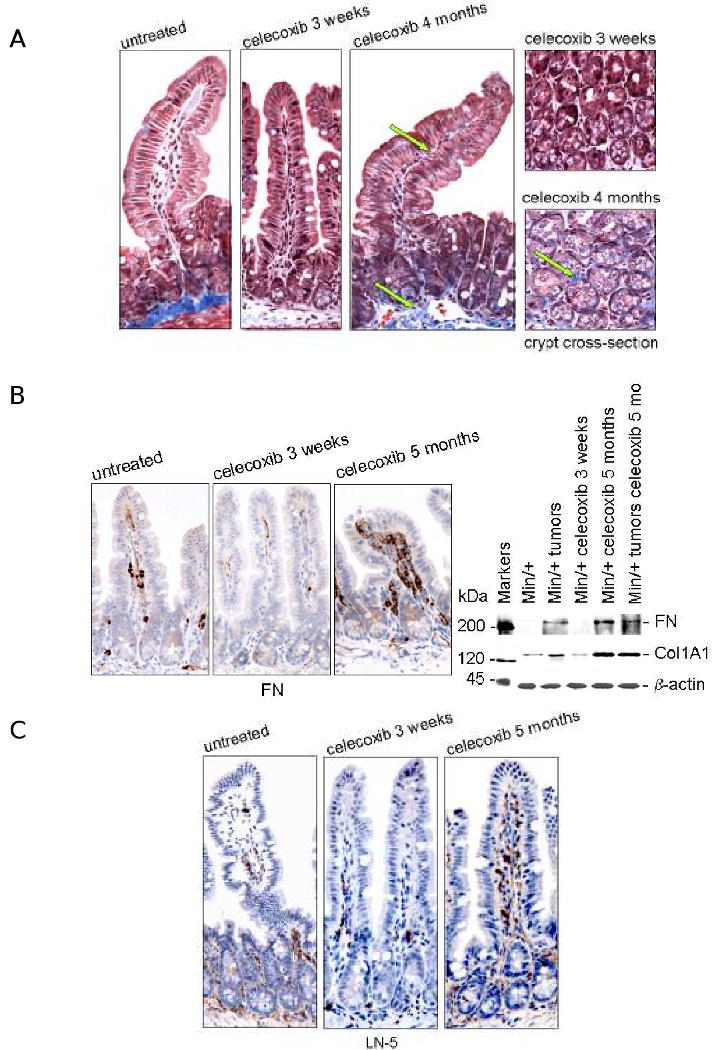
(A) Representative photomicrographs of Masson trichrome-stained ileum from untreated and treated Min/+ mice at 40× magnification. Images of tissues cross-sectioned at the base of crypts from short vs. long treatment groups are shown. Arrows point to the blue staining of the basement membranes indicative of enhanced collagen deposition. (B) IHC for fibronectin shown in representative photomicrographs of sectioned ileum from untreated and treated Min/+ mice revealed this ECM constituent was expressed in mesenchymal cells of the lamina propria. IB analysis for fibronectin expression used clone 10, and collagen IV used COL1A1. (C) Representative photomicrographs of IHC for laminin-5 γ2 in untreated and treated Min/+ mice. Control specimens processed in parallel that did not receive Proteinase XXIV treatment yielded no positive laminin-5 γ2 staining (data not shown).
Additional ECM constituents targeted by TGFβ include fibronectin (FN), laminin-5 (LN-5) and collagen type I, which are all proteins modulated during tissue remodeling, fibrosis, and tumor progression (9, 10). Chronic treatment with celecoxib dramatically increased FN expression in the submucosa, and the expanded population of myofibroblasts was strongly positive for this ECM protein (Fig. 5B). Consistent with increased TGFβ in the stromal compartment, IB analysis showed that FN and collagen 1A1 (COL1A1) were both up-regulated in chronically-treated intestine and in untreated tumors and in long term-treated Min/+ adenomas (Fig. 5B). LN-5 stimulates cell migration and invasion and has been implicated in tissue remodeling and colon cancer progression (33). IHC staining of 7 month-old Min/+ mice fed a normal diet without drug showed relatively low expression of COL1A1, FN, and LN-5, similar to the AIN-76A-fed Min/+ controls (Supplementary Data Fig. S5). Lastly, IHC to detect LN-5 levels showed increased expression in the pericryptal regions of the small bowel submucosa and in the lamina propria of villi in long term-treated Min/+ mice (Fig. 5C). These data collectively indicate that chronic exposure to celecoxib induced intestinal fibrosis.
