

Abstract
Background
Dosing frequency is an important determinant of regimen effectiveness.
Methods
To compare efficacy of once-daily (QD) versus twice-daily (BID) antiretroviral therapy, we randomized HIV-1 positive, treatment-naïve, patients to lopinavir/ritonavir (LPV/r) 400/100 mg BID (n=160) or LPV/r 800/200 mg QD (n=161), plus either emtricitabine 200 mg QD and extended-release stavudine (d4T-XR) 100 mg QD, or tenofovir 300 mg QD. Randomization was stratified by screening HIV-1 RNA </≥100,000 copies/mL. The primary efficacy endpoint was sustained virologic suppression/response (< 200 copies/mL, SVR) through week 48.
Results
Subjects were 78% male, 33% Hispanic, and 34% black. 82% of subjects completed the study, and 71% remained on initially assigned dose schedule. Probability of SVR did not differ significantly for the BID vs. QD comparison, with an absolute proportional difference (95% confidence interval [CI]) of 0.03 (−0.07, 0.12). The comparison depended on the screening RNA stratum (p=0.038); in the higher RNA stratum, the probability (95% CI) of SVR was significantly better in the BID arm: 0.89 (0.79, 0.94) compared to 0.76 (0.64, 0.84) in the QD arm; difference of 0.13 (0.01, 0.25). LPV trough plasma concentrations were higher with BID dosing. Adherence to prescribed doses of LPV/r was 90.6% in the QD arm versus 79.9% in the BID arm (p<0.001).
Conclusions
Although subjects assigned to QD regimens had better adherence, overall treatment outcomes were similar to the BID regimens. Subjects with HIV-1 RNA ≥100,000 copies/mL had better SVR on BID regimens at 48 weeks, suggesting a possible advantage in this setting for more frequent dosing.
Keywords: HIV, clinical trials, adherence, lopinavir, pharmacokinetics
INTRODUCTION
Daily dosing frequency is an important component of regimen complexity, and is generally regarded as a key contributor to treatment success or failure.1 Although patients and physicians consistently prefer once-a-day drugs to those administered more frequently,2,3 comparative trials suggest only a modest benefit in adherence to once-daily regimens as compared to twice-daily.4,5,6
Understanding the relative value of a simplified regimen is especially critical in chronic and life-threatening conditions such as tuberculosis and human immunodeficiency virus (HIV) infection. Paradoxically, although once-daily drug administration modestly improves the percent of doses taken as prescribed, drug concentrations at the end of a dosing interval are generally lower than when the same drug is administered more frequently.7,8 In addition, occasional missed or late doses may result in disproportionately reduced effectiveness if the drug is only administered once daily.4,5
Patients with chronic HIV infection can maintain prolonged control of virus replication, improve quality of health, and prolong life by taking combination antiretroviral therapy. Strategies to reduce the complexity of anti-HIV therapy have the potential to improve outcomes by increasing the likelihood of sustained virologic suppression, and by reducing the risk of drug resistance.2,9 While adherence to therapy has been shown to be a key factor in the success of a regimen,10,11 individual adherence may be difficult to predict and control.12,13,14
Lopinavir coformulated with ritonavir (LPV/r) was the first HIV protease inhibitor to be approved for use as either a QD or BID drug in treatment-naive patients. Our study, AIDS Clinical Trials Group (ACTG) Protocol A5073, was designed to compare the virologic efficacy of a once-daily versus twice-daily regimen containing LPV/r combined with once-daily antiretroviral nucleoside reverse transcriptase inhibitors (NRTI’s) in treatment-naïve HIV-1-infected patients. The study’s primary efficacy objective was to compare the probability of achieving a sustained virologic response (SVR) through 48 weeks. Once-a-day administration of LPV/r was investigational when our study was initiated, although the Food and Drug Administration approved this regimen for treatment-naïve patients three months after our study closed to accrual.
METHODS
Protocol A5073 was a multicenter, randomized, 48-week open-label trial combining two once-daily NRTI’s with: A) LPV/r 400 mg/100 mg BID self-administered (BID arm), B) LPV/r 800 mg/200 mg QD self-administered (QD arm) or C) LPV/r 800 mg/200 mg QD directly-observed therapy (results for this arm presented elsewhere15). 321 subjects were randomized (160 to BID, 161 to QD) from 23 U.S. sites and one site in Johannesburg, South Africa (16 subjects). Major entry criteria included age ≥13 years old, ≤7 days of prior ART, and screening plasma HIV-1 RNA ≥ 2000 copies/mL.
Eligible subjects were randomized via permuted blocks in a 2:2:1 ratio to the three study arms, with stratification by screening plasma HIV-1 RNA level: <100,000 and ≥100,000 copies/mL. Allocation was performed using a centralized-computer system requiring site personnel to enter subjects’ eligibility data to receive treatment assignment. Subjects received two NRTI’s: emtricitabine (FTC) 200 mg QD with either extended-release stavudine (d4T-XR, investigational formulation ultimately not developed) 100 mg QD or, beginning approximately 18 months after the enrollment started, tenofovir disoproxil fumarate (TDF) 300 mg QD. Subjects were then also allowed to switch from d4T-XR to TDF or vice versa. TDF or FTC were given as separate pills. All study drugs were provided through the protocol. Randomizations occurred between October, 2002 and January, 2005; follow-up ended in January, 2006.
Study visits were scheduled at weeks 2 (safety only), 4, 8, 16, 24, 32, 40, and 48. Visits included clinical assessments and laboratory testing, with plasma HIV-1 RNA measured by Roche UltraSensitive HIV-1 Monitor® assay (limit of quantification 50 copies/mL). LPV/r trough concentrations (Ctrough) were measured at study weeks 4, 16, 24 and 48. Adherence to LPV/r was assessed using electronic monitors (Medication Event Monitoring System [MEMS®, Aardex, Zug, Switzerland]), as well as self-reported adherence (results similar, not presented here).
SVR was defined as lack of: 1) confirmed HIV-1 RNA level ≥ 200 copies/mL after two consecutive HIV-1 RNA values < 200 copies/mL (confirmed viral relapse), 2) confirmed HIV-1 RNA level ≥ 200 copies/mL at or after visit week 24 without prior confirmed viral relapse or 3) HIV-1 RNA level ≥ 200 copies/mL at the Week 48 visit, which did not require confirmation (week 48 failure).
