

Abstract
Mycobacteriophages BPs, Angel and Halo are closely related viruses isolated from Mycobacterium smegmatis, and possess the smallest known mycobacteriophage genomes, 41 901 bp, 42 289 bp and 41 441 bp, respectively. Comparative genome analysis reveals a novel class of ultra-small mobile genetic elements; BPs and Halo each contain an insertion of the proposed mobile elements MPME1 and MPME2, respectively, at different locations, while Angel contains neither. The close similarity of the genomes provides a comparison of the pre- and post-integration sequences, revealing an unusual 6 bp insertion at one end of the element and no target duplication. Nine additional copies of these mobile elements are identified in a variety of different contexts in other mycobacteriophage genomes. In addition, BPs, Angel and Halo have an unusual lysogeny module in which the repressor and integrase genes are closely linked. The attP site is located within the repressor-coding region, such that prophage formation results in expression of a C-terminally truncated, but active, form of the repressor.
INTRODUCTION
Mycobacteriophages – viruses that infect mycobacterial hosts – can be readily isolated from environmental samples using Mycobacterium smegmatis as a host (Hatfull et al., 2006). The complete sequences of 32 mycobacteriophage genomes have been reported (Hatfull et al., 2006; Morris et al., 2007; Pham et al., 2007), revealing them to be genetically diverse and to harbour a large proportion of genes encoding products that are unrelated to other known proteins and are mostly of unknown function. The genomes are not homogeneously diverse and can be assorted into clusters of phages that are more closely related to each other than to other mycobacteriophages (Hatfull et al., 2006), although these clusters likely reflect the variation associated with a small sample size rather than a true representation of the larger population structure. Relatively few of these sequenced phages have been shown to infect Mycobacterium tuberculosis, with the notable exceptions of D29 (Ford et al., 1998a), TM4 (Ford et al., 1998b), L5 (Fullner & Hatfull, 1997) and Che12 (Hatfull et al., 2006; Kumar et al., 2008).
An unusual feature of mycobacteriophage genomes is their relatively large size. The average genome length is approximately 70 kbp, twice as large as the phages that infect dairy bacteria (Brussow, 2001; Pedulla et al., 2003). The reasons for this are unclear, but it does not appear to arise from systematic differences in the packaging constraints of the capsids or any obvious requirement to traverse the unusual cell walls of the mycobacteria. For example, there is relatively little difference in the genome length required to accommodate the virion structure and assembly proteins of these two groups of phages. The main differences in size correspond to those segments encoding non-structural genes. There is, however, substantial variation in genome size, with the largest being approximately 150 kbp (Bxz1, Catera), and the smallest being 41 441 bp (Angel) (Hatfull et al., 2006).
As in other bacteriophages, genetic mosaicism is a hallmark of the genome architectures of mycobacteriophages (Ford et al., 1998a; Hatfull et al., 2006; Pedulla et al., 2003). This is manifested by distinctly different evolutionary histories of different segments of the genomes, making it difficult to assemble meaningful reconstructions of whole phage genomes as phylogenetic units (Lawrence et al., 2002). This mosaicism can be illustrated by grouping mycobacteriophage genes into ‘phamilies’ of related sequences and representing the relationships as phamily circles (Hatfull et al., 2006). How this mosaicism is generated remains unclear, but it is proposed that it arises largely through a process of illegitimate recombination and selection for gene function and packagable-sized genomes (Hendrix, 2002). While transposition is expected to contribute to this process – and many mycobacterial transposons have been described – all of the currently sequenced mycobacteriophage genomes are devoid of known transposable elements. Interestingly, only a small proportion (∼15 %) of mycobacteriophage gene phamilies have significant database matches to sequences outside of the mycobacteriophage group, reflecting the abundance of novel genes seen in the phage population as a whole. Similar numbers of mycobacteriophage phamilies match phage and non-phage sequences, suggesting that new genes are acquired from both bacterial and other phage genomes during the course of their evolution (Hatfull et al., 2006).
Characterization of mycobacteriophage genomes not only offers insights into viral diversity and genome evolution, but also provides valuable resources for the development of tools for mycobacterial genetic manipulation (Hatfull, 2000). A variety of phage-derived tools have been described, including shuttle phasmids (Jacobs et al., 1987) that can be used to efficiently deliver transposons (Bardarov et al., 1997), reporter genes (Jacobs et al., 1993; Piuri et al., 2009), allelic-exchange substrates (Bardarov et al., 2002), and integration-proficient plasmid vectors (Freitas-Vieira et al., 1998; Kim et al., 2003; Lee et al., 1991; Morris et al., 2007; Pham et al., 2007). The general recombination functions encoded by Che9c genes 60 and 61 – homologues of recE and recT respectively – have also been exploited for an efficient recombineering system that simplifies construction of gene replacement mutants in both fast- and slow-growing mycobacteria (van Kessel & Hatfull, 2007, 2008), as well as facilitating mutagenesis of lytically replicating mycobacteriophage genomes (Marinelli et al., 2008; van Kessel et al., 2008).
Here we report the genome sequences of mycobacteriophages BPs and Angel, and comparative analysis with their close relative, mycobacteriophage Halo. Although the three genomes are almost identical in their nucleotide sequences, insertions/deletions in the right arms reveal a novel class of small mobile genetic elements that are present in many other mycobacteriophage genomes, but which had not been previously recognized. In addition, these phages contain an unusual lysogeny module, in which the phage attachment site, attP, is located within the repressor gene, leading to truncation of the repressor gene following phage integration. Finally, although these phages were isolated on lawns of M. smegmatis, they also infect M. tuberculosis, but at a greatly reduced plating efficiency, and host range mutants efficiently infecting both strains can be readily isolated.
METHODS
Bacterial strains and media.
M. smegmatis mc2155 was cultured on 7H10 agar (Difco), supplemented with 10 % albumin dextrose complex (ADC) and 0.5 % glycerol. Cultures were grown in 7H9 medium (Difco), containing 10 % ADC, 0.2 % glycerol and 0.05 % Tween 80. For phage infections, Tween 80 was omitted, and CaCl2 was added at a final concentration of 1 mM. M. tuberculosis mc27000 (Ojha et al., 2008) was cultured on 7H11 agar, supplemented with 10 % oleic acid albumin dextrose complex (OADC), 0.5 % glycerol and pantothenate (0.1 mg ml−1). Cultures were grown in 7H9 medium, supplemented with 10 % OADC, 0.2 % glycerol, pantothenate (0.1 mg ml−1) and 0.05 % Tween 80. For phage infections, cells were diluted approximately 1 : 10 in fresh medium and grown for approximately 24–36 h in medium lacking Tween 80, supplemented with 1 mM CaCl2. Phages were spotted onto top agar lawns seeded with either M. smegmatis or M. tuberculosis in 0.35 % mycobacterial top agar (MBTA) with 1 mM CaCl2 (and pantothenate for M. tuberculosis).
Phage isolation and purification.
Mycobacteriophages BPs and Angel were isolated from soil samples collected in the Oakland district of Pittsburgh, PA, and O'Hara Township, PA, respectively, by direct plating on lawns of M. smegmatis, without amplification, of soil extracts prepared with phage buffer (10 mM Tris/HCl pH 7.5, 10 mM MgSO4, 1 mM CaCl2, 68.5 mM NaCl). The extract was filtered through a 0.22 μm filter, and 50 μl of this sample was plated with 1 ml late-exponential-phase M. smegmatis mc2155 in 4.5 ml 0.35 % MBTA, supplemented with 1 mM CaCl2. Following several rounds of plaque purification, high-titres stocks were prepared and used for subsequent studies (Sarkis & Hatfull, 1998).
Genome sequence determination and analysis.
A library of BPs genomic DNA was generated by HydroShear (Gene Machines, Inc.) shearing and end-repair; 1–3 kbp DNA fragments were purified following gel electrophoresis and cloned into the EcoRV site in the pBluescript vector. DNAs from approximately 380 individual clones were prepared and sequenced from both ends of each insert using forward and reverse primers. The sequences of these clones assembled into a single contig, and ambiguous regions were resolved by sequencing directly from BPs DNA with 28 oligonucleotide primers. The GenBank accession numbers of BPs and Angel are EU568876 and FJ973624, respectively. The GenBank file of the Halo sequence (accession no. DQ398042) was updated and the new accession number is DQ398042.2.
Mycobacteriophage Angel was sequenced using Pyrosequencing technology at the University of Pittsburgh Genomics and Proteomics Core Laboratories (GPCL) as follows. Approximately 6 μg Angel genomic DNA was sheared by pressurized nitrogen nebulization into ∼200–800 bp fragments, which were purified on a Qiagen MinElute column and blunt-ended using T4 PNK and T4 DNA Polymerase. ‘A’ and ‘B’ 454 Inc. adaptors were ligated to the fragments, which were then denatured and annealed to primer-coated beads; the ‘A’ adaptor contained a 10 bp Multiplex Identifier (MID) tag to enable subsequent extraction of Angel-specific sequences from a multiplexed sequencing run. The beads were then used in an emulsion-based PCR, which in parallel amplifies the ssDNA fragment attached to each of the beads. A sequencing primer was added, and the beads were packed onto a 25×75 mm picotitre plate, then run on a 454 GS-FLX machine. Angel-specific sequencing reads from two separate sequencing runs were extracted from master standard flowgram format (sff) files using the command sfffile from the GS-FLX software package. Data from the two runs combined yielded a total of 7091 reads, averaging 230.8 bases per read, providing a total of 1 636 726 bases of raw sequencing information. The command sff2scf was used to generate trace files for each sequence reads, and these trace files were assembled using Phrap and viewed in Consed. The assembled data produced one large contig with consensus quality values of 60 or greater and average genome coverage of 39.5-fold. Defined genome ends were evident from a buildup of reads at either end of the contig, and confirmation of the predicted termini was in agreement with the closely related BPs and Halo genomes.