Discussion
Inflammation and tumor formation are closely linked, and these studies define some of the elements driving this association in the tumor-prone intestine of the Min/+ mouse. The normal inflammatory response exists to permit wound healing and microbial defense, and requires time-dependent coordination of both epithelial and stromal signaling elements. Tumor formation is promoted when inflammation persists. We found that a brief period of PGE2 inhibition both reduced tumor formation and induced changes in the intestine suggesting suppression of the stromal inflammatory response (7). In contrast, tumor production and COX-2 expression were increased during long term administration of celecoxib, which also produced a striking reduction of anti-inflammatory syndecan-1 in enterocyte membranes, coincident with upregulation of TGFβ signaling in the tissue stroma (Fig. 1). HSPGs such as syndecan-1 create a reservoir for TGFβ and locally-secreted Wnt proteins, providing important modulation of autocrine and paracrine signaling. Loss of HSPG function at enterocyte membranes should alter both epithelial and stromal cell differentiation and growth. In agreement with this, loss of syndecan-1 from the enterocyte membranes was associated with increased numbers of myofibroblasts in the lamina propria and submucosa, further evidence of increased TGFβ signaling and tissue remodeling. We conclude that chronic inhibition of COX-2 defeats the purpose of NSAID chemoprevention, likely because this condition results in a compensatory increase in stromal myofibroblasts, the source of COX-2, PGE2 and TGFβ in the intestinal mucosa.
These results from the Min/+ tumor model are supported by work in human tissues from the Adenoma Prevention with Celecoxib (APC) trial. The APC trial randomized patients at high risk for colorectal cancer to receive either placebo, celecoxib 200 mg twice daily, or celecoxib 400 mg twice daily. Patients treated with celecoxib had a significantly reduced risk of adenoma development over 1 year and 3 year surveillance intervals (3, 34). In a subset of patients, magnification chromoendoscopy was performed at baseline and after 8-12 months of treatment to identify rectal aberrant crypt foci (ACF) and to obtain biopsy specimens of both ACF and normal mucosa. These tissues were studied using IHC to characterize inflammatory mediators, including the TGFβ signaling partner, SMAD4. SMAD4 expression did not vary between normal rectal mucosa and ACF, however SMAD4 expression in baseline ACF was a predictor of the primary outcome measurement of the APC trial, i.e. adenoma recurrence at either the year 1 or the year 3 study colonoscopy (35). Patients who had reduced levels of SMAD4 in their ACF were less likely to have recurrent adenomas detected during the APC trial. In patients with intact nuclear SMAD4 expression at baseline, 80% developed recurrent adenomas, compared to 18% of those with reduced SMAD4 levels (risk ratio =0.23; p=0.01). When the effect of treatment was considered, the prognostic value of SMAD4 expression remained. This result from a human chemoprevention trial is consistent with the relationship between TGFβ signaling and celecoxib anti-tumor response observed in Min/+ mice.
