

Rapid-onset dystonia-parkinsonism (RDP) has been linked to mutations in the ATP1A3 gene.1 We present a case with onset of symptoms at age 4 years, with previously undescribed clinical features including episodes of flaccidity and lack of motion for hours, later evolving into episodes of stiffness. Sequencing of ATP1A3 revealed a novel heterozygous nucleotide substitution c.2767G>A, resulting in an aspartic acid (D) to asparagine (N) substitution at amino acid position 923. This mutation is predicted to impair the activity of the neuron-specific α3 subunit of Na,K-ATPase.
Diagnostic criteria for RDP include abrupt onset of dystonia with features of parkinsonism over a few minutes to 30 days, rostrocaudal progression, and prominent bulbar findings.2 Antecedent symptoms such as mild dystonia involving hands and arms may precede acute onset. Primary onset of symptoms is frequently associated with physical or emotional stress. Patients do not respond to l-dopa or dopamine agonists.3 The parkinsonian symptoms are limited to bradykinesia and truncal instability. The most common age at onset of symptoms is young adulthood.2 Onset at age 4 years was reported by Pittock et al.4 The described 38-year-old man had intermittent episodes of dysarthria and hemidystonia associated with stress and anxiety from an early age, lasting from hours to weeks. At baseline, he had only mild dysarthria and slight increase in tone in the left upper limb.
Case report.
We report a child with RDP who had very early onset of symptoms. Birth history was unremarkable. He had mild gross motor delay but excellent cognitive and language development. Hypotonia and left foot intoeing were noted at age 3 years. Ancestry is Caucasian (father) and Chinese (mother) with no family history of dystonia or Parkinson disease.
The patient had a typical initial presentation. On the day of onset, he sustained mild trauma to his forehead. Within 30 minutes he became mute, developed episodes of eye convergence, and was unable to walk. Over several hours he developed prominent hypotonia that later evolved into severe dystonia. He also developed mutism that subsequently evolved into severe dysarthria and drooling. All evaluations, including multiple metabolic tests, skin and muscle biopsies, and brain MRIs, were unrevealing. About 1 month after onset, F-18 fluorodeoxyglucose (FDG)-PET scanning demonstrated moderate hypermetabolism in the striatum involving caudate nuclei and putamen bilaterally. A recent FDG-PET study showed mildly decreased metabolic activity in both thalami and the left putamen. CSF neurotransmitter metabolites, including homovanillic acid, were normal in the acute period and several years later.
The patient’s condition became stable over several months with mild improvement over the subsequent 8 years. Dystonia became less prominent, particularly in the lower extremities. He started to walk with difficulty, but without support (video on the Neurology® Web site at www.neurology.org, seconds 36–58). About a year after onset, however, he developed exceptional symptoms: episodes of flaccidity lasting for hours, later replaced by episodes of stiffness that were shorter and less frequent than the “flaccid” episodes and lasted for 20 to 40 minutes. He did not respond to trials of l-dopa in either the acute period or later, or to dopamine agonists (pergolide, pramipexole) or anticholinergic medications (trihexyphenidyl, benztropine). Over the years, there has been no improvement in his bulbar symptoms, a very striking oromotor dystonia, and apraxia (video, seconds 5–34). He is unable to produce words and has significant swallowing difficulty.
DNA sequencing of the ATP1A3 gene revealed a heterozygous nucleotide substitution (c.2767G>A) in exon 20 (figure), resulting in an amino acid substitution from aspartic acid (D) to asparagine (N) at codon position 923 (p.D923N), which neither parent carried. False paternity was excluded using 5 DNA markers on 5 chromosomes (certainty >99.9%). This mutation was absent from 338 Caucasian control chromosomes screened by DHPLC.1 The finding of a de novo mutation in the α3 subunit gene of Na,K-ATPase in RDP is not uncommon, with 4 out of 10 previously reported cases being de novo.2
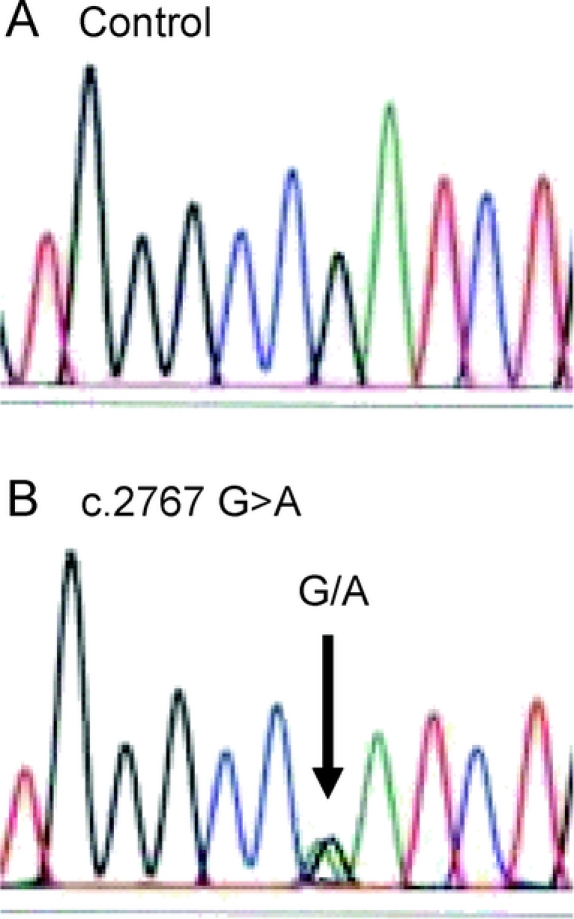
Figure Chromatograms illustrating a novel ATP1A3 gene mutation in a child with rapid-onset dystonia-parkinsonism
Sequence from the patient and a control. Chromatograms show the mutation found in the patient in the ATP1A3 gene. (A) Wild type sequence. The arrow in (B) shows the heterozygous mutant sequence at position c.2767 G>A that results in a D923N amino acid substitution in the protein.