Sequence analysis and annotation was performed using DNAMaster (http://cobamide2.bio.pitt.edu) and dotplot analysis using Gepard (http://mips.gsf.de/services/analysis/gepard). The Phamerator program automates the phamily organization described previously (Hatfull et al., 2006) and will be described in detail elsewhere. In brief, the 6858 predicted proteins encoded by 60 phage genomes (not including Angel) are compared pair-wise with each other by both clustal and blastp, and assorted into phamilies, such that proteins with similarity above a threshold level are grouped into the same phamily.
Electron microscopy.
A suspension of CsCl-purified virions was applied to a sample grid with a carbon-coated nitrocellulose film, stained with 2 % uranyl acetate, and examined in a FEI Morgagni 268 transmission electron microscope equipped with an AMT digital camera system.
Plasmid constructions.
Plasmids pYUB854 and pMH94 have been described previously (Bardarov et al., 2002; Lee et al., 1991). Plasmid pTRS1a was constructed as follows: a ∼1.5 kbp fragment containing BPs gene 32 and the attP core (BPs coordinates 27 590–29 061) was amplified from a BPs lysate using primers BPs27590 (5′-TACTTCATCGAGCGCACGCGCGTCT-3′) and BPs29061 (5′-AGGAGATGAAGAAGTGCGCCCGGAG-3′), and blunt-end cloned into the XhoI site of pYUB854. To construct plasmids pGWB37 and pGWB38, a ∼1.6 kbp fragment containing BPs gene 32, attP and 221 bp downstream of the attP core (BPs coordinates 27 696–29 270) was amplified using primers prmGB14 (5′-CGCTGCCAGACCCCAATTGCGGAAC-3′) and prmGB15 (5′-CTACTGATCGCGCGCCTTGAAGCTG-3′). This fragment was blunt-end cloned into the SalI fragment of pMH94 that lacked the original L5 int-attP insert, thus replacing the L5 int-attP of pMH94 with the BPs int-attP; pGWB37 and pGWB38 differ in regard to the orientation of the inserted fragment relative to the plasmid backbone. To construct pGWB40, a ∼1.8 kbp fragment containing BPs gene 32-attP-gene 33 and the gene 33–34 intergenic region (BPs coordinates: 27 696–29 522) was amplified using primers prmGB14 and prmGB28 (5′-CGGTTGGGGTCATGTGCACCAACATAG-3′) and blunt-end cloned into the same SalI fragment as pGWB37 and pGWB38.
PCR assays.
Site-specific integration between the putative BPs/Halo attP and M. smegmatis attB sites was confirmed in pGWB37 and pGWB40 transformants, as well as in Halo lysogens, by PCR amplification of the attL and attB sites. Pelleted cells were suspended in 500 μl 10 mM Tris (pH 8.0), 1 mM EDTA, heated for 20 min at 95 °C and 10 μl was used in PCRs with Pfu polymerase (Stratagene), 5 % DMSO and 10 nM dNTPs. Primers prmGB18 (5′-CCGGCACGAGATCAGCAGCTTCTCG-3′) and prmGB19 (5′-TGGCACAGACTCACCGATCCGCAGC-3′) were used to amplify attB, while primers prmGB18 and prmGB20 (5′-CGAGCGAGTCGAGATAGTCGTCCAG-3′) were used to amplify attL.
Immunity assays.
Immunity to mycobacteriophages Halo and BPs, as well as to D29 as a positive control, was tested by spotting serial dilutions of each phage onto lawns of M. smegmatis mc2155, mc2155 Halo lysogens, mc2155pGWB37 and mc2155pGWB40.
RESULTS
Isolation and genomic sequencing of mycobacteriophages BPs and Angel
Mycobacteriophages BPs and Angel were isolated from soil samples collected in the Oakland district of Pittsburgh, PA, and O'Hara Township, PA, respectively, by direct plating without amplification on a lawn of M. smegmatis. Following plaque purification and amplification, virion particles of BPs and Angel were examined by electron microscopy, which showed that this phage has an isometric head approximately 55 nm in diameter and a long flexible tail 210 nm long (Fig. 1); many other mycobacteriophages have a similar morphology, including mycobacteriophage Halo (Fig. 1). The BPs genome was sequenced by a shotgun approach as described previously (Hatfull et al., 2006), using forward and reverse sequencing primers on ∼384 clones and an ABI3730 automated sequencer. The cloned sequences assembled into contigs that were joined and polished using oligonucleotide primers on virion DNA template. Genome assembly suggested the presence of defined genome ends, which were determined to be 11-base 3′ extensions by sequencing off the end of phage genomes with closely positioned primers. The mycobacteriophage Angel genome was determined by pyrosequencing using a 454 genome analyser with an average redundancy of ∼40-fold. The Angel ends were shown by sequencing on template DNA to have identical 11-base 3′ extensions to those of BPs and Halo. Contiguous sequences of 41 901 bp for BPs and 41 441 bp for Angel were obtained, and both have a G+C content of 66.6 mol%. The GenBank accession numbers for BPs and Angel are EU568876 and FJ973624, respectively.
Fig. 1.

Morphologies of mycobacteriophages BPs, Angel and Halo. Purified phage particles were examined by electron microscopy; the scale marker corresponds to 100 nm.
Nucleotide similarities of BPs and Angel to other mycobacteriophage genomes
Comparison of the BPs and Angel sequences to all other mycobacteriophage genomes shows that they have strong and extended similarity to each other (99 % average nucleotide identity), as well as to mycobacteriophage Halo (Hatfull et al., 2006) (Fig. 2). The main differences between BPs and Halo are four small insertion/deletions in the rightmost 5 kbp of the genomes (Fig. 2). Although BPs and Angel do not have strong nucleotide similarity to other mycobacteriophages, weaker areas of similarity to TM4, Orion (and the closely related PG1) and Giles can be detected, with the TM4 similarity weak but extending over a substantial portion of the leftmost 20 kbp of the genomes. A small segment (∼450 bp) at the right end of the BPs genome – and whose absence from Angel is the primary difference between these two genomes – has high similarity to some other mycobacteriophage genomes and is described in further detail below.
Fig. 2.

Nucleotide sequence comparison of mycobacteriophages BPs and Halo. The nucleotide sequences of phage BPs and Halo were compared using the program Gepard, revealing them to be very closely related. The inset shows details of the rightmost 5 kbp of the two genomes and notes four specific insertions or deletions that are described further in the text.
Host range of mycobacteriophages BPs and Halo
Although mycobacteriophage BPs was isolated using M. smegmatis as a host, we have also tested whether it – and the other mycobacteriophages with completely sequenced genomes, most of which were also isolated using M. smegmatis as a host – also infects M. tuberculosis. We observed that lysates of BPs and Halo do not efficiently infect M. tuberculosis, but rather form plaques at a greatly reduced plating efficiency (approx. 10−5) compared to M. smegmatis (Fig. 3); Angel presumably behaves similarly. Interestingly, only a modest proportion (∼15 %) of all of the sequenced mycobacteriophages infect both fast- and slow-growing mycobacteria, including L5 (Fullner & Hatfull, 1997), D29 (Bardarov et al., 1997), Bxz2 (Pedulla et al., 2003), Che12 (Hatfull et al., 2006; Kumar et al., 2008) and TM4 (Jacobs et al., 1993). Most of the phages with sequenced genomes (with the notable exception of TM4; Timme & Brennan, 1984) were isolated using M. smegmatis as a host.
Fig. 3.

Host range of phages BPs and Halo. (a) Serial dilutions of BPs, Halo and D29 particles were spotted onto lawns of M. smegmatis and M. tuberculosis, as indicated. The phage titres were determined on M. smegmatis; and the following numbers of p.f.u. were used in the four spots from left to right: D29, 105, 103, 102, 10; BPs, 107, 106, 105, 104; Halo, 107, 106, 105, 104. Halo and BPs have a plating efficiency approximately 10−5-fold lower on M. tuberculosis than on M. smegmatis. (b) Isolation of Halo host-range mutants. A Halo plaque was picked from the M. tuberculosis lawn shown in (a), and 10-fold serial dilutions were plated on lawns of M. smegmatis and M. tuberculosis (upper parts of plates); this had equivalent efficiencies of plating on the two strains. A plaque was then picked from an M. smegmatis lawn similar to that shown in the upper part of the plate, serially diluted, and plated on M. tuberculosis and M. smegmatis lawns (lower parts of plates).