Crosstalk between critical signaling pathways in response to stress conditions is a likely basis for acquired resistance to celecoxib. The key pathway necessary for enterocyte proliferation, Wnt signaling, is co-dependent on PGE2 to effect tissue regeneration (36). Previously, we showed that PGE2 stimulated enterocyte growth and survival in Min/+ mice via transactivation of the epidermal growth factor receptor (37). Crosstalk also exists between PGE2 and TGFβ pathways in the intestine. For instance, mesenchymal loss of LKB1, the upstream regulator of TGFβ-dependent myofibroblast differentiation, caused polyp formation (38). Cox-2-PGE2 and TGFβ signaling pathways both activate the transcriptional pro-inflammatory and anti-apoptotic programs of NF-κB. Relevant to our work is the finding that TLR4 enhanced TGFβ signaling and fibrosis, as well as Cox-2 expression, in the normal intestine and in colitis-associated tumorigenesis (11, 15, 18). TLR4 signals with the adaptor Myeloid Differentiation Factor (MyD88) to activate NF-κB pro-inflammatory signaling, increasing Cox-2 expression. Suppressing this signaling pathway inhibited intestinal tumor growth, since MyD88-/-Min/+ mice survived longer and bore slower growing tumors with lower Cox-2 expression (11). However at sites of inflammation, or in tumors where concentrations of inflammatory cytokines and TGFβ ligands are high, dose-dependent cross-control of TGFβ, NF-κB, and c-Jun N-terminal kinase activation occurs (39). This signaling reciprocity, in turn, dictates biological outcome, including drug sensitivity or resistance (40). Consistent with this view, a selective IKKβ inhibitor, designed to inhibit NF-κB signaling, exacerbated intestinal inflammation upon prolonged administration by increasing cytokine IL-1β secretion (41).
In addition to the inflammatory mediators produced by stromal myofibroblasts, these data suggest other mechanisms for enhanced tumor formation in the Min/+ model. For example, intestinal barrier function is the gatekeeper blocking the development of inflammation, and is dependent on E-cadherin-mediated epithelial cell-cell adhesion. We showed previously that small bowel enterocytes in Min/+ mice displayed deficient E-cadherin-mediated cell-cell adhesion, a defect normalized by 3-week celecoxib treatment (31). Others have reported on the role of syndecan-1 on the basolateral surfaces of enterocytes in barrier function maintenance (26). Our results are consistent with this finding, since syndecan-1 expression was increased in the basolateral membranes of enterocytes from Min/+ treated short term with celecoxib. In addition, separate ex vivo treatments of WT small bowel with PGE2, TGFβ, or heparinase each induced syndecan-1 ectodomain loss, reproducing the appearance of ileum from chronically-treated Min/+ mice (Fig. 2D). This result indicates that PGE2, HPA-1, as well as TGFβ have negative effects on the anti-inflammatory functions of syndecan-1.
Our results have implications for chemoprevention of patients at high risk for CRC. Although the APC trial treated patients for 3 years with celecoxib without demonstrating an overall increase in tumor formation, it is possible that chronic use in humans will mimic results from the Min/+ model. Just as some patients are more prone to inflammatory conditions of the intestine, resistance to the anti-tumor activity of celecoxib may develop in patients at different rates, perhaps related to inter-individual differences in TGFβ signaling. These data suggest that the use of celecoxib for adenoma prevention should be limited to short term treatment intervals, for a minimum period allowing the tissue to return fully to a baseline state before re-starting medication. Two other observations argue for this approach. First, in patients with FAP, celecoxib induced the regression of existing adenomas (2), and as a result chronic administration should not be required for chemopreventive efficacy. In addition, celecoxib use at high doses was associated with cardiovascular adverse events, and regimens that limit treatment duration should minimize this risk. In summary, these data demonstrate long term consequences resulting from chronic COX-2 inhibition that should be considered in clinical settings warranting this treatment.
Supplementary Material
Figure S1. Short term celecoxib reduced visible intestinal tumors in Min/+ mice, but long-term drug exposure caused drug-resistant tumors, consistent with our previously reported result (7). All mice were fed the tumor promoting, high fat AIN-76A diet. Min/+ mice treated for 3 weeks and untreated controls were sacrificed at 3 months of age because untreated animals at this time become moribund due to tumor load. However, Min/+ mice treated for 5 months with celecoxib were sacrificed at ∼7 months of age because these animals maintained a healthy appearance and weight and the added time optimized drug-resistant tumor outgrowth and progression. All tumors were counted by an investigator who was blinded to the genotype and treatment status of the mice. N = 6 mice per treatment group. Error bars represent standard error of the mean (SEM).
