

Abstract
The zinc(II)-dependent matrix metalloproteinases (MMPs) are associated with a variety of diseases. Development of inhibitors to modulate MMP activity has been an active area of investigation for therapeutic development. Hydroxypyrones and hydroxythiopyrones are alternative zinc-binding groups (ZBGs) that, when combined with peptidomimetic backbones, comprise a novel class of MMP inhibitors (MMPi). In this report, a series of hydroxypyrone- and hydroxythiopyrone-based MMPi with aryl backbones at the 2-, 5-, and 6-positions of the hydroxypyrone ring have been synthesized. Synthetic routes for developing inhibitors with substituents at two of these positions (so-called double-handed inhibitors) are also explored. The MMP inhibition profiles and structure–activity relationship of synthesized hydroxypyrones and hydroxythiopyrones have been analyzed. The results here show that the ZBG, the position of the backbone on the ZBG, and the nature of the linker between the ZBG and backbone are critical for MMPi activities.
Keywords: Drug design, Inhibitors, Metalloproteinases, Pyrones, Zinc
Matrix metalloproteinases (MMPs) are a class of hydrolytic metalloenzymes involved in the degradation of the extracellular matrix (ECM).1–3 At the catalytic center of MMPs is a conserved tris(histidine)-bound zinc(II) ion. The protein matrix surrounding the zinc center is comprised of a series of subsite pockets designated as S1′, S2′, S3′, S1, S2, and S3 (Fig. 1). The different structures of the MMP subsites, and the amino acids comprising those subsites, lead to substrate selectivity for different MMP isoforms. MMPs are involved in tissue remodeling, wound healing, and growth. The misregulated activities of these enzymes are also implicated in a variety of diseases such as cancer, arthritis, atherosclerosis, and heart disease.1–3 Thus, a number of investigations in both academia and industry have been carried out to develop MMP inhibitors (MMPi) as therapeutics to treat MMP-related diseases.1–4 A typical MMPi has two parts (Fig. 1): a zinc-binding group (ZBG) able to chelate a zinc(II) ion thus blocking the access of the substrate to the catalytic center, and a peptidomimetic backbone that provides non-covalent interactions with the subsite pockets thus tuning inhibitor potency and selectivity. Often in an MMPi a linking group (L) can also be defined that connects the backbone substituent to the ZBG. Most reported MMPi employ a hydroxamic acid as the ZBG with at least part of the backbone directed toward the S1′ pocket.1–4 This strategy has been successful in providing potent MMPi, but hydroxamate inhibitors have limitations including in vivo hydrolysis and dose-limiting side effects. To overcome the drawbacks of hydroxamic acids, new MMPi with alternative ZBGs have been explored.
Figure 1.

Schematic interactions between MMPs and MMP inhibitors.
Hydroxypyrones and hydroxythiopyrones have versatile metal coordination chemistry.5–10 Among the most commonly studied hydroxypyrones are natural products maltol and kojic acid (Fig. 2), which are widely used as food and cosmetics additives, suggesting that they possess good biocompatibility.11 Hydroxypyrones have been explored for the development of new MMPi. Tris(pyrazolyl)borate zinc(II) complexes, used to mimic the MMP catalytic zinc(II) center, show that hydroxypyrones and hydroxythiopyrones can bind to the zinc(II) ion in a bidentate fashion.12 Maltol (hydroxypyrone) and thiomaltol (hydroxythiopyrone) are more effective ZBGs against MMP-3 (stromelysin) when compared to a simple hydroxamate ligand.13,14 Using hydroxypyrone as the ZBG, a series of potent and selective pyrone-based inhibitors of MMP-3 have been developed by attaching an aryl backbone to the 2-position of the pyrone ring.15,16 Simple hydroxythiopyrones have shown much higher inhibition activity (30–60 fold) than corresponding hydroxypyrones.14 This observation promoted us to explore the potential of a hydroxythiopyrone ZBG for development of full length MMP inhibitors. Unlike hydroxamate terminal chelators, in which the inhibitor backbone can only be extended in one direction, the hydroxypyrone ring has several positions (2-, 5-, an 6-) to attach backbones (Fig. 2). Hence, it is possible to develop novel hydroxypyrone-based inhibitors with multiple backbones to interact with MMP pockets (Fig. 1). In this report, we describe the syntheses of hydroxythiopyrone-based MMP inhibitors and their inhibition activities are compared with that of corresponding hydroxypyrones. The synthetic schemes for developing double-handed hydroxypyrone-based inhibitors have been explored. Hydroxypyrones and their hydroxythiopyrone analogues have also been widely reported in other biomedical applications such as iron balance in anemia and iron overload disorder,7,8 aluminium removal in Alzheimer’s disease,9,18,19 treatment of diabetes, 20–23 and contrast agents for medical imaging.24 As such, the synthetic studies and activity analysis provided here should be a valuable reference for development of new hydroxypyrones and hydroxythiopyrones for a wide range of medicinal applications.