To determine whether the reduced plating efficiency on M. tuberculosis is a result of either restriction/modification or a blockage in receptor association, a Halo plaque derived from a lawn of M. tuberculosis was picked and replated on lawns of both M. tuberculosis and M. smegmatis (Fig. 3). This plated with equal efficiencies on both hosts. A single plaque from the M. smegmatis plate was then replated on both strains, and was also found to have equivalent plating efficiencies on both strains (Fig. 3). This observation makes it unlikely that restriction is the cause of the reduced plating efficiency, and we prefer the explanation that Halo and BPs are normally unable to recognize their receptor in M. tuberculosis, but that mutants overcoming this defect arise at a frequency of approximately 10−5. Since these mutants plate with equal efficiency on both hosts, these correspond to an expansion rather than a switch of the host range.
Organization of the BPs, Angel and Halo genomes
Analysis of the BPs genome identified 63 ORFs, and no tRNA or tmRNA genes (Table 1). With the exception of genes 32 and 33, all of the BPs genes are transcribed rightwards. Given the strong DNA sequence similarity between BPs, Angel and Halo, it is not surprising that the genome maps are extremely similar, with the main differences occurring to the right of gene 51 and mostly deriving from small insertions and deletions (Figs 2 and 4); Angel contains a total of 61 predicted ORFs (Fig. 4).
Table 1.
Genes encoded on the mycobacteriophage BPs genome
| Gene | F/R | Start | Stop | Product size (kDa) | Comments | Closest match | Other significant matches |
|---|---|---|---|---|---|---|---|
| 1 | F | 43 | 387 | 12.2 | Halo gp1 | TM4 gp3 | |
| 2 | F | 455 | 1891 | 52.2 | Terminase | Halo gp2 | TM4 gp4 |
| 3 | F | 1888 | 3402 | 55.3 | Portal | Halo gp3 | Che9d gp4 |
| 4 | F | 3403 | 6192 | 101.8 | Halo gp4 | TM4 gp6 | |
| 5 | F | 6189 | 6395 | 7.5 | Halo gp5 | TM4 gp7 | |
| 6 | F | 6514 | 7056 | 19.4 | Scaffold | Halo gp6 | Che9d gp6 |
| 7 | F | 7103 | 8038 | 33.5 | Capsid | Halo gp7 | Che9d gp7, PA6 gp6 |
| 8 | F | 8075 | 8302 | 7.5 | Halo gp8 | ||
| 9 | F | 8314 | 8811 | 17.7 | Halo gp9 | PA6 gp7, Che9d gp9 | |
| 10 | F | 8811 | 9167 | 13.4 | Halo gp10 | TM4 gp11 | |
| 11 | F | 9154 | 9417 | 9.3 | Halo gp11 | TM4 gp12 | |
| 12 | F | 9414 | 9857 | 15.9 | Halo gp12 | TM4 gp13 | |
| 13 | F | 9854 | 10468 | 21.8 | Major tail subunit | Halo gp13 | TM4 gp14 |
| 15 | F | 10569 | 11464 | 33.4 | Halo gp14 | TM4 gp15 | |
| 14 | F | 10569 | 11078 | 18.7 | Halo gp15 | TM4 gp14 | |
| 16 | F | 11464 | 15477 | 137.7 | Tapemeasure protein | Halo gp16 | TM4 gp17, Che9d gp17 |
| 17 | F | 15477 | 16649 | 44.7 | Minor tail protein | Halo gp17 | TM4 gp18, Giles gp21 |
| 18 | F | 16649 | 18403 | 65.3 | Minor tail protein | Halo gp18 | TM4 gp19. Giles gp22 |
| 19 | F | 18403 | 18864 | 17.4 | Halo gp19 | TM4 gp20 | |
| 20 | F | 18861 | 19997 | 40.4 | Halo gp20 | TM4 gp21, Giles gp24 | |
| 21 | F | 20008 | 20442 | 16.5 | Halo gp21 | TM4 gp22 | |
| 22 | F | 20442 | 22835 | 82.1 | Minor tail protein | Halo gp22 | L5 gp32 |
| 23 | F | 22850 | 23170 | 12.0 | Halo gp23 | ||
| 24 | F | 23173 | 23355 | 6.5 | Halo gp24 | ||
| 25 | F | 23355 | 23630 | 9.4 | Halo gp25 | ||
| 26 | F | 23630 | 23824 | 7.1 | Halo gp26 | ||
| 27 | F | 23885 | 25201 | 47.4 | Lysin A | Halo gp27 | R. erythropolis peptidase |
| 28 | F | 25201 | 26409 | 42.9 | Lysin B | Halo gp28 | Giles gp32 |
| 29 | F | 26435 | 26785 | 11.8 | Halo gp29 | ||
| 30 | F | 26790 | 27110 | 11.3 | Halo gp30 | Giles gp34 | |
| 31 | F | 27107 | 27724 | 23.1 | Halo gp31 | Giles gp35 | |
| 32 | R | 27721 | 28917 | 42.5 | Integrase | Halo gp32 | Nocardia phage integrase |
| 33 | R | 28914 | 29324 | 14.7 | Repressor | Halo gp33 | Nocardia DNA-binding protein |
| 34 | F | 29498 | 29755 | 9.2 | Xis? | Halo gp34 | |
| 35 | F | 29752 | 30150 | 13.8 | Halo gp35 | ||
| 36 | F | 30150 | 30749 | 22.2 | Halo gp36 | PG1 gp57 etc. | |
| 37 | F | 30749 | 30931 | 7.0 | Halo gp37 | ||
| 38 | F | 30928 | 31278 | 13.9 | Halo gp38 | P2 gp91 | |
| 39 | F | 31275 | 31424 | 4.7 | Halo gp39 | ||
| 40 | F | 31424 | 31774 | 12.7 | Halo gp40 | ||
| 41 | F | 31758 | 31910 | 5.5 | Halo gp41 | ||
| 42 | F | 31907 | 32992 | 40.2 | RecE | Halo gp42 | Che9c gp60 |
| 43 | F | 33010 | 34398 | 49.3 | RecT | Halo gp43 | Giles gp53 |
| 44 | F | 34401 | 34949 | 19.8 | Halo gp44 | ||
| 45 | F | 34946 | 35164 | 7.9 | Halo gp45 | ||
| 46 | F | 35128 | 35520 | 14.6 | Halo gp46 | ||
| 47 | F | 35517 | 35906 | 14.3 | Halo gp47 | ||
| 48 | F | 35894 | 36139 | 9.1 | Halo gp48 | ||
| 49 | F | 36136 | 36480 | 12.7 | Halo gp49 | ||
| 50 | F | 36477 | 36917 | 16.0 | Halo gp50 | ||
| 51 | F | 36914 | 37504 | 21.5 | RuvC | Halo gp51 | Giles gp67 |
| 52 | F | 37506 | 38051 | 20.6 | Halo gp53 | ||
| 53 | F | 38048 | 38206 | 5.9 | Halo gp54 | ||
| 54 | F | 38203 | 38631 | 16.2 | Halo gp55/gp57 | ||
| 55 | F | 38688 | 38972 | 10.0 | Halo gp58 | ||
| 56 | F | 38975 | 39766 | 27.4 | Halo gp59 | Qyrzula gp8, Rosebush gp8, etc. | |
| 57 | F | 39766 | 39891 | 4.7 | Halo gp60 | ||
| 58 | F | 39937 | 40308 | 14.2 | Che8 gp89 | Halo gp56; PMC gp80 | |
| 59 | F | 40295 | 40393 | 3.6 | Halo gp60 | ||
| 60 | F | 40437 | 40760 | 10.9 | Halo gp61 | ||
| 61 | F | 40859 | 41101 | 8.3 | Halo gp62 | ||
| 62 | F | 41125 | 41451 | 12.2 | Halo gp63 | TM4 gp89; LambdaSa03 HNH | |
| 63 | F | 41545 | 41766 | 7.9 | Halo gp64 | PG1 gp69, etc. |
Fig. 4.

Genome organizations of mycobacteriophages BPs, Angel and Halo. Genome maps were generated using the program Phamerator, and each of the predicted ORFs are shown as coloured boxes. Rightwards-transcribed genes are shown above the genome (with 1 kbp scale markers) and leftwards-transcribed genes below it. Gene numbers are indicated within each box, and the phamily (Pham) to which that gene belongs is listed above each gene box. Genes are colour-coded according to their Pham designation, and those that represent unique genes are coloured white. The BPs, Angel and Halo genomes are near-identical (99 % nucleotide identity) for the leftmost 50 genes, and a single map is shown that represents all three genomes for these parts. The three genomes vary in the regions to the right of gene 50, and the individual genome organizations in these regions are shown.