HIV-1 genotyping was performed using the GeneSeq® HIV assay at ViroLogic, Inc. (South San Francisco, CA; now called Monogram Biosciences) for the 65 virologic failures. Genotypic resistance was assessed using the Stanford Algorithm (version 4.2.1, 7/31/06).
Safety and tolerability were assessed as time to the first new grade 3 or 4 sign, symptom, or laboratory toxicity as defined by the ACTG,16 that was at least one grade higher than at baseline, and as time from start of treatment until premature discontinuation of the originally assigned LPV/r dose schedule.
Plasma concentrations of lopinavir (LPV) and ritonavir (RTV) were determined using a validated high-performance liquid chromatography (HPLC) assay with ultraviolet detection as previously described.17 Calibration standard curves ranged from 25 to 5000 ng/mL. Intraday and interday coefficients of variation were less than 6% for all analytes.
The planned sample size was 300 subjects in the BID and QD arms based on the goal of estimating the difference in probability of SVR between the two arms with a 95% two-sided confidence interval width of 0.2 or less. Efficacy analyses were intent-to-treat according to subjects’ initially randomized LPV/r dose schedule. Two-sided 95% confidence intervals and 5% significance level tests were applied. Presented p-values are nominal. The study was reviewed by an ACTG appointed study monitoring committee on three occasions, and blinded efficacy comparisons were presented only at the first review, soon after study initiation. Time-to-event distributions were summarized with Kaplan-Meier (KM) estimated survival curves and compared with a log rank test stratified by screening plasma HIV-1 RNA (<100,000 versus ≥100,000 copies/mL). The primary efficacy analysis was based on KM estimates at 48 weeks of the probability of SVR based on the time from randomization to the initial virologic failure sample date. Subjects without observed virologic failure were right censored at the date of their last HIV-1 RNA measurement. Unconfirmed failures prior to week 48 without a subsequent confirmation sample were treated as failures in the primary analysis. Variance estimation used Greenwood’s formula.
Treatment interaction with screening plasma HIV-1 RNA was assessed with a Cox proportional hazards model on time to virologic failure. Supplemental analyses included virology related sensitivity analyses of as-treated (while on initially randomized LPV/r dose schedule) time to virologic failure and time to regimen completion (the first of virologic failure or discontinuation of initially randomized LPV/r dose scheduled), as well as cross-sectional secondary analyses of plasma HIV-1 RNA level with a focus on the week 48 time point. Safety analyses were based on data collected from treatment initiation to the earlier of 56 days after discontinuing assigned LPV/r dose schedule or completion of study follow-up.
Percent adherence based on electronic monitor data was calculated separately for each individual over visit weeks 0-24 and weeks 24-48, as 100 X the number of dosing intervals in which the pill bottle was opened at least once, divided by the number of dosing intervals. For subjects randomized to the BID and QD arms, respectively, dosing intervals were defined as consecutive 12-hour (03:00 to 14:59 and 15:00 to 02:59) and 24-hour (03:00 to 02:59) periods.
Comparisons of CD4+ cell count change from baseline, percent adherence and Ctrough of LPV and RTV at specific visit weeks were carried out with the Wilcoxon-Mann-Whitney test stratified by screening plasma HIV-1 RNA level. Ctrough data were analyzed as-treated, with concentrations below the limit of quantification (LLOQ, 0.025 μg/mL) imputed as 0.0125 μg/mL. Proportions of subjects with Ctrough<0.025 μg/mL were compared with Fisher’s exact test.
Analyses were carried out with Statistical Analysis System (SAS) version 9 (SAS Institute, Cary, NC) and S-plus version 6 (Insightful Corp., Seattle, WA).
RESULTS
Figure 1 displays the disposition of subjects. One subject from the BID arm was subsequently declared ineligible because of prohibited medications at entry. This subject’s data were excluded from analyses, leaving 159 subjects on the BID and 161 on the QD arm. Baseline characteristics appeared balanced across arms (Table 1).
Figure 1.

Subject randomization and disposition. Shown are numbers of subjects randomized, completing therapy, and included in the final analysis. LPV/r QD = lopinavir/ritonavir 800 mg/200mg once daily; LFU = lost to follow-up.
Table 1.
Baseline Characteristics of Subjects
| Characteristics | ||||
|---|---|---|---|---|
| LPV/r BID (n=159) |
LPV/r QD (n=161) |
Total (n=320) |
||
| Sex | Male | 122 (77%) | 127 (79%) | 249 (78%) |
| Female | 37 (23%) | 34 (21%) | 71 (22%) | |
| Age (yrs) | Mean (SD) | 38.2 (9.4) | 39.3 (10.4) | 38.8 (9.9) |
| Median (Q1, Q3) | 39.0 (31.0, 45.0) | 38.0 (32.0, 45.0) | 39.0 (31.5, 45.0) | |