Figure 2.

Structures of hydroxamate, hydroxypyrone, and hydroxythiopyrone chelators.
Previously, our laboratory has reported several potent and selective hydroxypyrone MMPi with aryl backbones at the 2-position. 15,16 Thus, we first synthesized corresponding 2-backbone hydroxythiopyrones and evaluated their inhibition activities (Scheme 1).15,16 Intermediate 1 was prepared according to a literature method.8 A coupling reaction of compound 1 with 4-methoxybenzylamine in the presence of EDCI and HOBt provided amide 2 in 71% yield. It was found that coupling reagent 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI) was preferable over dicyclohexylcarbodiimide (DCC), due to the difficulty in removing dicyclohexylurea (DCU) from reaction mixtures employing DCC. The benzyl protecting group on compound 2 was easily removed by stirring in CH3CO2H/HCl (v/v 1:1) at room temperature overnight to give the previously reported hydroxypyrone AM-4 (referred to here as compound 3) in 81% yield. The corresponding hydroxythiopyrone AM-4S (3-S) was synthesized in 61% yield by thionation of compound 3 with P4S10 in the presence of hexamethyldisiloxane (HMDO).25 The methyl group on the aryl backbone of compound 3 was removed by reaction with BBr3 resulting in the new hydroxypyrone 4 in 63% yield.
Scheme 1.

Synthetic scheme for 2-C backbone MMPi.
The inhibitory activities of synthesized hydroxypyrones and hydroxythiopyrones against MMPs were evaluated against human MMP catalytic domain using a fluorescent substrate assay.17 MMP-1 (collagenase), MMP-2 (gelatinase A), and MMP-3 (stromelysin) are examples of MMPs with shallow, intermediate, and deep S1′ pockets, respectively. For comparison of structural impact on the inhibitor activity, all inhibitors were evaluated at a concentration of 50 µM against the aforementioned MMPs (Table 1). IC50 values were determined in cases where an inhibitor showed relatively strong inhibition against a given MMP.
Table 1.