In the BPs, Angel and Halo genomes, a putative integrase (int) gene (32) is positioned about 67 % of the genome length from the left end and is transcribed leftwards. These genomes are thus composed of relatively long left arms (genes 1–31) and correspondingly short right arms (genes 34–63 and 34–64 in BPs and Halo, respectively), with the short right arms accounting for these being the smallest of the mycobacteriophage genomes. Database searching shows that several of the left-arm genes encode virion structure and assembly proteins (Table 1, Fig. 4), and these are generally syntenic with the virion structure operons of other siphoviral phages. The predicted lysis functions (genes 27 and 28) – which are located either to the left or to the right of the structural genes in other mycobacteriophages (Hatfull, 2006) – are positioned to the right of the structural genes in BPs and Halo (Fig. 4). Only four of the BPs right-arm genes (42, 43, 51 and 62), and their Angel counterparts, can be assigned putative functions based on database similarities, all of which are implicated in recombination; gp42 and gp43 as components of a RecE/T-like homologous recombination system, gp51 as a putative Holliday junction resolvase, and gp62 as an HNH endonuclease (Table 1, Fig. 4).
BPs, Angel, and Halo virion structure and assembly genes
The BPs, Angel and Halo virion structure and assembly genes are encoded in the left arms, spanning genes 1 to 31. The roles of 16 of these genes can be predicted from database matches and other organizational features (Table 1). In particular, a large subunit terminase, portal, scaffold head assembly protein, major capsid proteins, major tail subunit, and the tapemeasure protein (encoded by genes 2, 3, 6, 7, 11 and 16, respectively) can be identified by sequence similarities to known proteins. In addition, genes 14 and 15 are predicted to encode tail assembly proteins expressed as a gp14 product of gene 14, and a longer protein that derives from a −1 frameshift approximately 8 codons prior to the stop codon of gene 14. This organization is highly conserved among tailed bacteriophages (Xu et al., 2004), and BPs gp14 and gp15 have weak sequence similarity (45–50 % aa identity) to TM4 gp15 and gp16, which have been shown to be expressed in this manner (Ford et al., 1998b; Xu et al., 2004). The genes (17–22) immediately downstream of the tapemeasure protein gene are predicted to encode minor tail proteins, and these have sequence similarity to other putative mycobacteriophage minor tail proteins. Genes 23–26 are only found in BPs, Angel and Halo, and no homologues are readily identifiable. We predict that they are also involved in tail formation, although one or more could potentially associate with the lysis functions that are encoded by the adjacent gene 27 (lysin A) and 28 (lysin B). We note that the tapemeasure protein contains a Motif 3 sequence that has been identified in several other mycobacteriophage tapemeasure proteins (Pedulla et al., 2003), and which has been shown to play a role in the ability of phage TM4 to efficiently infect stationary-phase M. smegmatis cells (Piuri & Hatfull, 2006). The length of the tapemeasure gene correlates closely with the length of the phage tail (Fig. 1) – assuming a proportionality constant of 0.15 nm tail length per amino acid (Katsura & Hendrix, 1984; Katsura, 1987; Pedulla et al., 2003; Pham et al., 2007). The BPs, Angel and Halo genes at the extreme right end of the left-arm operon (29–31) likely also encode virion structural and assembly proteins. While there are no gp29 homologues in other mycobacteriophages, gp30 and gp31 have sequence similarity to Giles gp34 and gp35, respectively, and are encoded in a similar location in the Giles genome, to the right of the lysis genes at the extreme end of the left-arm operon (and the rightmost gene in Giles, 36, encodes a known virion protein) (Morris et al., 2008).
It is plausible that gene 1 of BPs, Angel and Halo encodes a small terminase subunit. While gp1 does not have sequence similarity to known terminase subunits, it is related to TM4 gp3 (34 % identity) and Corndog gp31 (26 % identity), both of which are located immediately upstream of a large terminase subunit. The function of gp4 is more puzzling. It is related to TM4 gp6 (38 % identity) and more weakly to Che9d gp5 (28 % identity), both of which are located – as in BPs, Angel and Halo – immediately downstream of portal genes. However, these relationships are complex. The predicted BPs, Angel and Halo gp4 proteins are much larger (929 residues) than any of the mycobacteriophage homologues and contain a central 380-residue portion (residues 240–620) that is absent from TM4 gp6. psi-blast searches indicate that the upstream portions of these proteins (residues 1–240) are related to a large group of proteins encoded by mycobacteriophages and other phages, including some predicted capsid morphogenesis proteins (e.g. Lactobacillus phage SPP1 gp7) and phage Mu F related proteins. We therefore predict that gp4 plays a role in head morphogenesis. The role of the central 380-residue insertion in gp4 relative to TM4 gp6 is unclear, but psi-blast searches show that it has weak similarity (∼25 % identity) to a large group of methyl-accepting chemotaxis proteins.
The viral determinants of mycobacteriophage host range – specifically the ability of mycobacteriophages to infect M. tuberculosis – have not been elucidated. We presume that it is primarily a function of the protein components at the tip of the tail, where direct interactions with the host cell wall are expected to be important. However, identification of the genes encoding these components is unclear. BPs and Halo mutants that efficiently infect M. tuberculosis can be readily isolated (see above), and it is of interest whether they share tail genes with TM4, L5, D29, Che12 and Bxz2, which also infect M. tuberculosis. Because the left arms of BPs, Angel and Halo have weak but detectable nucleotide sequence similarity to TM4, the details of this relationship are of interest (see Supplementary Fig. S1, available with the online version of this paper). The most closely related segments are those corresponding to the terminase gene and to tail genes, spanning from the major tail subunit gene (14) to the end of gene 19 (although the similarity is weak and the precise end points are unclear). This relationship is also reflected in the amino acid sequence similarities of these tail genes with the TM4 tail gene homologues (genes 14–19); we note that BPs gene products gp20–gp22 also have reasonable levels of amino acid sequence similarity to their TM4 counterparts (49 %, 40 % and 34 %, respectively) even though the nucleotide sequence similarity is weak (Supplementary Fig. S1). The similarity between the tail proteins of TM4 and BPs is consistent with these being involved in host-range determination. In contrast, several of the BPs, Angel and Halo head proteins, including the portal (gp3), scaffold (gp6) and capsid (gp7) are most closely related to those of phage Che9d (52 %, 31 % and 46 %, respectively), reflecting the mosaic architecture of the virion structural operon in BPs. SDS-PAGE analysis of BPs virion proteins shows the presence of high-molecular-mass bands, suggestive of covalent cross-linking of the capsid proteins, as shown in other mycobacteriophages such as L5 (Hatfull & Sarkis, 1993) and D29 (Ford et al., 1998a) (data not shown); the BPs and D29 capsid proteins are more distantly related and share 24 % amino acid sequence identity.
Lysogeny, immunity and integration of BPs, Angel and Halo
BPs, Angel and Halo form slightly turbid plaques on M. smegmatis lawns. To determine if these phages form lysogens, and at what frequency, serial dilutions of an M. smegmatis culture were plated on solid medium to which approximately 109 p.f.u. of BPs and Halo phage had been added. Colonies grew on the phage-seeded plates at a frequency approximately 5 % of that observed on a control plate. This 5 % lysogenization frequency is lower than that reported for L5 (Sarkis et al., 1995), but similar to that of phage Giles (Morris et al., 2008). Two individual colonies from the Halo-seeded plate were purified and tested for lysogeny by immunity and phage release (Fig. 5). Lysogens of Halo conferred strong immunity to infection to both BPs and Halo, but not to any of the other mycobacteriophages that we tested (data not shown). Lysogenization frequencies of Angel have not been determined, but given its genomic similarity to BPs and Halo, we predict that it behaves similarly.
Fig. 5.

Lysogeny and integration of phages BPs, Angel and Halo. (a) The sequence of the BPs genome spanning the start of gene 34 to the beginning of gene 32 (int) (corresponding to coordinates 29 500–28 901); BPs, Angel and Halo are identical in this region. Gene 33 is predicted to encode the phage repressor, and the amino acid sequence is shown in red type, with the putative helix–turn–helix motif boxed; the integrase protein (gp32) is shown in blue type. The core of the attP site that is near-identical to the predicted attB site in M. smegmatis is shown in bold type with the two differences between attP and attB boxed. For simplicity, the orientation of this genome segment is reversed relative to that shown in Fig. 4. (b) Integration and immunity functions. Plasmids pTRS1a, pGWB37, and pGWB40 containing the indicated BPs segments were electroporated into M. smegmatis, and transformants were selected. Plasmid pTRS1a, which contains gene 32 and the attP core site, but no prospective arm-type integrase binding sites, gave few if any transformants, whereas both pGWB37 and pGWB40 yielded approximately 102 transformants per μg DNA. The inset table summarizes whether each plasmid efficiently transforms M. smegmatis (trans) or confers immunity (imm). (c) PCR analysis of M. smegmatis Halo lysogens and pGWB40 transformants. Primers were used to generate PCR products corresponding to the M. smegmatis Halo attB site or to the predicted attL junction site generated by integration, as shown in the upper and lower panels, respectively. Lanes 1–2, two independent pGWB40 transformants; lanes 3–6, Halo lysogens of M. smegmatis mc2155; lane 7, a pMH94 transformant of M. smegmatis mc2155; lane 8, a pJV53 transformant of M. smegmatis mc2155; lane 9, M. smegmatis mc2155. (d) Immunity of Halo lysogens and transformants. Serial dilutions of phages D29, BPs and Halo were spotted onto lawns of either M. smegmatis mc2155, a Halo lysogen [mc2155(Halo)], or transformants generated with pGWB37 and pGWB40. Phage concentrations are highest in the leftmost spot.
