

Abstract
Amyloid precursor protein (APP) has been strongly implicated in the pathogenesis of Alzheimer's disease (AD). Although impaired synaptic function is believed to be an early and causative event in AD, how APP physiologically regulates synaptic properties remains poorly understood. Here, we report a critical role for APP in the regulation of L-type calcium channels (LTCC) in GABAergic inhibitory neurons in striatum and hippocampus. APP deletion in mice leads to an increase in the levels of Cav1.2, the pore-forming subunit of LTCCs, and subsequent increases in GABAergic calcium currents (ICa2+) that can be reversed by reintroduction of APP. Upregulated levels of Cav1.2 result in reduced GABAergic paired-pulse inhibition and increased GABAergic post-tetanic potentiation in both striatal and hippocampal neurons, indicating that APP modulates synaptic properties of GABAergic neurons by regulating Cav1.2. Furthermore, APP physically interacts with Cav1.2, suggesting a mechanism in which loss of APP leads to an inappropriate accumulation and aberrant activity of Cav1.2. These results provide a direct link between APP and calcium signaling and might help explain how altered APP regulation leads to changes in synaptic function that occur with AD.
Introduction
APP is an integral type I membrane protein highly expressed in the CNS. Genetic and biochemical studies have established pivotal roles of APP in Alzheimer's disease (AD). APP mutation and gene duplication are causal for a subset of early-onset familial Alzheimer's disease (FAD) cases (Hardy, 2006; Rovelet-Lecrux et al., 2006). APP processing generates β-amyloid (Aβ) peptides, which are the principal components of amyloid plaques. Although much focus has been given to Aβ in trying to understand the neuropathology of AD, less is known about how the endogenous function of APP relates to misfunction occurring in AD. Neuronal APP is targeted to presynaptic terminals (Koo et al., 1990; Sisodia et al., 1993) as well as dendrites (Yamazaki et al., 1995; Hoe et al., 2009) where it has been shown to potentiate the formation, maintenance and function of synapses (Seabrook et al., 1999; Wang et al., 2005; Priller et al., 2006; Zheng and Koo, 2006). Given the importance of synaptic deficits in the onset and progression of AD, an understanding of how APP functions at the synapse may reveal how misregulation of APP species leads to the development of disease.
Much of the understanding of the endogenous synaptic function of APP has come from the study of APP-deficient (APP−/−) mice (Zheng et al., 1995). APP−/− mice exhibit impairments in synaptic formation and function as well as in spatial learning and long-term potentiation (LTP) (Dawson et al., 1999; Phinney et al., 1999; Seabrook et al., 1999). Interestingly, a clue to how LTP is impaired in APP−/− mice has been provided by the observation that GABAergic paired-pulse inhibition (PPI) is significantly reduced in hippocampal slices from APP−/− mice (Seabrook et al., 1999), suggesting a role for APP in GABA-mediated inhibition. However, the mechanism by which APP regulates GABAergic activities is still not well understood.
We have demonstrated that N- and L-type calcium channels (NTCC, LTCC), in addition to P/Q type calcium channels (P/QTCC), mediate synaptic transmission in APP−/− neuromuscular junctions (NMJ) (Yang et al., 2007). Thus, one potential mechanism explaining differences in GABAergic transmission in the absence of APP is through the alteration of voltage-gated calcium channel (VGCC) activity. Calcium enters the cytoplasm across the plasma membrane via various calcium channels, including VGCCs, which are classified into distinct subtypes (L, N, P/Q, R and T) (for review, see Tsien et al., 1995), and is released from internal stores (for review, see Tsien and Tsien, 1990). Despite the tight coupling between calcium channels that release intracellular calcium stores and VGCCs in the cell membrane (Chavis et al., 1996; Ouardouz et al., 2003), and the critical role of VGCCs in neuronal activity (Jensen et al., 1999; Catterall, 2000; Jensen and Mody, 2001), the influence of APP on cell membrane calcium channels, particularly VGCCs, remains enigmatic.
Cav1.2 and Cav1.3 are the primary LTCCs expressed in the nervous system, with Cav1.2 accounting for ∼80% of the Cav1 channels in the brain (Calin-Jageman and Lee, 2008). We report here that the level of Cav1.2 protein is significantly higher in predominantly GABAergic regions of the CNS in APP−/− mice, leading to abnormal GABAergic PPI and GABAergic post-tetanic potentiation (PTP). APP and Cav1.2 proteins bind each other, suggesting a mechanism of regulation that involves their physical interaction. This study provides a new mechanism by which endogenous APP can modulate synaptic strength through the regulation of LTCC expression and activity.
Materials and Methods
Neuronal culture and whole-cell patch-clamp recordings.