Percent inhibition at inhibitor concentrations of 50 µM and select IC50 values (in parentheses)
| Compound ID | Structure | MMP-1 | MMP-2 | MMP-3 |
|---|---|---|---|---|
| 3-S (AM-4S) | 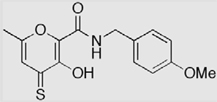 |
70 ± 1 (30) | 94 ± 1 (19) | 97 ± 1 (16.4 ± 2.3) |
| 3 (AM-4) | 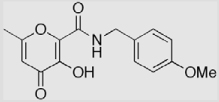 |
29 ± 6 | 26 ± 2 | 93 ± 2 (2.4)a |
| 4 | 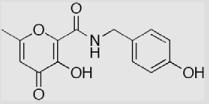 |
66 ± 4 | 25 ± 4 | 94 ± 1 (3.7 ± 1.2) |
| AM-2S | 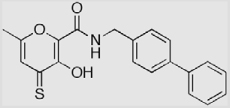 |
– | – | (5.6)b |
| AM-2a | 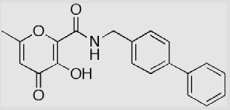 |
(>50) | (9.3) | (0.24) |
| 9a-S | 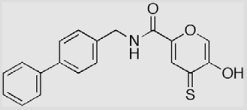 |
62 ± 2 (44) | 98 ± 1 (14.4) | 91 ± 1 (13.3) |
| 9a | 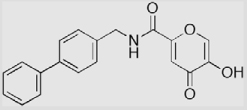 |
12 ± 2 | 14 ± 3 | 44 ± 2 |
| 9b-S | 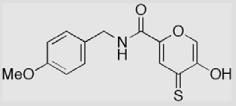 |
31 ± 1 | 85 ± 2 | 91 ± 1 (13.9 ± 0.9) |
| 9b | 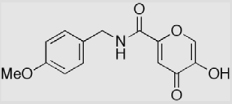 |
16 ± 4 | 11 ± 4 | 22 ± 4 |
| 14a-Sc | 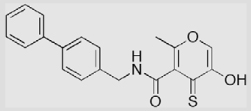 |
52 | 94 (23) | 45 |
| 14a | 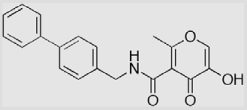 |
15 ± 4 | 21 ± 1 | 28 ± 1 |
| 14b-S | 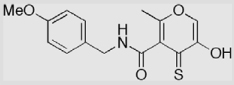 |
49 ± 5 | 69 ± 1 (30) | 36 ± 1 |
| 14b | 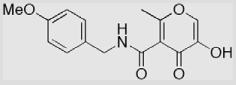 |
15 ± 1 | 33 ± 2 | 18 ± 1 |
| 15 | 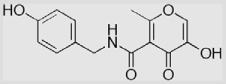 |
17 ± 5 | 24 ± 5 | 32 ± 1 |
| 21 | 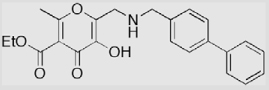 |
13 ± 1 | 18 ± 1 | <1 |
| 22 | 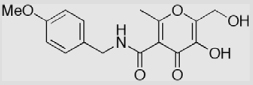 |
18 ± 3 | 24 ± 4 | 32 ± 1 |
| 26 | 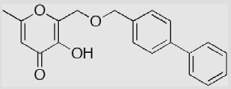 |
21 ± 4 | 15 ± 4 | 26 ± 2 |
| 30 | 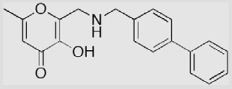 |
6 ± 1 | 63 ± 1 (37.7 ± 1.2) | 4 ± 1 |
Inhibition data comparison of 2-substituted thiopyrone 3-S and pyrone 3 and 4 shows that hydroxythiopyrone 3-S led to higher inhibition than hydroxypyrones 3 and 4 for all MMPs tested at 50 µM concentration. This outcome agrees with the earlier finding that O,S donor ligands are more potent ZBGs than O,O ligands.13,14 But it is interesting to note that thiopyrone 3-S and AM-2S have poorer IC50 values than corresponding pyrone 3 and AM-2 (Table 1) against MMP-3.15,16 X-ray structures of thiomaltol and maltol tris(pyrazolyl)borate zinc(II) complexes reveal that the hydroxythiopyrone and hydroxypyrone ligands coordinate to tris(pyrazolyl) borate zinc(II) in different geometries (Fig. 3),12 suggesting that substituents at the same positions of thiopyrone and pyrone rings may interact with MMP active sites in different fashions. These findings promote us to investigate 5- and 6-substituted hydroxypyrones and hydroxythiopyrones.
Figure 3.

Geometries of maltol- and thiomaltol-tris(pyrazolyl)borate zinc(II) complexes.