Figure S2. IB analysis showed the relative TGFβ expression in total cell lysates prepared from the non-tumor mucosa of untreated Min/+ mice vs. total cell lysates prepared from pooled small bowel tumors of untreated control and celecoxib long term-treated mice. This experiment was performed as detailed in Fig. 1A.
Figure S3. Representative photomicrographs of IHC for the mature macrophage marker, F4/80, showed that these myeloid cells were relatively rare in the small and large intestine of Min/+ mice at all treatment points.
Figure S4. (A) Representative photomicrographs of Masson trichrome-stained, age-matched female Min/+ control mouse ileum (Left) and 8 month-old male Apc1638/+ mouse ileum (Right) showed that increased collagen deposition observed in 7-month old Min/+ treated long term with celecoxib was drug-induced and not due to aging. (B) Representative photomicrographs of IHC for CD34, collagen IV, and fibronectin of 7-month old, female Min/+ mouse ileum.
Figure S5. Representative photomicrographs of IHC for collagen IV of sectioned ileum from untreated and short and long term-treated Min/+ mice is shown.
Table S1. List of antibodies used in this study, dilutions used for IHC, and vendors.
Abbreviations
- APC
Adenomatous polyposis coli
- Min/+
C57BL/6J-Min/+
- WT
C57BL/6J-Apc+/+
- HPA-1
heparanase-1
- TGFβ
Transforming growth factor-beta
- IHC
immunohistochemistry
- ECM
extracellular matrix
- NSAID
non-steroidal anti-inflammatory drug
- PGE2
prostaglandin E2
- CRC
colorectal cancer
- IBD
inflammatory bowel disease
- rTGFβ1
recombinant TGFβ1
- αSMA
α-smooth muscle actin
References
- 1.Waddell WR, Ganser GF, Cerise EJ, Loughry RW. Sulindac for polyposis of the colon. The American Journal of Surgery. 1989;157:175–9. doi: 10.1016/0002-9610(89)90442-x. [DOI] [PubMed] [Google Scholar]
- 2.Steinbach G, Lynch PM, Phillips RKS, et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. The New England Journal of Medicine. 2000;342(26):1946–52. doi: 10.1056/NEJM200006293422603. [DOI] [PubMed] [Google Scholar]
- 3.Arber N, Eagle CJ, Spicak J, et al. Celecoxib for the prevention of colorectal adenomatous polyps. N Engl J Med. 2006;355(9):885–95. doi: 10.1056/NEJMoa061652. [DOI] [PubMed] [Google Scholar]
- 4.Bertagnolli MM. Chemoprevention of colorectal cancer with cyclooxygenase-2 inhibitors: two steps forward, one step back. Lancet Oncology. 2007;8:439–43. doi: 10.1016/S1470-2045(07)70139-0. [DOI] [PubMed] [Google Scholar]
- 5.Solomon SD, McMurray JJ, Pfeffer MA, et al. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N Engl J Med. 2005;352(11):1071–80. doi: 10.1056/NEJMoa050405. [DOI] [PubMed] [Google Scholar]
- 6.Bresalier RS, Sandler RS, Quan H, et al. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med. 2005;352(11):1092–102. doi: 10.1056/NEJMoa050493. [DOI] [PubMed] [Google Scholar]
- 7.Carothers AM, Moran AE, Cho NL, Redston M, Bertagnolli MM. Changes in antitumor response in C57BL/6J-Min/+ mice during long-term administration of a selective cyclooxygenase-2-inhibitor. Cancer Research. 2006;66:6432–8. doi: 10.1158/0008-5472.CAN-06-0992. [DOI] [PubMed] [Google Scholar]
- 8.Sonoshita M, Takaku K, Sasaki N, et al. Acceleration of intestinal polyposis through prostaglandin receptor EP2 in Apc (Delta 716) knockout mice. Nat Med. 2001;7(n):1048–51. doi: 10.1038/nm0901-1048. [DOI] [PubMed] [Google Scholar]