Discussion.
Based on a crystal structure of Na,K-ATPase α1 subunit,5 the mutated residue is buried in the membrane and lies close to the ion-binding residue Q920. Prior mutations of Na,K-ATPase provide insight into the D923N mutation. In α2, mutations of the equivalent residue, D925L or D925N, had residual Na,K-ATPase activity; however, affinities for both Na+ and K+ were significantly reduced.6 In α1 subunit, mutation of the glutamine equivalent to Q920 in the human α3 subunit gene abolished activity, consistent with its role in ion binding.7 Thus D923 is close enough to the ion-binding cavity to affect enzyme activity when mutated. Based on the behavior of the same substitution in the α2 subunit, we expect Na,K-ATPase in patients with this mutation to have impaired activity and ion-binding affinities.
The novel mutation detected in our patient seems to be a causative one. This patient’s presentation has distinctive features and demonstrates that early childhood onset of RDP may occur.
Supplementary Material
Supplemental data at www.neurology.org
Dr. Sweadner was supported by the Bachmann-Strauss Dystonia and Parkinson Foundation. Drs. Sweadner and Ozelius were supported by NIH grant NS058949.
Disclosure: The authors report no disclosures.
Received October 17, 2008. Accepted in final form March 13, 2009.
Address correspondence and reprint requests to Dr. Basil T. Darras, Department of Neurology, Fegan 11, Children’s Hospital Boston, 300 Longwood Avenue, Boston, MA 02115; basil.darras@childrens.harvard.edu
&NA;
- 1.de Carvalho Aguiar P, Sweadner KJ, Penniston JT, et al. Mutations in the Na+/K+ -ATPase alpha3 gene ATP1A3 are associated with rapid-onset dystonia parkinsonism. Neuron 2004;43:169–175. [DOI] [PubMed] [Google Scholar]
- 2.Brashear A, Dobyns WB, de Carvalho Aguiar P, et al. The phenotypic spectrum of rapid-onset dystonia-parkinsonism (RDP) and mutations in the ATP1A3 gene. Brain 2007;130(Pt 3):828–835. [DOI] [PubMed] [Google Scholar]
- 3.Brashear A, Farlow MR, Butler IJ, Kasarskis EJ, Dobyns WB. Variable phenotype of rapid-onset dystonia-parkinsonism. Mov Disord 1996;11:151–156. [DOI] [PubMed] [Google Scholar]
- 4.Pittock SJ, Joyce C, O’Keane V, et al. Rapid-onset dystonia-parkinsonism: a clinical and genetic analysis of a new kindred. Neurology 2000;55:991–995. [DOI] [PubMed] [Google Scholar]
- 5.Morth JP, Pedersen BP, Toustrup-Jensen MS, et al. Crystal structure of the sodium-potassium pump. Nature 2007;450:1043–1049. [DOI] [PubMed] [Google Scholar]
- 6.Yamamoto S, Kuntzweiler TA, Wallick ET, Sperelakis N, Yatani A. Amino acid substitutions in the rat Na+, K(+)-ATPase alpha 2-subunit alter the cation regulation of pump current expressed in HeLa cells. J Physiol 1996;495(Pt 3):733–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arguello JM, Whitis J, Lingrel JB. Alanine scanning mutagenesis of oxygen-containing amino acids in the transmembrane region of the Na,K-ATPase. Arch Biochem Biophys 1999;367:341–347. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.