Bioinformatic analysis readily identified genes 32 of BPs, Angel and Halo – which share 100 % amino acid sequence identity – as members of the tyrosine-integrase family. These are distant relatives of all other mycobacteriophage integrases, with the nearest being Giles gp29, with which they share 18.2 % amino acid sequence identity. Among the non-mycobacteriophage integrases, the closest relatives are predicted integrases of putative prophages in the genomes of Nocardia farcinica and Corynebacterium diphtheriae (55 % and 40 % amino acid identity, respectively). We reported previously that a 35 bp segment of Halo is closely related to the M. smegmatis mc2155 genome (coordinates 6 410 365–6 410 399) and presumably corresponds to the common core of putative attP and attB sites, with attB overlapping a tRNAArg gene (Msmeg_6349); the same 35 bp segment is present in BPs (at coordinates 29 014–29 048), 5′ to the start of the int gene (32) (Fig. 5), and similarly in Angel.
A peculiar feature of the int-attP organization is that the putative attP core is situated within the coding region of the upstream gene (33) (Fig. 5a). If this is indeed used for integrase-mediated recombination then gene 33 would be split, such that the predicted prophage-encoded product is 32 amino acids shorter than the predicted phage-encoded protein. Support for the use of this putative attP site is provided by comparison with related proteins in Nocardia and Corynebacterium, in which the regions of similarity do not extend beyond the putative attP site. Of further interest is the observation that gp33 contains a pfam07022 motif associated with helix–turn–helix DNA-binding motifs that is common among phage repressors. BPs, Angel and Halo gp34 are good candidates for excise proteins acting as recombination directionality factors (RDFs), based on database matches to a large number of other predicted excise proteins.
To test for functionality of the BPs/Angel/Halo integration systems, vectors were constructed containing segments of the gene 31–34 region. Plasmid pTRS1a contains the complete int gene and the predicted attP common core of BPs, but lacks flanking sequences where arm-type integrase-binding sites are anticipated to be located. When pTRS1a was electroporated into M. smegmatis, few if any transformants were obtained (Fig. 5b). In contrast, plasmid pGWB37 contains an additional 221 bp flanking the attP common core and is therefore expected to have a functional attP site. Electroporation of M. smegmatis with pGWB37 generated approximately 102 transformants per μg DNA. While this transformation frequency is 103-fold lower than observed with an L5-integration vector in a control experiment, PCR analysis of the transformants showed the interruption of the BPs/Halo attB site and generation of an attL recombinant site, producing the same PCR products as seen with a Halo lysogen (data not shown). A plasmid (pGWB38) containing an identical DNA insert but in the opposite orientation relative to the vector backbone failed to transform M. smegmatis, suggesting that integrase expression may be dependent on vector sequences and providing a plausible explanation for the low transformation frequency. Plasmid pGWB40 containing genes 32 and 33, as well as the 33–34 intergenic region, also transformed at a low frequency (102 transformants per μg DNA), even though it seems highly likely that this contains all of the sequences required for attP function (Fig. 5). PCR analysis demonstrated that pGWB40 also integrates into the predicted attB site (Fig. 5c).
To test whether the putative repressor gene (33) confers immunity to superinfection, pGWB37 and pGWB40 transformants were compared to a Halo lysogen in immunity tests (Fig. 5d). A Halo lysogen confers high levels of immunity to both BPs and Halo, whereas pGWB37 does not. Plasmid pGWB40 transformants confer immunity to both BPs and Halo, although some plaques are seen at higher phage concentrations, and the degree of immunity is less than that of the Halo lysogen (Fig. 5d). These observations are consistent with gp33 having repressor activity, although its expression may be lower than from the lysogen.
A peculiar aspect of the immunity-integration functions in these phages is the location of the attP site within the repressor gene. Furthermore, because the base in the M. smegmatis genome immediately to the right of the 34 bp common core is an A, then integration results in the generation of a termination codon and C-terminal truncation of the repressor by 32 residues. The truncated form confers immunity as seen in pGWB40, which is also predicted to be made from a prophage. The longer form made from a replicating viral genome could have different or additional functions, and its expression could potentially affect the translation of gpInt, whose initiation codon overlaps the stop codon of gene 33. We also note that the two base differences between the attP and attB common cores correspond to the innermost base pair of the stem of the D arm of the tRNAArg, such that if strand exchange occurs to the left of the first base difference (as shown in Fig. 5a) then the tRNAArg structure would be conserved following integration. While the positions of strand exchange are not known, we note that the tRNA structure would be conserved even if strand exchange occurs between the positions of the two base differences between the attP and attB cores. A similar tRNA gene (NT02MT4110) and putative attB site are present in the M. tuberculosis genome, and while there are additional differences in the core sequences, we predict that integration into this site should occur with maintenance of tRNA function. As noted previously (Pham et al., 2007), the putative BPs/Angel/Halo attB site is the same as that used for integration of the tox-containing phage in Corynebacterium diphtheriae.
BPs and Angel non-structural genes
BPs, Angel and Halo differ from other mycobacteriophages with a siphoviral morphotype in that the right arm encoding non-structural functions is relatively short. Genes 34–64 are all transcribed in the rightwards direction (Fig. 4), and the function of only four can be predicted from database matches, including RecE/T homologues (43, 44) involved in homologous recombination, a RuvC family Holliday junction resolvase (51) and an HNH endonuclease (62). All of these have related genes in one or more other mycobacteriophages, and the HNH endonucleases are very common. About 40 % of the right-arm genes (including 37, 38, 40, 41, 44, 45, 46, 47, 49, 50, 52, 55, 60 and 61) do not have database matches to other mycobacteriophage proteins other than those encoded by Halo, and of these, only gp38 has significant database matches to non-mycobacteriophage proteins, with similarity to a group of bacterial and phage-related proteins of unknown function.
The right arms of BPs and Angel are slightly smaller than Halo primarily because neither have Halo gene 52; the function of this gene is not known, although it is a distant relative of Barnyard gp102. The differences at the nucleotide level are of interest and – as shown in Fig. 2 (inset, difference no. 1) and Supplementary Fig. S2 – involve the replacement of a small segment (92 bp deletion; coordinates 37 505–37 597) in BPs with a 645 bp insertion (coordinates 37 499–38 144); Angel is identical to BPs in this region. This corresponds precisely to the first nucleotide after the termination codons of genes 51 in both genomes BPs and Halo, and the first nucleotide prior to gene 53 in Halo. While this corresponds to codon 34 in BPs gene 52, the annotation of this gene is ambiguous, in that translation initiation could occur at the start codon immediately adjacent to the stop codon of gene 51, or at the ATG codon corresponding to position 35 in the annotated gene (see Supplementary Fig. S2 for further clarification). In the latter explanation, Halo gene 52 would replace a gene 51–52 intergenic gap in BPs. As such, these genomic differences cannot be accounted for by a simple insertion or deletion event. At the rightmost insertion/deletion (indicated as no. 4 in the inset in Fig. 2) a 182 bp segment in BPs (coordinates 41 453–41 635) is absent from Halo and replaced by a 24 bp segment (coordinates 42 001–42 025), resulting in an N-terminal 31 codon deletion. The remaining two differences (nos 2 and 3 in the inset in Fig. 2) are discussed below.
Most of the genes found in non-structural regions of mycobacteriophages are of unknown function. We recently described a simple method for manipulating mycobacteriophage genomes and have used this to demonstrate that BPs genes 44, 50, 52, 54 and 58, as well as Halo genes 49 and 52, are not required for plaque formation (Marinelli et al., 2008). While we do not yet know if the recE/T genes are essential in BPs and Halo, it is noteworthy that the recT homologue in phage Che9c (gene 61) also is not required for plaque formation (Marinelli et al., 2008). Thus even though the genomes of BPs, Angel and Halo are considerably smaller than other mycobacteriophage genomes, a substantial proportion of the right-arm genes are not essential for plaque formation.
Identification of a new small mobile genetic element
At the right ends of the BPs and Halo genomes there is an insertion in each of the genomes relative to the other, although at different locations (corresponding to the insertion/deletions 2 and 3 in the inset of Fig. 2). In Halo, the insertion is within the homologue of BPs gene 54, and in BPs it is in the homologue of Halo gene 60 (Fig. 6); Angel contains neither of the insertions. The DNA segments present as insertions in BPs and Halo are distantly related but can be seen as weak similarity in Fig. 2. However, each encodes an ORF (Halo gene 56 and BPs gene 58), and the 123-residue predicted products share 67 % amino acid identity. Several lines of evidence support the hypothesis that these insertions result from transposition of a new class of small mobile genetic elements.
Fig. 6.