- 9.Powell DW, Adegboyege PA, Di Mari JF, Mifflin RC. Epthelial cells and their neighbors. I. Role of intestinal myofibroblasts in development, repair, and cancer. Am J Physiol. 2005;289:G2–G7. doi: 10.1152/ajpgi.00075.2005. [DOI] [PubMed] [Google Scholar]
- 10.Otte JM, Rosenberg IM, Podolsky DK. Intestinal myofibroblasts in innate immune responses. Gastroenterology. 2003;124:1866–78. doi: 10.1016/s0016-5085(03)00403-7. [DOI] [PubMed] [Google Scholar]
- 11.Takeda H, Miyoshi H, Tamai Y, Oshima M, Taketo MM. Simultaneous expression of COX-2 and mPGES-1 in mouse gastrointestinal hamartomas. British Journal of Cancer. 2004;90:701–4. doi: 10.1038/sj.bjc.6601584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsujii M, D RN. Alterations in cellular adhesion and apoptosis in epithelial cells overexpressing prostaglandin endoperoxide synthase 2. Cell. 1995:493–501. doi: 10.1016/0092-8674(95)90127-2. [DOI] [PubMed] [Google Scholar]
- 13.Enders G. Cyclooxygenase 2 overexpression abrogates the antiproliferative effects of TGF-β. Br J Cancer. 2007;7:1388–92. doi: 10.1038/sj.bjc.6604048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pardali K, Moustakas A. Actions of TGF-β as tumor suppressor and pro-metastatic factor in human cancer. Biochim Biophys Acta. 2007;1775:21–62. doi: 10.1016/j.bbcan.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 15.Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFβ activation. J Cell Sci. 2003;116:217–24. doi: 10.1242/jcs.00229. [DOI] [PubMed] [Google Scholar]
- 16.Zhu Y, Richardson J, Parada L, Graff J. Smad3 mutant mice develop metastatic colorectal cancer. Cell. 1998:703–14. doi: 10.1016/s0092-8674(00)81730-4. [DOI] [PubMed] [Google Scholar]
- 17.Becker C, Fantini M, Neurath M. TGF-beta as a T cell regulator in colitis and colon cancer. Cytokine Growth Factor Rev. 2006;17:97–106. doi: 10.1016/j.cytogfr.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 18.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodeling. Nat Rev Mol Cell Biol. 2002;3:349–63. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- 19.McKaig BC, Hughes K, Tighe PJ, Mahida TR. Differential expression of TGFβ isoforms by normal and inflammatory bowel disease intestinal myofibroblasts. Am J Physiol. 2002:C172–C82. doi: 10.1152/ajpcell.00048.2001. [DOI] [PubMed] [Google Scholar]
- 20.Di Sabatino A, Jackson CL, Pickard KM, et al. Transforming growth factor-β signaling and matrix metalloproteinases in the mucosa overlying Crohn's disease strictures. Gut. 2009 doi: 10.1136/gut.2008.149096. In press. [DOI] [PubMed] [Google Scholar]
- 21.Grady W, Rajput A, Meyeroff L, et al. Mutation of the type II transforming growth factor beta receptor is coincident with the transformation of human colon adenomas to malignant carcinomas. Cancer Res. 1998;58:3101–4. [PubMed] [Google Scholar]
- 22.Saisekharan R, Shriver Z, Narayanasami U. Roles of heparan-sulphate glycosaminoglycans in cancer. Nat Rev Cancer. 2002;2:521–8. doi: 10.1038/nrc842. [DOI] [PubMed] [Google Scholar]
- 23.Hashimoto Y, Skacel M, Adams JC. Association of loss of epithelial syndecan-1 with stage and local metastasis of colorectal adenocarcinomas: An immunohistochemical study of clinically annotated tumors. BMC Cancer. 2008;8:185. doi: 10.1186/1471-2407-8-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alexander CM, Reichsman F, Hinkes MT, et al. Syndecan-1 is required for Wnt-1-induced mammary tumorigenesis in mice. Nat Genet. 2000;25:329–32. doi: 10.1038/77108. [DOI] [PubMed] [Google Scholar]