| Min, Max | 17.0, 64.0 | 18.0, 66.0 | 17.0, 66.0 | |
| Race/ethnicity | White Non-Hispanic | 43 (27%) | 54 (34%) | 97 (30%) |
| Black Non-Hispanic | 58 (36%) | 51 (32%) | 109 (34%) | |
| Hispanic (Regardless of Race) |
51 (32%) | 54 (34%) | 105 (33%) | |
| Asian, Pacific Islander | 5 (3%) | 1 (1%) | 6 (2%) | |
| American Indian, Alaskan Native |
1 (1%) | 1 (1%) | 2 (1%) | |
| Other/unknown/more than one race |
1 (1%) | 0 (0%) | 1 (0%) | |
| IV drug history | Never | 137 (86%) | 143 (89%) | 280 (88%) |
| Currently | 0 (0%) | 1 (1%) | 1 (0%) | |
| Previously | 22 (14%) | 17 (11%) | 39 (12%) | |
| NRTI Started | Never started Rx | 1 (1%) | 0 (0%) | 1 (0%) |
| d4T XR | 95 (60%) | 100 (62%) | 195 (61%) | |
| TDF | 63 (40%) | 61 (38%) | 124 (39%) | |
|
History of AIDS Defining Diagnosis |
Yes | 26 (16%) | 32 (20%) | 58 (18%) |
| No | 133 (84%) | 129 (80%) | 262 (82%) | |
| Hepatitis C antibody | Positive | 20 (13%) | 21 (13%) | 41 (13%) |
| Negative | 136 (87%) | 138 (86%) | 274 (87%) | |
| Indeterminate | 0 (0%) | 1 (1%) | 1 (0%) | |
|
HIV-1 RNA (log10(cp/mL))* |
Mean (SD) | 4.9 (0.7) | 4.8 (0.6) | 4.9 (0.6) |
| Median (Q1, Q3) | 4.8 (4.5, 5.4) | 4.8 (4.5, 5.2) | 4.8 (4.5, 5.3) | |
| Min, Max | 3.0, 6.5 | 3.1, 6.6 | 3.0, 6.6 | |
| Screening HIV-1 RNA | < 100,000 | 77 (48%) | 79 (49%) | 156 (49%) |
| >= 100,000 | 82 (52%) | 82 (51%) | 164 (51%) | |
| CD4 Count (cells/mm3)* | Mean (SD) | 221 (187) | 233 (179) | 227 (183) |
| Median (Q1, Q3) | 194 (65.5, 314) | 218 (76.0, 336) | 203 (67.8, 321) | |
| Min, Max | 0.0, 890 | 0.0, 869 | 0.0, 890 | |
| (cells/mm3) | 0-50 | 33 (21%) | 35 (22%) | 68 (21%) |
| 51-100 | 18 (11%) | 14 (9%) | 32 (10%) | |
| 101-200 | 34 (21%) | 25 (16%) | 59 (18%) | |
| 201-350 | 45 (28%) | 51 (32%) | 96 (30%) | |
| 351-500 | 17 (11%) | 24 (15%) | 41 (13%) | |
| >500 | 12 (8%) | 12 (7%) | 24 (8%) | |
Baseline HIV RNA level and CD4 cell count were calculated as the geometric and arithmetic means, respectively, of pre-entry and entry evaluations.
Treatment was dispensed to 319 subjects, and 226 (71%) remained on their initial LPV/r dose assignment throughout follow-up: 117 (74%) on BID and 109 (68%) on QD arms, (Figure 1). Reasons for early discontinuation are summarized in Table 2. Only one subject on LPV/r QD switched dosing to a BID schedule. There were no significant differences in the distribution of the time to premature discontinuation of assigned LPV/r dose schedule (p-value=0.17), and the treatment effect did not differ significantly by screening HIV-1 RNA stratum (p-value=0.62). Sixteen of the 195 subjects initiating treatment with d4T-XR switched to TDF, with 7 (7%), and 9 (9%) in the BID and QD arms. No subject switched from TDF to d4T-XR. More subjects discontinued LPV/r because of toxicity on the QD than on the BID arm (17 versus 10), but this difference was not statistically significant (p-value=0.20, post hoc analysis).
Table 2.
LPV/r disposition among eligible randomized subjects
| Status (Reason) | LPV/r BID (n=159) |
LPV/r QD (n=161) |
|---|---|---|
| Completed Study on LPV/r | 117 (74%) | 109 (68%) |
| Never Started LPV/r | 1 (1%) | 0 (0%) |
| Discontinued LPV/r | 41 (26%) | 52 (32%) |
| Toxicity | 10 | 17 |
| Non-compliant* | 10 | 14 |
| Lost to follow-up | 6 | 5 |
| Incarcerated | 5 | 3 |
| Unable to Attend Clinic | 4 | 2 |
| Severely Debilitated | 3 | 3 |
| Died** | 1 | 1 |
| Switched dose schedule | 0 | 1 |
| Other† | 2 | 6 |
Non-compliant with study visits (3), with study medication (8), with both study visits and medication (13).
Three additional subjects died after LPV/r discontinuation (see Figure 1).
Volume/timing of meds (3), pregnancy, non-protocol defined clinical event, treatment not working, withdrew consent, disallowed medication (1 each).
Sixty-five subjects had virologic failure, with 29 and 36 in the BID and QD arms, respectively. Of these, 42 occurred while subjects were on their initially assigned LPV/r dose schedule; 17 (26%) were confirmed virologic relapses. Kaplan-Meier plots for the probability of SVR are presented in Figure 2, overall (A) and by screening HIV-1 RNA stratum (B). The estimated probability (95% confidence interval [CI]) of SVR beyond 48 weeks was 0.81 (0.73, 0.86) and 0.78 (0.70, 0.84) in the BID and QD arms, respectively, a difference of 0.03 (−0.07, 0.12) (Table 3).
Figure 2.
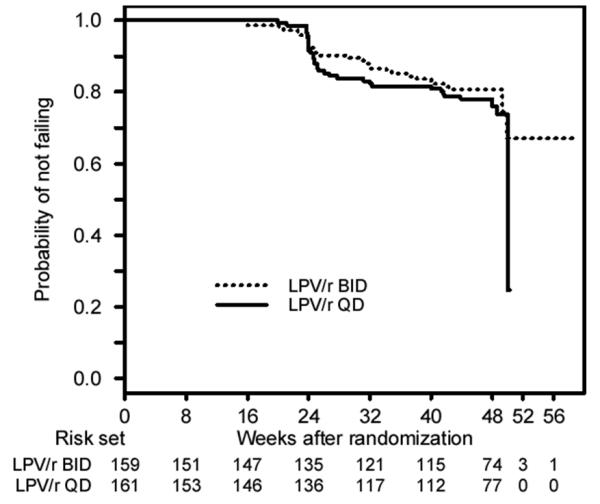
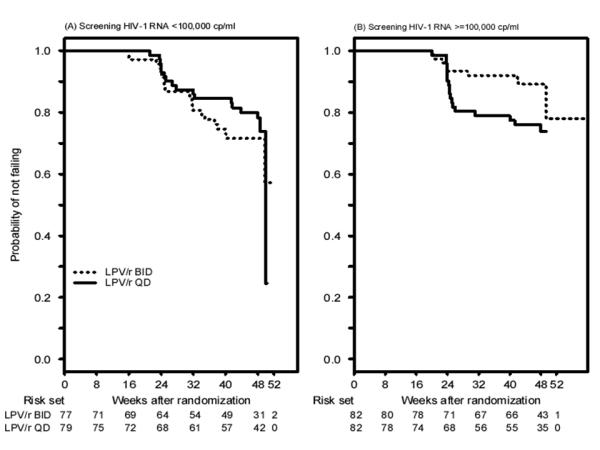
Time to virologic failure overall (A) and by screening plasma HIV-1 RNA level (B). Subjects whose final visit occurred after 48 weeks were included in the proportional hazards model analyses and are shown in the figure based on the actual time of last visit. The decline in the probability of not failing after 48 weeks is due to an event occurring after sharp decrease in the number of subjects at risk in this 48 week study. Only 10 subjects completed their final follow-up visit after 50 week and most subjects came in for their final visit by that time.