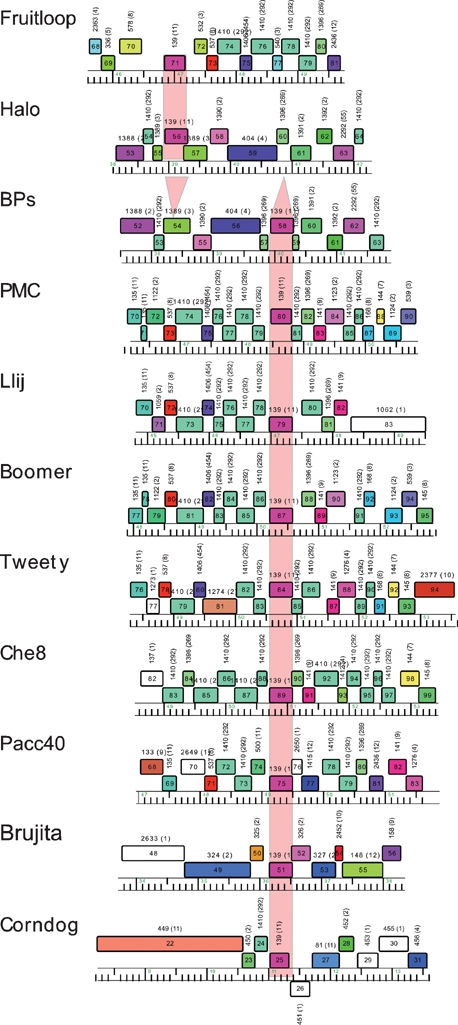
Mycobacteriophage genomes encoding protein members of Pham139. Phages Halo and BPs both contain genes that are members of Pham139, each of which appears in a location in one phage in which it is absent from the other. Of the 60 mycobacteriophage genomes in the current Phamerator database, nine other genomes also encode Pham139 phamily members. Approximately 5 kbp segments of each of the genomes are shown, centred about their Pham139 members. Genome segments are labelled as described for Fig. 4.
First, the genome maps in Fig. 4 show that the putative gene products Halo gp56 and BPs gp58 are members of a phamily (Pham139) that has an additional nine members; these correspond to Boomer gp87, PMC gp80, Llij gp79, Che8 gp89, Tweety gp84, Pacc40 gp75, Fruitloop gp71, Brujita gp51 and Corndog gp25 (Fig. 6). The sequence similarity between these is very high, and all except Halo gp56 and Fruitloop gp71 are 100 % identical to BPs gp58; this includes Corndog but extends over only approximately the first 100 residues due to a C-terminal deletion. Halo gp56 and Fruitloop gp71 are 100 % identical to each other, but only 67 % identical to BPs gp68 and its relatives. When examined at the nucleotide sequence level, BPs, PMC, Llij, Boomer, Tweety, Che8, Pacc40 and Brujita all contain an identical segment of 439 bp, with the exception of Brujita, which contains a single base difference (at coordinate 36 374); Corndog has a 363 bp segment that is 100 % identical to these (Fig. 7). A related segment is identical in Halo and Fruitloop but is one base longer and shares only 79 % nucleotide identity with BPs. The finding of multiple copies of these two related segments in several independent contexts is unusual. We propose that these relationships derive from the presence of two different members of a novel class of mobile genetic elements, which we will refer to as MPME1 (mycobacterium phage mobile element 1) present in BPs, PMC, Llij, Boomer, Tweety, Che8, Pacc40 and Brujita and as a truncated copy in Corndog, and MPME2, which is present in Halo and Fruitloop (Fig. 7).
Fig. 7.

Location and structure of MPME1 and MPME2. Segments of 11 mycobacteriophage genomes are shown, indicating the positions of mycobacterium phage mobile elements 1 (MPME1) and 2 (MPME2). All nine copies of MPME1 are 100 % identical at the nucleotide sequence level – with the exception of a single base difference in Brujita – and are 439 bp long; MPME1 in Corndog is truncated at its right end. MPME1 and MPME2 are bordered by left and right 11 bp inverted repeats (IR-L and IR-R, respectively), shown as black triangles. MPME2 has 78 % nucleotide sequence identity with MPME1 and is 440 bp long; the MPME1 elements in Halo and Fruitloop are identical. The MPME elements code for 123-residue products that are candidates for providing transposase (TnpA) activity. The genome coordinates for the ends of the elements are shown. It should be noted that in each genome the assigned ORFs immediately upstream and downstream of the insertion are unlikely to be expressed, since they correspond to the two remnants of the gene into which the insertion occurred. It should also be noted that some of the genes flanking the insertion are members of a very large phamily (Pham1410), and since any gene has only to be significantly related to one other phamily member to be in that Pham, any genes in the same phamily may not necessarily be related. For example, the gene upstream of the Che8 insert is not related at the sequence level to those upstream of the insertions in PMC, Llij, Boomer, Tweety or Corndog.
Examination of MPME1 reveals short-terminal inverted repeats (IR), characteristic elements of bacterial mobile genetic elements (5′-TTATC[a/t]GGGGT) (Fig. 8). The length of the IR is 11 bp, of which 10 of the 11 bp are symmetrically related; the non-symmetrical base is at position 6 (where position 1 corresponds to the outermost base of the IR; Fig. 8). In Corndog, only the left IR (IR-L) is identifiable, and the right IR (IR-R) is lost due to a 76 bp deletion relative to BPs at the right end of the element. MPME2 has similar IRs to MPME1, with the notable exception that IR-R differs at position 8 (Fig. 8).
Fig. 8.

Sequences of insertion sites of the MPME1 and MPME2. (a) The MPME1 insertion in phage BPs and the MPME2 insertion in phage Halo are at different genome locations, and the near-identity of the two genomes in these locations enables elucidation of the pre-integration target sequences. The boxed sequences in BPs and Halo correspond to the DNA sequences that are present in one genome but absent in the other, with the sequences of the junctions shown. Each insertion contains 11 bp inverted repeats (IR-L and IR-R; bold type) of which 10 bp are symmetrically positioned. Phages PMC (and the very close relative Boomer, not shown), Tweety (and its very close relative Llij, not shown), Pacc40, Che8 and Corndog all contain identical copies of the MPME1 present in BPs, although the right of Corndog is deleted. PMC and Tweety are related genomes, and the left-side target sequences are identical, suggesting that these are derived from the same initial integration event; the right-side sequences are different, which further suggests that there have been additional rearrangements. Corndog contains a truncated copy of the element in which the 3′ end of the element is missing 76 bp. Fruitloop contains a copy of MPME2 that is identical to that in Halo and also shares the same 6 bp immediately left of IR-L. The sequence of the element is shown in italics, and the flanking TGA codon in Corndog that terminates the putative TnpA protein is underlined. (b) Brujita and Che9c are related genomes, and Brujita contains an MPME1 element 99 % identical to that in BPs. The flanking sequences in Che9c are near-identical (with the exception of the position immediately to the right of IR-R in the insertion in Brujita), and the boxed sequence represents that which is present in Brujita and absent from Che9c. (c) The pre-integration site corresponding to the insertion in Che8 is not known. However, phage Bxz1 contains a common 110 bp region (shaded box) ∼90 % identical to the left side of the Che8 insertion, and the sequence alignment stops precisely at a position corresponding to 6 bp to the left of the left inverted repeat. At the right end, Che8 and Omega share ∼200 bp of near sequence identity that matches precisely to the end of the right inverted repeat (shaded box). Omega coordinates: 50 716-50 497; Che8 coordinates: 51 089–51 308.
Since the Halo and BPs genomes are very closely related, and because each represents an independent insertion, comparison of the genomes provides two examples of the pre-integration site and the structures following integration; Angel contains the same pre-integration sites for both insertions. The total length of the insertion is 445 bp in BPs and 446 bp in Halo, corresponding to the 439 bp MPME1 and the 440 bp MPME2 elements, respectively, plus an additional 6 bp immediately adjacent to IR-L (Fig. 8). In both examples, the right junction results from joining of position 1 of IR-R to the putative target sequence, whereas at IR-L there are insertions of 6 bp between the left IR-L and target sequence. This 6 bp is different in the Halo and BPs insertions, and the 6 bp sequence in BPs is not present elsewhere in the BPs genome; there are a number of other occurrences of the Halo 6 bp sequence elsewhere in Halo. Direct repeats corresponding to a target duplication are not observed.
Comparison of Brujita and the related Che9c genome provides an additional example of a pre-integration site and the post-insertion structure (Fig. 8b). The total size of the insertion is 445 bp, encompassing MPME1 and six bases to the left of IR-L, although there is also a single base change immediately adjacent to IR-R. These observations suggest strongly that the 6 bp insertion occurs when the elements are mobilized, although its origins are unclear. It is plausible that it derives from the donor, and it is noteworthy that the two MPME2 insertions contain identical 6 bp segments (Fig. 8a). PMC and Tweety share these 6 bp in common, although the sequence similarity extends beyond that in the flanking sequences, and thus they might reflect a single insertion event. Pacc40, Che8 and Corndog all contain the same 6 bp to the left of IR-L – and could therefore have arisen from the same donor – but this sequence is different to that found in the PMC/Tweety, Brujita and BPs insertions.