The synthesis of inhibitors with backbones in the 6-C position was performed according to Scheme 2, resulting in compounds 9a, 9b, 9a-S, and 9b-S. The ring hydroxyl of kojic acid (5) was selectively protected with a benzyl group by treatment with BnBr in the presence of NaOH to give compound 6. The hydroxymethyl group of 6 was oxidized to carboxylic acid 7 in 65% yield using Jones reagent. The aryl backbones were introduced by coupling reactions using EDCI/HOBt which afforded compounds 8a and 8b in 73–74% yield. The removal of the benzyl protecting groups from compound 8a and 8b was conducted in mixed-solvents HCl/CH3CO2H/CF3CO2H (v/v 10:10:1) at 60 °C, providing compounds 9a and 9b in 76–80% yield. In contrast to the benzyl group deprotection of compound 2 (Scheme 1), the benzyl group in 8a and 8b survived in CH3CO2H/HCl (v/v 1:1) at room temperature, but addition of CF3CO2H with increased temperature promoted its removal. This observation indicates that the substitution pattern on the pyrone can significantly change the electronic properties of the 3-benzyloxy group on the ring. Thionation of 9a and 9b by P4S10 in the presence of HMDO provided 9a-S and 9b-S in 61–68% yield.25
Scheme 2.

Synthetic scheme for 6-C backbone MMPi.
Syntheses of 5-C backbone inhibitors 14a, 14b, 14a-S, and 14b-S followed a scheme recently developed in our laboratory (Scheme 3),26 with some modifications. In the intramolecular condensation of activated bromo ester 10 to produce 3,3-diethyoxypyran-4-one 11, previously we used 2 equiv each of ethyl β-keto ester and NaH in order to make a homogeneous anion solution. Ethyl β-keto ester has an Rf value similar to that of product 11 and is difficult to separate in large-scale preparations. In the improved procedure, 1.0 equiv of ethyl β-keto ester and 2.0 equiv of NaH were employed. The reaction was first stirred at room temperature for 1 h to form intermediate I then refluxed to generate product 11. In the preparation of amide 13a and 13b, EDCI/HOBt was employed as the coupling system instead of DCC/DMAP or DCC/NHS to avoid DCU contamination during product purification. After coupling of the backbone, the remaining synthetic procedures were performed as previously reported.26 In contrast to the high yields (60–70%) in formation of the hydroxythiopyrones 3-S, 9a-S, and 9b-S from the corresponding hydroxypyrones, thionation of 14a and 14b provided 14a-S and 14b-S in relatively low yields (16–18%).25 The low yields in this transformation were tentatively explained by a steric effect at the keto position of 14a and 14b which arose from the neighboring 3-hydroxy and 5-amido groups. Demethylation of 14b by BBr3 affords 15 in 63% yield.
Scheme 3.

Synthetic scheme for 5-C backbone MMPi.
In vitro evaluation of 5- and 6-substituted inhibitors shows that hydroxythiopyrones such as 9a-S, 9b-S, 14a-S, and 14b-S were consistently more potent than their hydroxypyrone counterparts 9a, 9b, 14a, and 14b for all MMPs tested. For hydroxypyrone inhibitors, only inhibitors with backbones at the 2-position (e.g., 3, 4, and AM-2) were selective against MMP-3 over MMP-1 and MMP-2; and all 5- and 6-backbone hydroxypyrones 9a–b, 14a–b, and 15 were overall less potent for all MMPs and generally lacked isoform selectivity. The high potency and selectivity of these 2-C backbone hydroxypyrone inhibitors for MMP-3 is consistent with the favorable orientation of the 2-substituent of the hydroxypyrone toward the MMP-3 S1′ pocket.15 The low inhibition of MMP-1 by the 2-C backbone inhibitors confirms the incompatibility between the bulky aryl backbones and the smaller S1′ pocket in this MMP. 9a showed higher inhibition against MMP-3 over other MMPs, while 9a-S showed comparable inhibition for MMP-2 and MMP-3; compound 14a was not potent and lacked selectivity for all MMPs, but 14a-S showed some preference against MMP-2. These findings further highlight the interplay between the ZBG structure and the position of the backbone on the ZBG can be optimized to improve both potency and selectivity.