Table 3.
Estimated probabilities (95% confidence interval) of virologic outcome
| Outcome* | ||||
|---|---|---|---|---|
| LPV/r BID | LPV/r QD | Difference | ||
| SVR† (Intent-to-treat) | Overall | 0.81 ( 0.73, 0.86) | 0.78 (0.70, 0.84) | 0.03 (−0.07, 0.12) |
| <100,000 cp/mL | 0.72 ( 0.59, 0.81) | 0.80 ( 0.69, 0.88) | −0.09 (−0.23, 0.06) | |
| ≥100,000 cp/mL | 0.89 ( 0.79, 0.94) | 0.76 ( 0.64, 0.84) | 0.13 (0.01, 0.25) | |
| SVR† (As-treated) | Overall | 0.85 ( 0.78, 0.90) | 0.85 ( 0.78, 0.90) | 0 (−0.09, 0.09) |
| <100,000 cp/mL | 0.80 ( 0.67, 0.88) | 0.86 ( 0.75, 0.93) | −0.07 (−0.20, 0.07) | |
| ≥100,000 cp/mL | 0.90 ( 0.80, 0.95) | 0.84 ( 0.73, 0.91) | 0.06 (−0.06, 0.17) | |
| Regimen completion‡ | Overall | 0.66 ( 0.58, 0.73) | 0.61 ( 0.53, 0.68) | 0.05 (−0.05, 0.16) |
| <100,000 cp/mL | 0.58 ( 0.47, 0.69) | 0.61 ( 0.49, 0.71) | −0.02 (−0.18, 0.13) | |
| ≥100,000 cp/mL | 0.73 ( 0.62, 0.81) | 0.61 ( 0.50, 0.71) | 0.12 (−0.02, 0.26) | |
| HIV-1 RNA <200 cp/mL | Overall | 0.87 ( 0.81, 0.93) | 0.81 ( 0.74, 0.88) | 0.06 (−0.03, 0.15) |
| <100,000 cp/mL | 0.82 ( 0.72, 0.91) | 0.80 ( 0.70, 0.90) | 0.02 (−0.12, 0.15) | |
| ≥100,000 cp/mL | 0.91 ( 0.84, 0.98) | 0.82 ( 0.73, 0.91) | 0.09 (−0.02, 0.20) | |
Based on Kaplan-Meier estimates and of having an HIV-1 RNA result <200 copies/mL at week 48 based on a cross-sectional analysis (missing data excluded), and corresponding differences with 95% CI overall and by screening HIV-1 RNA level
Sustained virologic response (SVR) defined as remaining free of virologic failure. Virologic failure = any one of the following three occurrences: confirmed HIV-1 RNA ≥200 cp/mL after two consecutive HIV-1 RNA <200 cp/mL (confirmed relapse); confirmed HIV-1 RNA ≥200 cp/mL at or after visit week 24 without prior viral relapse; HIV-1 RNA ≥200 cp/mL at visit week 48, which did not require confirmation
Regimen completion = virologic failure or discontinuation of initially randomized LPV/r dose schedule, whichever occurred earlier
The QD versus BID treatment effect differed significantly by subjects’ screening HIV-1 RNA level (p-value=0.038). In the <100,000 copies/mL stratum, the estimated probabilities (95% CI) of SVR beyond 48 weeks were 0.72 (0.59, 0.81) and 0.80 (0.69, 0.88) in the BID and QD arms, with a difference of -0.09 (95% CI -0.23, 0.06). In the ≥100,000 copies/mL stratum these proportions were 0.89 (0.79, 0.94) and 0.76 (0.64, 0.84) with a difference of 0.13 (95% CI 0.01, 0.25), consistent with a higher probability of SVR in the BID arm compared to the QD arm. Similar results were obtained in sensitivity analyses, where initial failures prior to week 48 in participants lacking a confirmation sample were treated as non-failures, and where time to virologic failure was based on the scheduled visit week (results not shown).
In as-treated analyses, the 95% CI’s on the difference in the probability of SVR did not exclude a difference of 0, overall or in each screening HIV-1 RNA stratum (Table 3). The secondary endpoint of failure to complete the regimen completion occurred in 122 subjects (55 and 67 in the BID and QD arms). Of these, 42 were virologic failures, with 20 and 22 on the BID and QD arms. The 95% CIs on the difference in the probability of SVR and remaining on LPV/r beyond 48 weeks did not exclude a difference of 0 overall, or in each screening HIV-1 RNA stratum (Table 3).
In secondary cross-sectional analyses, the 95% confidence interval on the difference in the probability of having a plasma HIV-1 RNA level below 200 copies/mL at visit week 48 did not exclude a difference of 0, overall or when analyzed within each screening HIV-1 RNA stratum (Table 3). Analyses with a <50 copies/mL HIV-1 RNA threshold, analyses with missing values treated as failures, and as-treated analyses showed similar results for the treatment comparisons (not shown). CD4 cell counts increased by a median (25th-75th percentile) of 161 (97, 261) cells/μL (BID and QD arms combined) at visit week 48, and there were no significant treatment differences (p-value=0.77).
Eighteen subjects reported 23 new AIDS defining diagnoses during follow-up, with 6 cases of esophageal candidiasis, and 3 each of cryptococcal meningitis, mucocutaneous Kaposi sarcoma and wasting syndrome. A total of 22 subjects developed new AIDS defining diagnoses or died without new AIDS defining diagnosis, with 9 (of 159) and 13 (of 161) subjects on the BID and QD arms, respectively. There was no significant difference in the time to this outcome between the two arms (p-value=0.41).