For the insertions in PMC, Tweety, Che8 and Corndog there is not a known sequence context that simply corresponds to a pre-integration site. However, a ∼110 bp sequence to the left of Che8 IR-L has sequence similarity to part of the otherwise unrelated mycobacteriophage Bxz1 (Fig. 8c). Interestingly, the sequence similarity ends 6 bp to the left of the Che8 IR-L, as seen in the other MPME structures. Furthermore, the sequence to the right of the Che8 element shares sequence similarity with a segment of the mycobacteriophage Omega genome, and the discontinuity in the relationship ends precisely at the junction of Che8 IR-R, consistent with this being the position of joining of the element to the target sequence.
The shared nucleotide sequences shown in Fig. 7 are unrelated to other mobile elements, and the predicted 123-residue protein product is not closely related to any known proteins. We have not been able to identify any putative DNA-binding or other transposase-associated motifs in the protein sequence, but presume that it is the transposase mediating mobility of the elements. The termination codon of this putative transposase gene corresponds to the outside 3 bp of IR-R (Figs 7 and 8) and the start codon is 68 bp inside of IR-L; we do not know if transcription signals reside within this segment.
DISCUSSION
Mycobacteriophages BPs and Angel are newly isolated phages with several features of interest, particularly their small genome size, their host range, an unusual organization of the integration and immunity functions, and a new small mobile genetic element. Importantly, the near-identity of the BPs, Angel and Halo genomes is critical for identification of the putative new mobile elements, providing both pre-integration and post-integration sequences. This ability to compare closely related phages isolated from a population with such wide diversity emphasizes the value of finding new phages that are close relatives of those previously isolated.
While the majority of sequenced mycobacteriophage genomes encode a recognizable integrase gene, only L5 (Donnelly-Wu et al., 1993), Che12 (Kumar et al., 2008), Bxb1 (Jain & Hatfull, 2000) and closely related phages produce obviously turbid plaques and form lysogens at relatively high frequencies (∼20 %; Sarkis et al., 1995). Presumably the other integrase-containing phages can also form lysogens, either at lower frequencies or perhaps in other mycobacterial hosts. Notably, M. smegmatis lysogens of mycobacteriophage Giles are formed at low frequency (∼5 %; Morris et al., 2008), and similarly, BPs and Halo also form lysogens at approximately 5 %. However, the only mycobacteriophages for which repressor genes have been identified are L5 (Donnelly-Wu et al., 1993) and Bxb1 (Jain & Hatfull, 2000), and in most other genomes the repressors have yet to be identified. BPs and Halo represent only the second type of mycobacteriophage repressors to be described, although this protein phamily is restricted to BPs and Halo. While the L5 and Bxb1 genomes contain multiple copies (>30) of repressor-binding sites that play a role in transcriptional silencing of these genomes (Brown et al., 1997), we have not been able to identify any similar repeated sequences in BPs or Halo, and none are apparent in the analysis shown in Fig. 2.
Although the integration and immunity functions are not closely linked either in the well-studied phage lambda genome or in mycobacteriophages L5 (Hatfull & Sarkis, 1993) and Bxb1 (Mediavilla et al., 2000), in other temperate phage genomes – such as many of the dairy phages (Brussow, 2001) – the lysogeny functions are tightly clustered. However, we are not aware of any previous example where not only are the immunity and integration functions very tightly linked, but the attP site is located within the repressor gene itself. This gives rise to the odd situation where the repressor gene product is different when expressed from a prophage or from a viral genome. We presume that a promoter for repressor synthesis lies within the gene 33–34 intergenic interval, although the incomplete immunity conferred by plasmid pGWB40 (Fig. 5) suggests that other BPs/Halo genes may be required for proper repressor expression or for repressor stability.
The identification of MPME1 and MPME2 as novel mobile genetic elements is of considerable interest. The elements have not been found elsewhere, in either other phage or bacterial genomes, and yet are present as at least eight independent insertion events in the mycobacteriophages (in Halo; in BPS; in PMC, Boomer, Llij and Tweety; in Fruitloop; in Brujita, in Pacc40; in Che8; and in Corndog). Phages PMC, Llij, Boomer and Tweety are closely related, and the sequences flanking each IR-L are the same, strongly suggesting that they were all derived from the same insertion event. However, the sequences to the right of IR-R are the same in PMC and Boomer, but different in Llij and Tweety, suggesting that additional rearrangements have occurred. The observation that rearrangements in addition to the insertion event can occur emphasizes the considerable value of having pre-integration target sequences in BPs, Angel and Halo. It therefore seems highly unlikely that the lack of direct repeats results either from recombination between two different MPME copies (either intra- or intermolecularly) or by adjacent deletion.
The MPME1 and MPME2 elements are distinct from previously described mobile genetic elements in several important ways. First, they are smaller than any known family of mobile elements in prokaryotes, of which the smallest are the IS6 family (750–900 bp) and the IS1031 family (850–950 bp). Second, MPME1 and MPME2 do not generate direct repeats of the target, a common feature of bacterial transposons; the exceptions among known IS elements are the IS91, IS110 and IS200/IS605 families, all of which are non-canonical IS elements that do not possess inverted repeats at their termini. MPME1 and MPME2 clearly do have IRs, and these appear to define the positions of strand exchange, at least at IR-R. A most unusual aspect of these elements is the generation of a 6 bp insertion between IR-L and the target sequence. At least in BPs, this 6 bp sequence does not appear elsewhere in the genome, and it seems likely that this genetic information is derived from the donor during insertion. This would indicate an unusual transposition mechanism, since the presumed transposase would need to cleave the element from the donor differently at the two IRs. We note that MPME1 and MPME2 share some features of the IS200/IS605 family (Kersulyte et al., 2002) in that these transposases are also small (IS608 TnpA is 155 aa; Guynet et al., 2008), and there is asymmetry in the terminal sequences (assuming that the additional 6 bp in MPME insertions are derived from the donor).
A potential explanation for the small size of these elements is that they are non-functional and represent defective transposons that are incapable of independent movement. Although we cannot eliminate this possibility, it seems unlikely given the relationships of the elements that we have identified. For example, while MPME1 and MPME2 share only 79 % nucleotide sequence similarity, the overall structures are very similar, differ by only 1 bp in length and contain both IRs and the left-end adjacent 6 bp insertion; it is unlikely that both elements would be similarly defective given their subsequent divergence.
Comparative analysis of mycobacteriophage genomes has shown that they are genetically diverse and characterized by architectural mosaicism. Several mechanisms have been proposed to contribute to these structures, including homologous recombination between similar sequences (Hatfull et al., 2008), illegitimate recombination between dissimilar sequences (Pedulla et al., 2003), and integrase-mediated site-specific recombination between attachment site sequence and secondary sites (Morris et al., 2008). While transposons are found in phages of other bacterial hosts, as well as in the chromosome of mycobacteria, none had been previously identified in mycobacteriophage genomes. In view of the high genetic diversity of the mycobacteriophages, it is perhaps not surprising that the first mobile genetic element to be described from these is unlike other prokaryotic mobile elements, even though thousands have been reported and there are more than 20 well-defined families. Thus even though no previously reported transposons have been found in mycobacteriophage genomes, the finding of these new mobile elements suggests that transposition does indeed play a role in evolution of mycobacteriophage genomes.
Acknowledgments
This work was supported by a grant to the University of Pittsburgh from the Howard Hughes Medical Institute (HHMI) in support of Graham Hatfull under HHMI's Professors program, and by grants from NIH to R. W. H. (GM51975), and to G. F. H (AI28927). We thank Tom Harper for microscopy help, and Steve Cresawn and Craig Peebles for useful discussions.
Footnotes
References
- Bardarov, S., Kriakov, J., Carriere, C., Yu, S., Vaamonde, C., McAdam, R. A., Bloom, B. R., Hatfull, G. F. & Jacobs, W. R., Jr (1997). Conditionally replicating mycobacteriophages: a system for transposon delivery to Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 94, 10961–10966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardarov, S., Bardarov, S., Jr, Pavelka, M. S., Jr, Sambandamurthy, V., Larsen, M., Tufariello, J., Chan, J., Hatfull, G. & Jacobs, W. R., Jr (2002). Specialized transduction: an efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG and M. smegmatis. Microbiology 148, 3007–3017. [DOI] [PubMed] [Google Scholar]
- Brown, K. L., Sarkis, G. J., Wadsworth, C. & Hatfull, G. F. (1997). Transcriptional silencing by the mycobacteriophage L5 repressor. EMBO J 16, 5914–5921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brussow, H. (2001). Phages of dairy bacteria. Annu Rev Microbiol 55, 283–303. [DOI] [PubMed] [Google Scholar]
- Donnelly-Wu, M. K., Jacobs, W. R., Jr & Hatfull, G. F. (1993). Superinfection immunity of mycobacteriophage L5: applications for genetic transformation of mycobacteria. Mol Microbiol 7, 407–417. [DOI] [PubMed] [Google Scholar]
- Ford, M. E., Sarkis, G. J., Belanger, A. E., Hendrix, R. W. & Hatfull, G. F. (1998a). Genome structure of mycobacteriophage D29: implications for phage evolution. J Mol Biol 279, 143–164. [DOI] [PubMed] [Google Scholar]
- Ford, M. E., Stenstrom, C., Hendrix, R. W. & Hatfull, G. F. (1998b). Mycobacteriophage TM4: genome structure and gene expression. Tuber Lung Dis 79, 63–73. [DOI] [PubMed] [Google Scholar]
- Freitas-Vieira, A., Anes, E. & Moniz-Pereira, J. (1998). The site-specific recombination locus of mycobacteriophage Ms6 determines DNA integration at the tRNAAla gene of Mycobacterium spp. Microbiology 144, 3397–3406. [DOI] [PubMed] [Google Scholar]
- Fullner, K. J. & Hatfull, G. F. (1997). Mycobacteriophage L5 infection of Mycobacterium bovis BCG: implications for phage genetics in the slow-growing mycobacteria. Mol Microbiol 26, 755–766. [DOI] [PubMed] [Google Scholar]
- Guynet, C., Hickman, A. B., Barabas, O., Dyda, F., Chandler, M. & Ton-Hoang, B. (2008). In vitro reconstitution of a single-stranded transposition mechanism of IS608. Mol Cell 29, 302–312. [DOI] [PubMed] [Google Scholar]
- Hatfull, G. F. (2000). Molecular genetics of mycobacteriophages. In Molecular Genetics of the Mycobacteria, pp. 37–54. Edited by G. F. Hatfull & W. R. Jacobs, Jr. Washington, DC: American Society for Microbiology.