Most reported MMPi interact with MMPs at the primed side of the active site (i.e., right-handed inhibitors). Inhibitors with backbones designed to interact with both the primed and unprimed pockets, so-called ‘double-handed’ inhibitors, may have higher selectivity and potency (Fig. 1).3,27,28 Hydroxypyrones and hydroxythiopyrones have several positions (2-, 5-, and 6-) for derivatization (Fig. 2), and thus are ideal for development of MMPi with multiple backbones. In Scheme 4, a structural extension at both sides of the hydroxypyrone chelator was achieved. Intermediate 16 was obtained in 60% yield by refluxing compound 11 in formic acid in the presence of water. A hydroxymethyl group was introduced at the 2-position of compound 16 by reaction with formaldehyde in the presence of NaOH aqueous solution. This hydroxymethylation reaction takes advantage of the ring hydroxyl of 16 resulting in compound 17 in 60% yield. Compared with the natural products kojic acid and maltol (Fig. 2), synthon 17 is highly versatile and provides synthetic handles for further functional extension at both sides of the hydroxypyrone chelator. The structure of 17 was unambiguously assigned by determining a single X-ray crystal structure of the compound complexed with iron (see Supplementary data). The ring hydroxyl was selectively benzylated using benzyl bromide in the presence of K2CO3 providing intermediate 18 in 61% yield. Mesylation of 18 provided sulfonic acid ester 19 in 67% yield. The 4-biphenylmethyl backbone was introduced by refluxing mesylate 19 with 4-biphenylmethylamine in CH3CN resulting in compound 20 in 73% yield. The benzyl group was removed by refluxing compound 20 in HCO2H/CF3CO2H mixed solvents, which afforded compound 21 in 51% yield. As the 5-ester and 6-methyl groups provide attachment points for further functionalization of compound 21, it is possible to synthesize novel hydroxypyrone inhibitors with multiple backbones to interact with MMP active sites. Similarly, compound 14b, with an amido backbone at the 5-position, could also be hydroxymethylated at the 2-position generating compound 22 and thus providing a second route for introducing a new backbone at this position.
Scheme 4.

Synthetic scheme for double-handed (2-C, 5-C) MMPi intermediates.
The simple double-handed intermediates 21 and 22 were not potent against the MMPs screened. Comparing the structure of 21 with potent inhibitor AM-2, which uses an amido linkage to attach the backbone, we reason that the backbone linkage might be another important factor governing the interaction between an inhibitor and MMPs. Thus two analogous compounds of AM-2, compounds 26 and 30 with ether and amino linkages, respectively, were synthesized to investigate the impact of the linker on inhibitor potency.
The synthesis of compound 26 is shown in Scheme 5. Intermediate 23 was prepared from kojic acid (5) following a reported method.8 The hydroxyl group on the pyrone ring was selectively protected with a PMB (p-methoxybenzyl) group using PMBCl to give compound 24. The introduction of a 4-biphenylmethyl backbone was attained by treatment of compound 24 with 4-bromomethyl-biphenyl in the presence of NaH that led to the formation of compound 25. The selective removal of the PMB group was achieved by treatment of compound 25 with CF3CO2H at room temperature in CH2Cl2 providing inhibitor 26 in 84% yield. The use of a benzyl (BnBr) group instead of a PMB group to protect the pyrone hydroxyl group was also examined. It was found that the benzyl protecting group was not cleaved with CF3CO2H at room temperature. The benzyl group could be removed by refluxing in CH3CO2H/CF3CO2H (v/v 1:1) or hydrogenation in the presence of Pd/C, but under such conditions the 4-biphenylmethyl backbone was also partially removed, resulting in a low yield and difficulty in purifying product 26.
Scheme 5.

Synthetic scheme for compound 26.
The preparation of compound 30 is shown in Scheme 6. Treatment of intermediate 24 with methanesulfonyl chloride at 0 °C provided the mesylate ester 27 in 50% yield. The major side product in this transformation was the corresponding chloride 28 (21% yield). The 4-biphenylmethyl backbone was introduced by refluxing of 27 with 4-biphenylmethylamine in CH3CN in the presence of triethylamine, resulting in compound 29. Again, the PMB protecting group was easily removed by treatment with CF3CO2H at room temperature to give compound 30. In the preparation of 30, a benzyl protecting group was tested instead of the PMB protecting group, but again the 4-biphenylmethyl backbone was partially removed, as was found in the preparation of compound 26.
Scheme 6.

Synthetic scheme for compound 30.