Cultured striatal and hippocampal neurons were prepared as described (Ventimigilia and Lindsay, 1998) with modification. Briefly, dissection solution consisted of Hank's balanced salt solution supplemented with 25 mm HEPES, pH 7.2. Striatum was isolated from P0 to P1 pups of wild-type (WT) and APP−/− mice. The tissue pieces were trypsinized and digested at room temperature for 15 min, and were then gently triturated in culture medium containing 10% heat-inactivated fetal bovine serum using fire-polished pipettes and centrifuged at 2000 rpm for 10 min. Cell suspensions were plated at 1–2 × 104 cells/ml onto 12 mm Matrigel-coated coverslips. Neurons were cultured in Neurobasal Medium supplemented with B27 and l-glutamine (Invitrogen). One day after plating, 4 μm cytosine β-d-arabinofuranoside (Ara-C) (Sigma) was added to prevent astrocyte proliferation. ICa2+, carried by Ba2+, was recorded from cultured striatal and hippocampal neurons [10–14 days in vitro (DIV)] at 28 ± 3°C using a MultiClamp 700B amplifier (Molecular Devices). The recording chamber was perfused with Tyrode's solution of the following composition (in mm): 100 NaCl, 20 tetraethylammonium-Cl, 5 4-AP, 4 KCl, 1 MgCl2, 10 BaCl2, 0.01 glycine, 25 HEPES, 30 glucose, 0.001 TTX. The glass micropipettes had a resistance of 3–5 MΩ when filled with internal solution containing (in mm): 135 CsCl, 4 MgCl2, 10 HEPES, 10 EGTA, 4 MgATP, 0.3 Na3GTP. A prepulse protocol consisting of 300–700 ms to −30 mV followed by 50 ms to −50 mV, before each test pulse was used to inactivate T-type Ca2+ channels and Na+ channels. Inward Ca2+ currents were elicited by 200 ms test pulses of variable amplitude (−40 to +60 mV at a step of 10 mV) from a holding potential of –60 mV. The interval between test pulses was 10 s. Calcium channel blockers were used at the following concentrations: 10 min, 10 μm nifedipine (L-blocker); 10 min, 1 μm ω-CgTx-GVIA (N-blocker); 10 min, 100 nm ω-Agatoxin-IVA (P/Q blocker). GABAergic PTP was evaluated in striatal and hippocampal cultures in the presence of 50 μm APV and 20 μm CNQX. Single and paired-pulse responses were induced by local electrical stimulation at a rate of 0.05 Hz using a concentric bipolar electrode (WPI, Inc). The amplitude of single IPSCs was evaluated by measuring the amplitude of single pulse-evoked IPSCs or amplitude of the first IPSC in a pair. After single or paired-pulse recordings, tetanization at a frequency of 20 Hz for 1 s was applied. Single or paired pulse response was recorded again 20 s after the tetanic stimulation. Membrane currents were filtered at 1 kHz, digitized at 10 kHz, recorded with Clampex 9 and analyzed with Clampfit 9 software (Molecular Devices) and OriginPro 7.5 (OriginLab).
Western blotting.
Striatum and hippocampus of P21-P30 mice were dissected on ice and homogenized in lysis buffer containing 50 mm Tris, pH 7.5, 150 mm NaCl, 1% Nonidet P40, 1 mm EDTA and protease inhibitors (Complete Mini; Roche). Cellular debris was removed by centrifugation and supernatant was collected for analysis. Tissue lysates were subjected to SDS-PAGE, transferred to nitrocellulose membranes, and probed with specific antibodies against the α1 subunits of P/QTCC, NTCC, or LTCC (all from Millipore). Control antigen of LTCC together with α1C antibody was used in some experiments to check the specificity of α1C antibody. Anti-tubulin antibody hybridization was used as loading control. Bound antibodies were detected using horseradish peroxidase-coupled secondary antibodies (Vector) and enhanced chemiluminescence. The relative optical density of immunoreactive bands was quantified using Scion Image software. For the γ-secretase inhibitor experiment, 1 μm N-[N-(3,5-difluorophenacetyl-l-alanyl)]-S-phenylglycine t-butyl ester (DAPT) (Calbiochem) was added to the culture medium on the day of plating. Medium was changed every week with new inhibitor added. Neurons were collected after 11 DIV for Western blot analysis.
Lentiviral infection.