- 25.Chen Q, Sivakumar P, Barley C, Peters DM, Gomes RR, Farach-Carson MC. Potential role of heparan sulfate proteoglycans in regulation of Transforming Growth Factor-β (TGF-β) by modulating assembly of Latent TGF-β-binding Protein-1. J Biol Chem. 2007;282:26418–30. doi: 10.1074/jbc.M703341200. [DOI] [PubMed] [Google Scholar]
- 26.Bode L, Salvestrini C, Park PW, et al. Heparan sulfate and syndecan-1 are essential in maintaining murine and human intestinal epithelial barrier function. J Clin Invest. 2008;118:229–38. doi: 10.1172/JCI32335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fitzgerald ML, Wang Z, Park PW, Murphy G, Bernfield M. Shedding of syndecan-1 and -1 ectodomains is regulated by multiple signaling pathways and mediated by a TIMP-3-sensitive metalloproteinase. J Cell Bio. 2000;148:811–24. doi: 10.1083/jcb.148.4.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kliment CR, Englert JM, Gouchuico BR, et al. Oxidative stress alters syndecan-1 distribution in lungs with pulmonary fibrosis. J Biol Chem. 2009;284:3537–45. doi: 10.1074/jbc.M807001200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang Y, MacLeod V, Miao HQ, et al. Heparanase enhances syndecan-1 shedding: A novel mechanism for stimulation of tumor growth and metastasis. J Biol Chem. 2007;282:13326–33. doi: 10.1074/jbc.M611259200. [DOI] [PubMed] [Google Scholar]
- 30.Waterman M, Ben-Izhak O, Eliakim R, Groisman G, Vlodavsky I, Ilan N. Heparanase upregulation by colonic epithelium in inflammatory bowel disease. Mod Pathol. 2007;20:8–14. doi: 10.1038/modpathol.3800710. [DOI] [PubMed] [Google Scholar]
- 31.Carothers AM, Melstrom KAJ, Mueller JD, Weyant MJ, Bertagnolli MM. Progressive changes in adherens junction structure during intestinal adenoma formation in Apc mutant mice. J Biol Chem. 2001;276(42):39094–102. doi: 10.1074/jbc.M103450200. [DOI] [PubMed] [Google Scholar]
- 32.Bellin A, Mattoli S. The role of the fibrocyte, a bone marrow-derived mesenchymal progenitor, in reactive and reparative fibroses. Lab Invest. 2007;87:858–70. doi: 10.1038/labinvest.3700654. [DOI] [PubMed] [Google Scholar]
- 33.Pyke C, Salo S, Ralfkiaer E, Romer J, Dano K, Tryggvason K. Laminin-5 is a marker of invading cancer cells in some human carcinomas and is coexpressed with the receptor for urokinase plasminogen activator in budding cancer cells in colon adenocarcinoma. Cancer Research. 1995;55:4132–9. [PubMed] [Google Scholar]
- 34.Bertagnolli MM, Eagle CJ, Zauber AG, et al. A Randomized Trial of Celecoxib to Prevent Sporadic Colorectal Adenomas. N Engl J Med. 2006;355(9):873–84. doi: 10.1056/NEJMoa061355. [DOI] [PubMed] [Google Scholar]
- 35.Cho NL, Zauber A, Redston M, et al. Aberrant Crypt Foci in the Adenoma Prevention with Celecoxib (APC) Trial. Cancer Prevention Research. 2008;1(1):21–31. doi: 10.1158/1940-6207.CAPR-07-0011. [DOI] [PubMed] [Google Scholar]
- 36.Shao J, Jung C, Liu C, S H. Prostaglandin E2 stimulates the β-catenin-T cell factor-dependent transcription in colon cancer. J Biol Chem. 2005;280:26565–72. doi: 10.1074/jbc.M413056200. [DOI] [PubMed] [Google Scholar]
- 37.Moran AE, Hunt DH, Javid SH, Redston M, Carothers AM, Bertagnolli MM. Apc deficiency is associated with increased Egfr activity in the intestinal enterocytes and adenomas of C57BL/6J-Min/+ mice. J Biol Chem. 2004:43261–72. doi: 10.1074/jbc.M404276200. [DOI] [PubMed] [Google Scholar]