The inhibition data for compounds AM-2, 26, and 30 reveals the importance of the backbone linkage. The three inhibitors have the same ZBG and 4-biphenylmethyl backbone, but each has a different linker, namely amido (AM-2), ether (26), and amino (30). Compounds 26 and 30 are much less potent than AM-2 against MMP-3, which may be partially explained by favorable hydrogen bonding between the amido carbonyl group of AM-2 and the amino acid L164 in the MMP.15 In addition, the ether linkage of 26 and the amino linkage of 30 are electron-donating, while the amido linkage of AM-2 is electron-withdrawing. The differing electronic effects of linkers at the 2-position leads to significant differences in the pKa values of the hydroxypyrone chelator,30 with the amide compound being more acidic than either the ether- or amino-linked compound (unpublished results). These findings suggest that the linking group between the backbone and the ZBG can significantly influence the efficacy of an inhibitor through both direct (e.g., H-bonding) and inductive (e.g., ligand acidity) effects that must be carefully considered for successful MMPi design.
In summary, diverse arrays of hydroxypyrone and hydroxythiopyrone derivatives with different substitution patterns were synthesized and their inhibitory activity against three MMPs were examined. Our results suggest that hydroxypyrones and hydroxythiopyrones have different conformations with the same backbones leading to different selectivity and potency against MMPs. Our findings also show that an amide linkage between the chelator and the backbone is also essential for potent inhibition in this system. These synthetic approaches for manipulating hydroxypyrones and the structure–activity relationship information obtained from these MMPi should provide guidance for future design and optimization of potent and selective hydroxypyrone-based MMPi with multiple interactions with MMPs.
Supplementary Material
Acknowledgments
The authors thank Dr. Yongxuan Su for mass spectral analyses of all compounds, Dr. J.R. Stork, Dr. A.G. DiPasquale, and Professor A.L. Rheingold for assistance with the X-ray structure determination. This material is based upon work supported in part by the NIH (R01 HL00049-01) and the American Heart Association (0430009N).
Footnotes
Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.bmcl.2009.02.044.
References and notes
- 1.Puerta DT, Cohen SM. Curr. Top. Med. Chem. 2004;4:1551. doi: 10.2174/1568026043387368. [DOI] [PubMed] [Google Scholar]
- 2.Skiles JW, Gonnella NC, Jeng AY. Curr. Med. Chem. 2004;11:2911. doi: 10.2174/0929867043364018. [DOI] [PubMed] [Google Scholar]
- 3.Whittaker MF, Floyd CD, Brown P, Gearing AJH. Chem. Rev. 1999;99:2735. doi: 10.1021/cr0100345. [DOI] [PubMed] [Google Scholar]
- 4.Matziari M, Dive V, Yiotakis A. Med. Res. Rev. 2007;27:528. doi: 10.1002/med.20066. [DOI] [PubMed] [Google Scholar]
- 5.Ahmet MT, Frampton CS, Silver J. Dalton Trans. 1988:1159. [Google Scholar]
- 6.Ellis BL, Duhme AK, Hider RC, Hossain MB, Rizvi S, van der Helm D. J. Med. Chem. 1996;39:3659. doi: 10.1021/jm960220g. [DOI] [PubMed] [Google Scholar]
- 7.Liu ZD, Hider RC. Med. Res. Rev. 2002;22:26. doi: 10.1002/med.1027. [DOI] [PubMed] [Google Scholar]