- Hatfull, G. F. (2006). Mycobacteriophages. In The Bacteriophages, pp. 602–620. Edited by R. Calendar. New York: Oxford University Press.
- Hatfull, G. F. & Sarkis, G. J. (1993). DNA sequence, structure and gene expression of mycobacteriophage L5: a phage system for mycobacterial genetics. Mol Microbiol 7, 395–405. [DOI] [PubMed] [Google Scholar]
- Hatfull, G. F., Pedulla, M. L., Jacobs-Sera, D., Cichon, P. M., Foley, A., Ford, M. E., Gonda, R. M., Houtz, J. M., Hryckowian, A. J. & other authors (2006). Exploring the mycobacteriophage metaproteome: phage genomics as an educational platform. PLoS Genet 2, e92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatfull, G. F., Cresawn, S. G. & Hendrix, R. W. (2008). Comparative genomics of the mycobacteriophages: insights into bacteriophage evolution. Res Microbiol 159, 332–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrix, R. W. (2002). Bacteriophages: evolution of the majority. Theor Popul Biol 61, 471–480. [DOI] [PubMed] [Google Scholar]
- Jacobs, W. R., Jr, Tuckman, M. & Bloom, B. R. (1987). Introduction of foreign DNA into mycobacteria using a shuttle phasmid. Nature 327, 532–535. [DOI] [PubMed] [Google Scholar]
- Jacobs, W. R., Jr, Barletta, R. G., Udani, R., Chan, J., Kalkut, G., Sosne, G., Kieser, T., Sarkis, G. J., Hatfull, G. F. & Bloom, B. R. (1993). Rapid assessment of drug susceptibilities of Mycobacterium tuberculosis by means of luciferase reporter phages. Science 260, 819–822. [DOI] [PubMed] [Google Scholar]
- Jain, S. & Hatfull, G. F. (2000). Transcriptional regulation and immunity in mycobacteriophage Bxb1. Mol Microbiol 38, 971–985. [DOI] [PubMed] [Google Scholar]
- Katsura, I. (1987). Determination of bacteriophage lambda tail length by a protein ruler. Nature 327, 73–75. [DOI] [PubMed] [Google Scholar]
- Katsura, I. & Hendrix, R. W. (1984). Length determination in bacteriophage lambda tails. Cell 39, 691–698. [DOI] [PubMed] [Google Scholar]
- Kersulyte, D., Velapatino, B., Dailide, G., Mukhopadhyay, A. K., Ito, Y., Cahuayme, L., Parkinson, A. J., Gilman, R. H. & Berg, D. E. (2002). Transposable element ISHp608 of Helicobacter pylori: nonrandom geographic distribution, functional organization, and insertion specificity. J Bacteriol 184, 992–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, A. I., Ghosh, P., Aaron, M. A., Bibb, L. A., Jain, S. & Hatfull, G. F. (2003). Mycobacteriophage Bxb1 integrates into the Mycobacterium smegmatis groEL1 gene. Mol Microbiol 50, 463–473. [DOI] [PubMed] [Google Scholar]
- Kumar, V., Loganathan, P., Sivaramakrishnan, G., Kriakov, J., Dusthakeer, A., Subramanyam, B., Chan, J., Jacobs, W. R., Jr & Paranji Rama, N. (2008). Characterization of temperate phage Che12 and construction of a new tool for diagnosis of tuberculosis. Tuberculosis (Edinb) 88, 616–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence, J. G., Hatfull, G. F. & Hendrix, R. W. (2002). Imbroglios of viral taxonomy: genetic exchange and failings of phenetic approaches. J Bacteriol 184, 4891–4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, M. H., Pascopella, L., Jacobs, W. R., Jr & Hatfull, G. F. (1991). Site-specific integration of mycobacteriophage L5: integration- proficient vectors for Mycobacterium smegmatis, Mycobacterium tuberculosis, and bacille Calmette-Guérin. Proc Natl Acad Sci U S A 88, 3111–3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinelli, L. J., Piuri, M., Swigonova, Z., Balachandran, A., Oldfield, L., van Kessel, J. C. & Hatfull, G. F. (2008). BRED: a simple and powerful tool for constructing mutant and recombinant bacteriophage genomes. PLoS One 3, e3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mediavilla, J., Jain, S., Kriakov, J., Ford, M. E., Duda, R. L., Jacobs, W. R., Jr, Hendrix, R. W. & Hatfull, G. F. (2000). Genome organization and characterization of mycobacteriophage Bxb1. Mol Microbiol 38, 955–970. [DOI] [PubMed] [Google Scholar]
- Morris, P., Marinelli, L. J., Jacobs-Sera, D., Hendrix, R. W. & Hatfull, G. F. (2008). Genomic characterization of mycobacteriophage Giles: evidence for phage acquisition of host DNA by illegitimate recombination. J Bacteriol 190, 2172–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojha, A. K., Baughn, A. D., Sambandan, D., Hsu, T., Trivelli, X., Guerardel, Y., Alahari, A., Kremer, L., Jacobs, W. R., Jr & Hatfull, G. F. (2008). Growth of Mycobacterium tuberculosis biofilms containing free mycolic acids and harbouring drug-tolerant bacteria. Mol Microbiol 69, 164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedulla, M. L., Ford, M. E., Houtz, J. M., Karthikeyan, T., Wadsworth, C., Lewis, J. A., Jacobs-Sera, D., Falbo, J., Gross, J. & other authors (2003). Origins of highly mosaic mycobacteriophage genomes. Cell 113, 171–182. [DOI] [PubMed] [Google Scholar]
- Pham, T. T., Jacobs-Sera, D., Pedulla, M. L., Hendrix, R. W. & Hatfull, G. F. (2007). Comparative genomic analysis of mycobacteriophage Tweety: evolutionary insights and construction of compatible site-specific integration vectors for mycobacteria. Microbiology 153, 2711–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piuri, M. & Hatfull, G. F. (2006). A peptidoglycan hydrolase motif within the mycobacteriophage TM4 tape measure protein promotes efficient infection of stationary phase cells. Mol Microbiol 62, 1569–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piuri, M., Jacobs, W. R., Jr & Hatfull, G. F. (2009). Fluoromycobacteriophages for rapid, specific, and sensitive antibiotic susceptibility testing of Mycobacterium tuberculosis. PLoS One 4, e4870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkis, G. J. & Hatfull, G. F. (1998). Mycobacteriophages. Methods Mol Biol 101, 145–173. [DOI] [PubMed] [Google Scholar]
- Sarkis, G. J., Jacobs, W. R., Jr & Hatfull, G. F. (1995). L5 luciferase reporter mycobacteriophages: a sensitive tool for the detection and assay of live mycobacteria. Mol Microbiol 15, 1055–1067. [DOI] [PubMed] [Google Scholar]
- Timme, T. L. & Brennan, P. J. (1984). Induction of bacteriophage from members of the Mycobacterium avium, Mycobacterium intracellulare, Mycobacterium scrofulaceum serocomplex. J Gen Microbiol 130, 2059–2066. [DOI] [PubMed] [Google Scholar]
- van Kessel, J. C. & Hatfull, G. F. (2007). Recombineering in Mycobacterium tuberculosis. Nat Methods 4, 147–152. [DOI] [PubMed] [Google Scholar]
- van Kessel, J. C. & Hatfull, G. F. (2008). Efficient point mutagenesis in mycobacteria using single-stranded DNA recombineering: characterization of antimycobacterial drug targets. Mol Microbiol 67, 1094–1107. [DOI] [PubMed] [Google Scholar]
- van Kessel, J. C., Marinelli, L. J. & Hatfull, G. F. (2008). Recombineering mycobacteria and their phages. Nat Rev Microbiol 6, 851–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, J., Hendrix, R. W. & Duda, R. L. (2004). Conserved translational frameshift in dsDNA bacteriophage tail assembly genes. Mol Cell 16, 11–21. [DOI] [PubMed] [Google Scholar]