Drug resistance results were obtained for 61/65 subjects experiencing virologic failure (one of the 61 subjects did not have a baseline genotype). Mutations associated with emergence of resistance did not appear different between QD and BID regimens (Table 4).
Table 4.
Known Resistance Mutations in Subjects with Virologic Failure
| Screening HIV-1 RNA <100,000 copies/mL | |||
|---|---|---|---|
| LPV/r BID (n=18) {n=77} |
LPV/r QD (n=17) {n=79} |
||
| PI* | Number of subjects with mutations at failure | 8 (44%) {10%} | 10 (59%) {13%} |
| Number of subjects with new mutations at failure | 2 (11%) {3%} | 1 (6%) {1%} | |
| NRTI† | Number of subjects with mutations at failure | 1 (6%) {1%} | 4 (24%) {5%} |
| Number of subjects with new mutations at failure | 1 (6%) {1%} | 3 (18%) {4%} | |
| Screening HIV-1 RNA >=100,000 copies/mL | |||
|---|---|---|---|
| LPV/r BID (n=8) {n=82} |
LPV/r QD (n=18) {n=82} |
||
| PI* | Number of subjects with mutations at failure | 3 (38%) {4%} | 7 (39%) {9%} |
| Number of subjects with new mutations at failure | 1 (13%) {1%} | 1 (6%) {1%} | |
| NRTI† | Number of subjects with mutations at failure | 3 (38%) {4%} | 8 (44%) {10%} |
| Number of subjects with new mutations at failure | 3 (38%) {4%} | 5 (28%) {6%} | |
(% of failures with resistance data) {% of randomized subjects}
(% of failures with resistance data) {% of randomized subjects}
New protease inhibitor (PI) mutations at failure included A71T, I47V, I54V, L10I, Q58E, N83D.
New nucleoside analog reverse transcriptase inhibitor (NRTI) mutations at failure included K219E, K219Q, L74V, M184V, T69A, V75L. Note that T69A and V75L were included in the resistance algorithm used for analysis. The mutation, T69A, is selected by NRTIs, but its effect on NRTI susceptibility is not known [Stanford HIV Resistance Database. http://hivdb.stanford.edu/, accessed 5/11/09]; T69A was identified at failure in one subject who also had M184V. The V75L mutation is is no longer considered to be associated with reduced NRTI susceptibility. V75L was identified at failure in one subject who also had M184V and K219Q.
Fifty-one subjects (16%) reported grade 3 or 4 signs or symptoms while on LPV/r that were at least one grade higher than at baseline. Most frequently reported were general bodyache (16 subjects, all of grade 3) and nausea (13 subjects, 9 from the QD arm). Eighty-eight subjects (28%) experienced grade 3 or higher laboratory abnormalities that were at least one grade higher than at baseline. Most frequently reported were increased creatine phosphokinase (22 subjects), lipase (17 subjects) and ALT (14 subjects). The distribution of the time to the first of either of such sign, symptom or laboratory toxicity was not significantly different for QD versus BID (p-value=0.80; 62 events in each arm).
A total of 94 subjects (29%), 45 (28%) and 49 (30%) in the BID and QD arms, respectively, experienced at least one a priori targeted diagnosis; the most frequently reported was gastrointestinal disorder in 23 (7%). Three subjects from the QD arm were diagnosed with appendicitis. Three subjects developed malignancies: one from the BID arm and two from the QD arm. A total of five subjects died during follow-up: one from the BID arm and four from the QD arm. Primary causes of death were hepatic failure (BID arm), advanced progressive multifocal leukoencephalopathy, pneumocystis pneumonia, Hodgkin lymphoma, and metastatic small cell carcinoma. The remaining subject with a malignancy had squamous cell carcinoma of the conjunctiva.
Adherence to LPV/r over weeks 0-24 as measured by electronic monitor was available for 151 of 158 (96%) and 154 of 161 (96%) subjects on the BID and QD arms, and for somewhat fewer subjects over weeks 24-48 (Table 5). Adherence was high, with 0-24 week medians of 82% and 91% on the BID and QD arms, and corresponding 24-48 week medians of 80% and 91%. For both periods, median adherence was higher among subjects on the QD vs. BID arm, and these differences were all statistically significant.
Table 5.
Electronic monitor-based adherence assessment by study week and treatment arm
| Study weeks |
Treatment arm (LPV/r frequency) |
Percent adherence | Number of dosing intervals per subject |
||||
|---|---|---|---|---|---|---|---|
| Number of subjects |
Median | Range | P-value | Median | Range | ||
| Weeks 0-24 |
BID | 151 | 82.1% | (8.9%, 100.0%) | 0.002 | 391 | (9, 393) |
| QD | 154 | 90.8% | (5.5%, 100.0%) | 195 | (4, 196) | ||
| Weeks 24-48 |
BID | 120 | 79.9% | (0.0%, 98.6%) | <0.001 | 281 | (49, 281) |
| QD | 114 | 90.6% | (0.0%, 100.0%) | 140 | (19, 140) | ||
LPV Ctrough distributions at visit Weeks 16 and 48 are summarized in Figure 3. At visit Week 48, the median (25th, 75th percentile) Ctrough for LPV was 5.6 (3.3, 8.2) and 3.4 (0.7, 8.6) μg/mL in the BID (n=103) and QD (n=99) arms; the corresponding numbers for RTV were 0.3 (0.2, 0.5) and 0.2 (0.05, 0.5) μg/mL. Compared to the BID arm, concentrations of LPV and RTV were lower and more variable in the QD arm. Concentration distribution differences for both LPV and RTV were statistically significant comparing BID and QD arms (p-values=0.013 and 0.005, respectively). At Week 48, the LPV Ctrough was below the LLOQ in 5.8 and 5.1% of subjects on the BID and QD arms, (p-value=1.0), and RTV Ctrough was below the LLOQ in 5.8% and 14.1% of subjects (p-value=0.06). When analysis was restricted to samples obtained within 2 hours of the scheduled dose, visit week 48 results were similar for the BID (n=63) versus QD (n=49) comparisons. LPV Ctrough distributions at weeks 4 and 24 were very similar to those at weeks 16 and 48 (not shown).