- 8.Liu ZD, Piyamongkol S, Liu DY, Khodr HH, Lu SL, Hider RC. Bioorg. Med. Chem. 2001;9:563. doi: 10.1016/s0968-0896(00)00273-x. [DOI] [PubMed] [Google Scholar]
- 9.Santos MA. Coord. Chem. Rev. 2002;228:187. [Google Scholar]
- 10.Thompson KH, Barta CA, Orvig C. Chem. Soc. Rev. 2006;35:545. doi: 10.1039/b416256k. [DOI] [PubMed] [Google Scholar]
- 11.Bentley R. Nat. Prod. Rep. 2006;23:1046. doi: 10.1039/b603758p. [DOI] [PubMed] [Google Scholar]
- 12.Puerta DT, Cohen SM. Inorg. Chem. 2003;42:3423. doi: 10.1021/ic026029g. [DOI] [PubMed] [Google Scholar]
- 13.Puerta DT, Griffin MO, Lewis JA, Romero-Perez D, Garcia R, Villarreal FJ, Cohen SM. J. Biol. Inorg. Chem. 2006;11:131. doi: 10.1007/s00775-005-0053-x. [DOI] [PubMed] [Google Scholar]
- 14.Puerta DT, Lewis JA, Cohen SM. J. Am. Chem. Soc. 2004;126:8388. doi: 10.1021/ja0485513. [DOI] [PubMed] [Google Scholar]
- 15.Puerta DT, Mongan J, Tran BL, McCammon JA, Cohen SM. J. Am. Chem. Soc. 2005;127:14148. doi: 10.1021/ja054558o. [DOI] [PubMed] [Google Scholar]
- 16.Agrawal A, Romero-Perez D, Jacobsen JA, Villarreal FJ, Cohen SM. ChemMedChem. 2008;3:812. doi: 10.1002/cmdc.200700290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Knight CG, Willenbrock F, Murphy G. FEBS Lett. 1992;296:263. doi: 10.1016/0014-5793(92)80300-6. [DOI] [PubMed] [Google Scholar]
- 18.Finnegan MM, Lutz TG, Nelson WO, Smith A, Orvig C. Inorg. Chem. 1987;26:2171. [Google Scholar]
- 19.Finnegan MM, Rettig SJ, Orvig C. J. Am. Chem. Soc. 1986;108:5033. [Google Scholar]
- 20.McNeill JH, Yuen VG, Hoveyda HR, Orvig C. J. Med. Chem. 1992;35:1489. doi: 10.1021/jm00086a020. [DOI] [PubMed] [Google Scholar]
- 21.Saatchi K, Thompson KH, Patrick BO, Pink M, Yuen VG, McNeill JH, Orvig C. Inorg. Chem. 2005;44:2689. doi: 10.1021/ic048186g. [DOI] [PubMed] [Google Scholar]
- 22.Song B, Saatchi K, Rawji GH, Orvig C. Inorg. Chim. Acta. 2002;339:393. [Google Scholar]
- 23.Thompson KH, Liboiron BD, Sun Y, Bellman KD, Setyawati IA, Patrick BO, Karunaratne V, Rawji G, Wheeler J, Sutton K, Bhanot S, Cassidy C, McNeill JH, Yuen VG, Orvig C. J. Biol. Inorg. Chem. 2003;8:66. doi: 10.1007/s00775-002-0388-5. [DOI] [PubMed] [Google Scholar]
- 24.Puerta DT, Botta M, Jocher CJ, Werner EJ, Avedano S, Raymond KN, Cohen SM. J. Am. Chem. Soc. 2006;128:2222. doi: 10.1021/ja057954f. [DOI] [PubMed] [Google Scholar]
- 25.Curphey TJ. J. Org. Chem. 2002;67:6461. doi: 10.1021/jo0256742. [DOI] [PubMed] [Google Scholar]
- 26.Yan YL, Cohen SM. Org. Lett. 2007;9:2517. doi: 10.1021/ol0707665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cuniasse P, Devel L, Makaritis A, Beau F, Georgiadis D, Matziari A, Yiotakis A, Dive V. Biochimie. 2005;87:393. doi: 10.1016/j.biochi.2004.09.025. [DOI] [PubMed] [Google Scholar]
- 28.Matziari M, Beau F, Cuniasse P, Dive V, Yiotakis A. J. Med. Chem. 2004;47:325. doi: 10.1021/jm0308491. [DOI] [PubMed] [Google Scholar]
- 29.Lewis JA, Mongan J, McCammon JA, Cohen SM. ChemMedChem. 2006;1:694. doi: 10.1002/cmdc.200600102. [DOI] [PubMed] [Google Scholar]
- 30.Gordon AEV, Xu J, Raymond KN, Durbin P. Chem. Rev. 2003;103:4207. doi: 10.1021/cr990114x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.