For construction of lentiviral expression vectors, the APP695 was PCR amplified with primers: 5′-ATGCTAGCGCCACCATGCTGCCCGGTTTGGCA-3′ and 5′-GATTAATTAACTAGTTCTGCATCTGCTCAAAGAA-3′ and cloned into the Nhe I–Pac I sites of FUGW (cmv)-RBN vector (Wang et al., 2009). The production of recombinant lentivirus was done by cotransfection of the expression vector FUGW (cmv)-APP and packaging vectors CMVΔ8.9 and pVSVG into HEK293T cells by Lipofectamine 2000 (Invitrogen). For each T75 flask, 10 μg of FUGW (cmv)-APP, 7.5 μg of CMVΔ8.9 and 5 μg of pVSVG plasmid were used for transfection. Sixty hours after transfection, the supernatant was collected and filtered with a 0.45 μm filter, aliquoted and stored at −80°C. Striatal neurons were infected 5 d after plating using unconcentrated viral supernatant (50–100 μl/ml media). Whole-cell recordings were performed in APP695-expressing neurons identified by GFP fluorescence. Nearby noninfected or GFP vector-infected neurons were recorded as controls for the rescue by APP695. Western blot analysis of rescue of LTCCs by APP695 infection was performed using antibodies against α1C, 6E10 (Covance) and APPc (Wang et al., 2009).
Coimmunoprecipitation.
Brain striatum tissue was dissected from C57BL/6 mice and homogenized in cold lysis buffer (20 mm Tris-HCl [pH 7.5], 150 mm NaCl, 1 mm EDTA, 5 mm DTT, 0.5% NP40, 50 mm NaF, and complete protease inhibitor). To remove cell debris the lysates were centrifuged for 15 min at 14,000 rpm at 4°C. Protein amount was determined using the BCA method. For the immunoprecipitation, 400 μg of protein in a volume of 400 μl was incubated with anti-α1C antibody (Millipore) at 4°C overnight. Then protein G plus protein A-coupled agarose beads (Calbiochem) were added and samples continued to incubate at 4°C for 4 h. To control for nonspecific binding, protein lysates incubated with rabbit IgG and beads were processed in parallel. Subsequently, the beads were washed three times each with 500 μl of cold lysis buffer [(in mm): 20 Tris-HCl, pH 7.5, 150 NaCl, 1 EDTA, 5 DTT, 0.5% NP40, 50 NaF, and complete protease inhibitor). Immunoprecipitates were eluted from the beads by adding 30 μl of SDS-PAGE sample buffer and heating for 10 min at 75°C. The eluates (10 μl) were analyzed by Western blotting.
Immunohistochemistry.
APP−/− and WT mice were perfused with 4% paraformaldehyde (for standard staining) or 4% paraformaldehyde with 0.5% glutaraldehyde (for immunohistochemistry for GABA). Brains were removed, postfixed for 3 h, and sucrose-protected in 30% sucrose overnight. Brains were sectioned at 20 μm into cryoprotectant medium. Free-floating sections were washed three times with PBS and incubated with the appropriate primary antibodies diluted in PBS + 0.4% Triton X-100 + 2% normal goat serum. For single labeling for CaV1.2, a rabbit anti-α1C antibody (Millipore, 1:500) was used. For double labeling, rabbit anti-α1C antibodies along with mouse anti-GABA antibodies (Sigma, 1:1000) were used. After primary antibody incubation, sections were washed in PBS 3 times for 10 min each and incubated in the appropriate species-specific secondary antibodies (Invitrogen, 1:600) diluted in PBS + 0.4% Triton X-100. After 1 h in secondary antibodies, sections were washed three times in PBS and mounted onto gelatin-coated slides, coverslipped with mounting medium containing 4′,6′-diamidino-2-phenylindole dihydrochloride (DAPI) (Vectashield), and imaged on a Leica TCS SP5 confocal microscope. For comparison in intensities between stained specimens, the identical confocal settings (gain, pinhole diameter) were used to image each specimen. Similarly, in postprocessing of images, identical contrast and brightness manipulations were made to the images simultaneously.
Quantitative RT-PCR.
Total RNA was isolated from striatum and hippocampus of WT and APP−/− mice using the RNeasy Lipid Tissue Mini Kit (Invitrogen) and subjected to DNase I digestion to remove contaminating genomic DNA. Reverse transcription was performed using a Superscript III reverse transcriptase (Invitrogen), and the reaction mix was subjected to quantitative real-time PCR using ABI PRISM Sequence Detection System 7000 (Applied Biosystems). Primers were designed with Primer Express Version 2.0 software (Applied Biosystems) using sequence data from NCBI. GAPDH primers were used as an internal control for each specific gene amplification. The relative levels of expression were quantified and analyzed by using ABI PRISM Sequence Detection System 7000 software. The real-time value for each sample was averaged and compared using the comparative CT method. The relative amount of target RNA was calculated relative to the expression of endogenous reference RNA and relative to a calibrator, which was the mean CT of control samples.
Statistical analysis.
Data were analyzed using Student's t test and nonparametric Kolmogorov–Smirnov test with p < 0.05 as statistically significant.