Figure 3.

LPV trough concentrations as a function of regimen assignment. Shown are median and range (box 25th-75th percentiles and whiskers 5th-95th percentiles) for lopinavir trough (C12hours) concentrations, expressed in micrograms/mL, obtained from study subjects at weeks 16 and 48 of treatment.
DISCUSSION
In this randomized trial, the BID and QD arms did not show significant differences in the probability of time to AIDS or death, LPV/r discontinuation, safety and immunologic outcomes. Although we observed no significant differences in the overall primary efficacy analysis of SVR through 48 weeks, the treatment effect of dose schedule on SVR depended on the screening plasma HIV-1 RNA level. Among subjects with HIV-1 RNA ≥100,000 copies/mL, the estimated probability of SVR after 48 weeks was 13% higher in the BID arm and the 95% CI excluded zero; in the lower screening HIV-RNA stratum, the corresponding CI did not exclude zero.
These results could be explained by differences in LPV/r pharmacokinetics, as the BID regimen produced significantly higher trough concentrations compared to the QD arm. LPV and RTV Ctrough’s did not differ as a function of screening HIV-1 RNA (p-values≥0.10). It is unlikely that this result reflected differential adherence, as adherence was significantly higher in the QD arm. However, as has been discussed by others, BID dosing may be more forgiving of missed or late doses than QD dosing, especially when comparing drugs with effective half-lives of a few hours like LPV.7 Higher trough concentrations are achieved when such drugs are dosed every 12 hours as compared to every 24 hours.
Having a higher pre-treatment viral load may increase the likelihood of treatment failure in the face of suboptimal drug concentrations, although this was not corroborated by viral resistance data. In virologic failure patients with high pre-treatment HIV-1 RNA, there were few patients who discontinued the originally assigned LPV/r dose schedule because of toxicity (two on BID and five on QD); this is unlikely to explain the difference in probability of achieving SVR.
Three published studies have evaluated the virologic efficacy and safety of QD versus BID LPV/r, although one of these was not a randomized controlled trial.18,19,20 These studies found no overall difference in virologic benefit in patients followed for up to 48 weeks. The most recently published of these studies found no significant difference in virologic benefit as a function of baseline plasma HIV-1 RNA, in contrast to our results.20 However, this finding was based on direct subgroup analyses, and used different primary endpoints as compared to our study.
There were two additional important differences between this study and ours. Our study used LPV/r soft-gel capsules, which was the only formulation available at the time. The study of Gathe et al.20 used LPV/r tablets, which have the potential advantages of a lower pill burden (four tablets versus six soft-gel capsules per day), and produce slightly higher LPV plasma concentrations.21 It is possible that a more convenient regimen producing higher drug concentrations overcomes potential disadvantages of the QD regimen in patients with higher HIV-1 RNA.
As expected, LPV and RTV trough concentrations were higher in subjects randomized to the BID arm. These results were similar to those previously reported in patients taking the same LPV/r regimens.18 Variability in trough concentrations was smaller for the BID regimen. Both of these factors may have contributed to the better virologic outcome seen in patients with high screening HIV-1 RNA assigned to BID LPV/r.
Adherence to both regimens was high, although patients randomized to the QD regimen reported taking a higher percentage of prescribed doses. Previous studies have suggested that adherence is better in patients who participate in clinical trials than in those who do not.1,8 The differences in treatment effect seen here could be magnified outside a clinical trial environment, where the frequency of missed or late drug doses is likely to be higher.
Patients and physicians generally prefer once-daily medications for chronic diseases. However, we found that the probability of achieving SVR was significantly better in patients with higher screening HIV-1 RNA randomized to the BID arm. This study demonstrates that while once daily administration improves adherence, the risk of lower trough concentrations may offset this benefit in selected patients. It may be possible to overcome this disadvantage by choosing a regimen or formulation that achieves higher trough concentrations of LPV.
Summary.
A randomized trial of once-daily versus twice-daily combination antiretrovirals for initial HIV-1 treatment showed no significant difference overall, but patients with higher HIV plasma RNA had a significantly greater probability of achieving a sustained virologic response on the twice-daily arm.
ACKNOWLEDGEMENTS
Supported by the AIDS Clinical Trials Group funded by the National Institute of Allergy and Infectious Diseases, including U01 AI68636 and AI068634. Abbott Laboratories, Bristol-Myers Squibb, and Gilead Pharmaceuticals provided study medications. The pharmaceutical sponsors monitored the development of the protocol and provided input into the design. They also reviewed earlier drafts of the manuscript prior to submission and suggested modifications. The decision to incorporate sponsors’ and supporters’ suggestions was exclusively the purview of the authors. All authors had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. This work was presented in part at the 14th Conference on Retroviruses and Opportunistic Infections, February 2007. The authors wish to thank the study participants for their contributions. We would also like to acknowledge the following individual site grant support, study team members, and site personnel:
Jorge L Santana Bagur, MD and Olga Mendez, MD- Puerto Rico- AIDS CRS (Site 5401) CTU Grant # 5 U01 A1069415-02
Margie Vasquez, RN and Karen Cavanagh, RN- NYU/NYC HHC at Bellevue (Site 401) ACTU Grants # AI -27665 and #AI069532, GCRC Grant # M01-RR00096
Kathleen Squires, MD and Bartolo Santos, RN-University of Southern California CRS (Site 1201) CTU Grant # 5U01AI069428-02
Andrea Weiss, RPh and Robin McKenzie, MD- Johns Hopkins Adult AIDS CRS (Site 201) CTU Grant #AI069465, GCRC Grant #RR025005
Carl J. Fichtenbaum, MD and Fran Hyc, RN, BSN- University of Cincinnati CRS (Site 2401) CTU Grant # AI-069513
Martha Greenwald, RN,MSN and Mitchell Goldman, MD-Indiana University AIDS CTU (Site 2601) CTU Grant # AI25859
Karen Tashima, MD and Pamela Poethke, RN- The Miriam Hospital ACTG CRS (Site 2951) CTU Grant #AI069472
Cathi Basler and Monica Carten- University of Colorado Hospital CRS (Site 6101) GCRC Grant #RR00051, CTU Grant # AI69450, NIH Grant #AA54907
Cindy Firnhaber, MD and Ian Sanne, MD- Wits HIV CRS (Site 11101) CTU Grant # AI069463
Aleshia Thomas, RN- Hospital of the University of Pennsylvania (Site 6201) CTU Grant #U01-AI 032783-14, CFAR Grant # 5-P30-AI-045008-09
David Perlman, MD, Gwen Costantini, FNP and Sondra Middleton, PA- Beth Israel Medical Center (Site 2851) CTU Grant # AI 46370
Ann Conrad, RN and Kim Whitely, RN- MetroHealth CRS (Site 2503) CTU Grant # U01-AI069501
Dr. Susan L. Koletar, MD and Mark D. Hite, RN- The Ohio State University AIDS CRS (Site 2301) CTU Grant # AI069474
Joseph Eron, MD, and David Currin, RN, CCRC-Wake County HHS CRS (Site 3206).