- 38.Vaahtomeri K, Ventelä E, Laajanen K, et al. Lkb1 is required for TGFβ-mediated myofibroblast differentiation. J Cell Sci. 2008;121:3531–40. doi: 10.1242/jcs.032706. [DOI] [PubMed] [Google Scholar]
- 39.Shim JH, Xiao C, Paschal AE, et al. TAK1, but not TAB1 or TAB2, plays an essential role in multiple signaling pathways in vivo. Genes Dev. 2005;19:2668–81. doi: 10.1101/gad.1360605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu T, Tian L, Han Y, Vogelbaum M, Stark GR. Dose-dependent cross-talk between the transforming growth factor-β and interleukin-1 signaling pathways. Proc Natl Acad Sci USA. 2007;104:4365–70. doi: 10.1073/pnas.0700118104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Greten FR, Arkan MC, Bollrath J, et al. NF-κB is a negative regulator of IL-1β secretion as revealed by genetic and pharmacological inhibition of IKKβ. Cell. 2007;130:918–31. doi: 10.1016/j.cell.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moran AE, Carothers AM, Weyant MJ, Redston M, Bertagnolli MM. Carnosol inhibits β-catenin tyrosine phosphorylation and prevents adenoma formation in the C57BL/6J-Min/+ (Min/+) mouse. Cancer Research. 2005;65:1097–104. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Short term celecoxib reduced visible intestinal tumors in Min/+ mice, but long-term drug exposure caused drug-resistant tumors, consistent with our previously reported result (7). All mice were fed the tumor promoting, high fat AIN-76A diet. Min/+ mice treated for 3 weeks and untreated controls were sacrificed at 3 months of age because untreated animals at this time become moribund due to tumor load. However, Min/+ mice treated for 5 months with celecoxib were sacrificed at ∼7 months of age because these animals maintained a healthy appearance and weight and the added time optimized drug-resistant tumor outgrowth and progression. All tumors were counted by an investigator who was blinded to the genotype and treatment status of the mice. N = 6 mice per treatment group. Error bars represent standard error of the mean (SEM).
Figure S2. IB analysis showed the relative TGFβ expression in total cell lysates prepared from the non-tumor mucosa of untreated Min/+ mice vs. total cell lysates prepared from pooled small bowel tumors of untreated control and celecoxib long term-treated mice. This experiment was performed as detailed in Fig. 1A.
Figure S3. Representative photomicrographs of IHC for the mature macrophage marker, F4/80, showed that these myeloid cells were relatively rare in the small and large intestine of Min/+ mice at all treatment points.
Figure S4. (A) Representative photomicrographs of Masson trichrome-stained, age-matched female Min/+ control mouse ileum (Left) and 8 month-old male Apc1638/+ mouse ileum (Right) showed that increased collagen deposition observed in 7-month old Min/+ treated long term with celecoxib was drug-induced and not due to aging. (B) Representative photomicrographs of IHC for CD34, collagen IV, and fibronectin of 7-month old, female Min/+ mouse ileum.
Figure S5. Representative photomicrographs of IHC for collagen IV of sectioned ileum from untreated and short and long term-treated Min/+ mice is shown.
Table S1. List of antibodies used in this study, dilutions used for IHC, and vendors.