Richard Pollard, MD and Nancy Fitch, ANP-UC Davis Medical Center (Site 3852)
Brenda Jackson, RN & Rebecca Basham- Vanderbilt University (Site 3652) CTU Grant # AI069439
Christine Hurley, RN; Mary Adams, RN- University of Rochester ACTG CRS (Site 1101) and AIDS Community Health Center (Site 1108) CTU Grant # U01AI069511-02, GCRC Grant # 5-MO1 RR00044
Ann C. Collier, MD and Beck A. Royer, PA-C- University of Washington AIDS CRS (Site 1401) CTU Grant #AI 069434
Kim Epperson and Tim Lane- Moses H. Cone Memorial Hospital CRS (Site 3203) CTU Grant # 5 U01 AI069423-02
David Currin, RN and Sue Richard, PA- University of North Carolina AIDS CRS (Site 3201) CTU Grant #AI69423-01, CFAR Grant #AI50410, GCRC Grant #RR00046
Henry H. Balfour, Jr. MD and Christine Fietzer, RN- University of Minnesota ACTU (Site 1501)
Hector Bolivar, MD and Margaret A. Fischl, MD- University of Miami AIDS CRS (Site 901) CTU Grant #5U01AI069477
Richard Pollard, MD and Abby Olusanya, NP- UC Davis Medical Center ACTU (Site 3851) CTU Grant # 1U01AI069483-03
Robert Redfield, MD and Charles Davis, MD- University of Maryland CRS (Site 4651)
Jack Degnan and Dee Dee Pacheco- University of California San Diego AVRC CRS (Site 701) CTU Grant # AI69432
Karen Tashima, MD, and Joan Gormley, BSN-SSTAR Family Healthcare Center (Site 2954).
Sharon Riddler, MD, MPH and Carol Oriss, BSN, RN- Pittsburgh CRS (Site 1001) CTU Grant # 1 U01-AI 069494-01
Lorna Nagamine, RN and Scott Souza, PharmD- University of Hawaii at Manoa (Site 5201)
Nayef El-Daher, MD, and Carol Greisberger, RN, BS- McCree McCuller Wellness Center (Site 1107).
John Black, MD, and Beth Zwickl, RN, CS, MSN- Methodist Hospital of Indiana (Site 2602).
Timothy Flanigan, MD, and Joan Gormley, BSN- Rhode Island Hospital (Site 2953) CTU Grant # AI046381.
Andrea Weiss, RPh and Robin McKenzie, MD- Johns Hopkins Adult AIDS CRS (Site 201) CTU Grant #AI069465, GCRC Grant #RR025005
Primary funding source: National Institutes of Health.
Footnotes
Potential conflicts of interest: C.F. reports receiving grant support from Boehringer-Ingelheim and GlaxoSmithKline for research unrelated to this study; has served as a consultant to Bristol-Myers Squibb, Boehringer-Ingelheim, GlaxoSmithKline, Merck, Roche, Schering-Plough, and Tibotec; and has received honoraria for presentations at meetings sponsored in part by Abbott Laboratories. C.T. reports serving as a member of a DSMB for Tibotec. R.G. reports receiving grant support from Bristol-Myers Squibb and Abbott for research unrelated to this study. A.A. reports receiving grant support from GlaxoSmithKline for research unrelated to this study, and has served on an advisory board for Abbott. S.H.E. is a member of the clinical advisory board of Monogram Biosciences (formerly ViroLogic, Inc.). J.A. has served on advisory boards for Gilead, Tibotec, Roche, Bristol-Myers Squibb, Pfizer, and Merck, and has received research grants from Gilead, Tibotec, Roche, Pfizer, Merck, and Schering-Plough for research unrelated to this study. I.S. has served on advisory boards for GlaxoSmithKline, Bristol-Myers Squibb, Pfizer, and Triangle Pharmaceuticals. A.K. reports receiving grant support from Tibotec, Gilead, Merck, Abbott, and Pfizer for research unrelated to this study, and has served as a consultant to Bristol-Myers Squibb and Boehringer-Ingelheim. D.M. reports receiving grant support from Abbott, Bristol-Myers Squibb, Boehringer-Ingelheim, Merck, Pfizer, Roche, Schering Plough, and Tibotec for research unrelated to this study, and has received honoraria from Bristol-Myers Squibb, Boehringer-Ingelheim, GlaxoSmithKline, and Tibotec. All other authors have no potential conflicts of interest to disclose.
Clinical trial registration: Clinicaltrials.gov registration number: NCT00036452
REFERENCES
- 1.Claxton AJ, Cramer J, Pierce C. A systematic review of the associations between dose regimens and medication compliance. Clin Ther. 2001;23:1296–1310. doi: 10.1016/s0149-2918(01)80109-0. [DOI] [PubMed] [Google Scholar]
- 2.Moyle G. Overcoming obstacles to the success of protease inhibitors in highly active antiretroviral therapy regimens. AIDS Patient Care STDS. 2002;16:585–97. doi: 10.1089/108729102761882125. [DOI] [PubMed] [Google Scholar]
- 3.Maitland D, Jackson A, Osorio J, Mandalia S, Gazzard BG, Moyle GJ, Epivir-Ziagen (EZ) Switch Study Team Switching from twice-daily abacavir and lamivudine to the once-daily fixed-dose combination tablet of abacavir and lamivudine improves patient adherence and satisfaction with therapy. HIV Med. 2008;9:667–72. doi: 10.1111/j.1468-1293.2008.00618.x. [DOI] [PubMed] [Google Scholar]
- 4.Cramer JA, Mattson RH, Prevey ML, Scheyer RD, Ouellette VL. How often is medication taken as prescribed? A novel assessment technique. JAMA. 1989;261:3273–7. [PubMed] [Google Scholar]
- 5.Hughes D. Less is more: medicines that require less frequent administration improve adherence, but are they better? Pharmacoeconomics. 2006;24:211–213. doi: 10.2165/00019053-200624030-00001. [DOI] [PubMed] [Google Scholar]
- 6.Iskedjian M, Einarson TR, MacKeigan LD, et al. Relationship between daily dose frequency and adherence to antihypertensive pharmacotherapy: Evidence from a meta-analysis. Clin Ther. 2002;24:302–316. doi: 10.1016/s0149-2918(02)85026-3. [DOI] [PubMed] [Google Scholar]
- 7.Comté L, Vrijens B, Tousset E, Gérard P, Urquhart J. Estimation of the comparative therapeutic superiority of QD and BID dosing regimens, based on integrated analysis of dosing history data and pharmacokinetics. J Pharmacokinet Pharmacodyn. 2007;34:549–58. doi: 10.1007/s10928-007-9058-0. [DOI] [PubMed] [Google Scholar]
- 8.Vrijens B, Urquhart J. Patient adherence to prescribed antimicrobial drug dosing regimens. J Antimicrob Chemother. 2005;55:616–627. doi: 10.1093/jac/dki066. [DOI] [PubMed] [Google Scholar]
- 9.Flexner C. HIV drug development: the next 25 years. Nature Rev Drug Disc. 2007;6:959–966. doi: 10.1038/nrd2336. [DOI] [PubMed] [Google Scholar]
- 10.Paterson DL, Swindells S, Mohr J, Brester M, Vergis EN, Squier C, Wagener MM, Singh N. Adherence to protease inhibitor therapy and outcomes in patients with HIV infection. Ann Intern Med. 2000;133:21–30. doi: 10.7326/0003-4819-133-1-200007040-00004. [DOI] [PubMed] [Google Scholar]
- 11.Gross R, Bilker WB, Friedman HM, Strom BL. Effect of adherence to newly initiated antiretroviral therapy on plasma viral load. AIDS. 2001;15:2109–2017. doi: 10.1097/00002030-200111090-00006. [DOI] [PubMed] [Google Scholar]
- 12.Carrieri MP, Leport C, Protopopescu C, et al. Factors associated with nonadherence to highly active antiretroviral therapy: a 5-year follow-up analysis with correction for the bias induced by missing data in the treatment maintenance phase. J Acquir Immune Defic Syndr. 2006;41:477–485. doi: 10.1097/01.qai.0000186364.27587.0e. [DOI] [PubMed] [Google Scholar]
- 13.Mills EJ, Nachega JB, Bangsberg DR, et al. Adherence to HAART: a systematic review of developed and developing nation patient-reported barriers and facilitators. PLoS Med. 2006;3:e438. doi: 10.1371/journal.pmed.0030438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gross R, Bilker WB, Friedman HM, Coyne JC, Strom BL. Provider inaccuracy in assessing adherence and outcomes with newly initiated antiretroviral therapy. AIDS. 2002;16:1835–1837. doi: 10.1097/00002030-200209060-00021. [DOI] [PubMed] [Google Scholar]
- 15.Gross R, Tierney C, Andrade A, et al. Modified directly observed antiretroviral therapy compared with self-administered therapy in treatment naïve HIV-1 infected patients: a randomized trial. Arch Intern Med. 2009 doi: 10.1001/archinternmed.2009.172. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.National Institute of Allergy and Infectious Diseases (NIAID) AIDS Clinical Trials Group (ACTG) toxicity tables, appendix II-B. NIAID; Bethesda, MD: 2004. Table for grading the severity of adult and pediatric adverse events, Version 1.0, December 2004. Available at: http://rcc.tech-res-intl.com. [Google Scholar]
- 17.Rezk NL, Tidwell RR, Kashuba AD. High-performance liquid chromatography assay for the quantification of HIV protease inhibitors and non-nucleoside reverse transcriptase inhibitors in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 2004;805:241–247. doi: 10.1016/j.jchromb.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 18.Eron JJ, Feinberg J, Kessler HA, Horowitz HW, Witt MD, Carpio FF, Wheeler DA, Ruane P, Mildvan D, Yangco BG, Bertz R, Bernstein B, King MS, Sun E. Once-daily versus twice-daily lopinavir/ritonavir in antiretroviral-naive HIV-positive patients: a 48-week randomized clinical trial. J Infect Dis. 2004;189:265–72. doi: 10.1086/380799. [DOI] [PubMed] [Google Scholar]
- 19.Johnson MA, Gathe JC, Jr, Podzamczer D, Molina JM, Naylor CT, Chiu YL, King MS, Podsadecki TJ, Hanna GJ, Brun SC. A once-daily lopinavir/ritonavir-based regimen provides noninferior antiviral activity compared with a twice-daily regimen. J Acquir Immune Defic Syndr. 2006;43:153–60. doi: 10.1097/01.qai.0000242449.67155.1a. [DOI] [PubMed] [Google Scholar]
- 20.Gathe J, Silva BA, Cohen DE, Loutfy MR, Podzamczer D, Rubio R, Gibbs S, Marsh T, Naylor C, Fredrick L, Bernstein B. A once-daily lopinavir/ritonavir-based regimen is noninferior to twice-daily dosing and results in similar safety and tolerability in antiretroviral-naive subjects through 48 weeks. J Acquir Immune Defic Syndr. 2009;50:474–81. doi: 10.1097/QAI.0b013e31819c2937. [DOI] [PubMed] [Google Scholar]
- 21.Klein CE, Chiu Y-L, Awni W, et al. The tablet formulation of lopinavir/ritonavir provides similar bioavailability to the soft gelatin capsule formulation with less pharmacokinetic variability and diminished food effect. J Acquir Immune Defic Syndr. 2007;44:401–410. doi: 10.1097/QAI.0b013e31803133c5. [DOI] [PubMed] [Google Scholar]