Results
Upregulated Cav1.2 protein levels and increased calcium currents in APP−/− striatum
Previous studies have described an upregulation of LTCCs and NTCCs, in addition to P/QTCC activation, mediating synaptic transmission at the NMJ of APP−/− mice (Yang et al., 2007). Therefore, we tested whether the same type of upregulation occurs at central GABAergic synapses, perhaps explaining previously described changes in the GABAergic paired-pulse response (Seabrook et al., 1999). Taking advantage of the striatum as a source of GABAergic neurons in the CNS, we examined the striatal levels of neuronal LTCC, NTCC and P/QTCC expression in WT and APP−/− mice (Fig. 1A). We found that the immunoreactivity of the α1C subunit of the LTCC (CaV1.2) was significantly higher in APP−/− mice compared with WT littermates while there were no appreciable differences between genotypes in expression of α1A (P/QTCC) and α1B (NTCC) (Fig. 1B), suggesting that Cav1.2 expression is specifically increased in the striatum, a GABAergic brain area, of APP−/− animals.
Figure 1.

Upregulated LTCC expression in APP−/− striatal tissue and enhanced ICa2+ in APP−/− striatal cultures. A, Representative immunoblots of tissue extracts from striatum of 3-week-old APP−/− (−/−) and WT (+/+) littermates. The blots were probed with anti-α1C, α1A and α1B subunit antibodies representing LTCC, P/QTCC and NTCC, respectively. α-Tubulin was used as loading control. B, Quantification of the immunoblots revealed a significant increase in LTCC expression, but not P/QTCC or NTCC in APP−/− mice. Each value represents the mean ± SEM of six samples/genotype. *p < 0.05. C, Plot of peak current density vs voltage in WT (N = 24) and APP−/− (N = 39) striatal neurons and example traces of whole-cell Ca2+ currents elicited by a series of depolarizing steps in WT and APP−/− striatal neurons. D, Plot of peak current density vs test pulse voltage under control conditions (WT, N = 9; APP−/−, N = 21), or in the presence of LTCC blocker (WT, N = 9; APP−/−, N = 21), LTCC blocker + P/QTCC blocker (WT, N = 9; APP−/−, N = 18), or LTCC blocker + P/QTCC blocker + NTCC blocker (WT, N = 8; APP−/−, N = 14). E, Bar graphs illustrate the blockade of peak current exerted by L-, N-, and P/Q-blockers, respectively, in WT and APP−/− striatal cultures. L-blocker-sensitive components increased significantly in APP−/− striatal cultures compared with WT controls. Error bars indicate SEM. **p < 0.01. Calibration: 100 pA/100 ms.
To understand whether changes in α1C levels observed in the APP−/− animals alter membrane calcium influx, whole-cell patch recordings were conducted in striatal cultures of APP−/− and WT mice at 11–16 DIV. Consistent with changes in LTCC expression, we found that the peak calcium current (ICa2+) density was dramatically increased in APP−/− striatal neurons compared with WT controls (Fig. 1C), while the voltage-dependent properties of ICa2+ and input resistance were unaltered (data not shown). Next, we attempted to determine which particular classes of VGCCs are altered in APP−/− striatal neurons based on measurements of current density. Type-specific Ca2+ channel blockers were applied sequentially [10 μm nifedipine (L-blocker), 1 μm ω-conotoxin GVIA (N-blocker) and 100 nm ω-agatoxin-IVA (P/Q blocker)] and the contribution of each VGCC type was calculated (Fig. 1D,E). The pharmacological analysis revealed a significantly increased sensitivity of APP−/− striatal neurons to the dihydropyridine antagonist, nifedipine, which blocks Cav1 LTCC channels (Fig. 1E). In contrast, we found no significant differences in ICa2+ carried by NTCCs and P/QTCCs (Fig. 1E) between the two genotypes when tested in the presence of NTCC and P/QTCC blockers. These results indicate that the increase in the total ICa2+ in APP−/− striatal neurons is mediated by a selective increase in Ca2+ influx through LTCCs.
To confirm that the upregulation of Cav1.2 LTCC expression and ICa2+ in GABAergic neurons of APP−/− mice is due to altered levels of APP, we restored APP expression by lentiviral infection of APP−/− striatal cultures with human APP695, the APP subtype predominantly expressed in neurons. Western blot analysis showed that reintroduction of APP into APP−/− striatal neurons restored levels of the Cav1.2 to levels observed in WT cells (Fig. 2A). Consistent with Western blot analysis, the peak ICa2+ density was also rescued in lentiviral APP695-infected APP−/− neurons to wild-type levels (compare Fig. 2B, APP−/− + APP695 with Fig. 1C, WT). These results establish that the upregulation of Cav1.2 LTCCs in striatal neurons of APP−/− mice results from the loss of APP.
Figure 2.
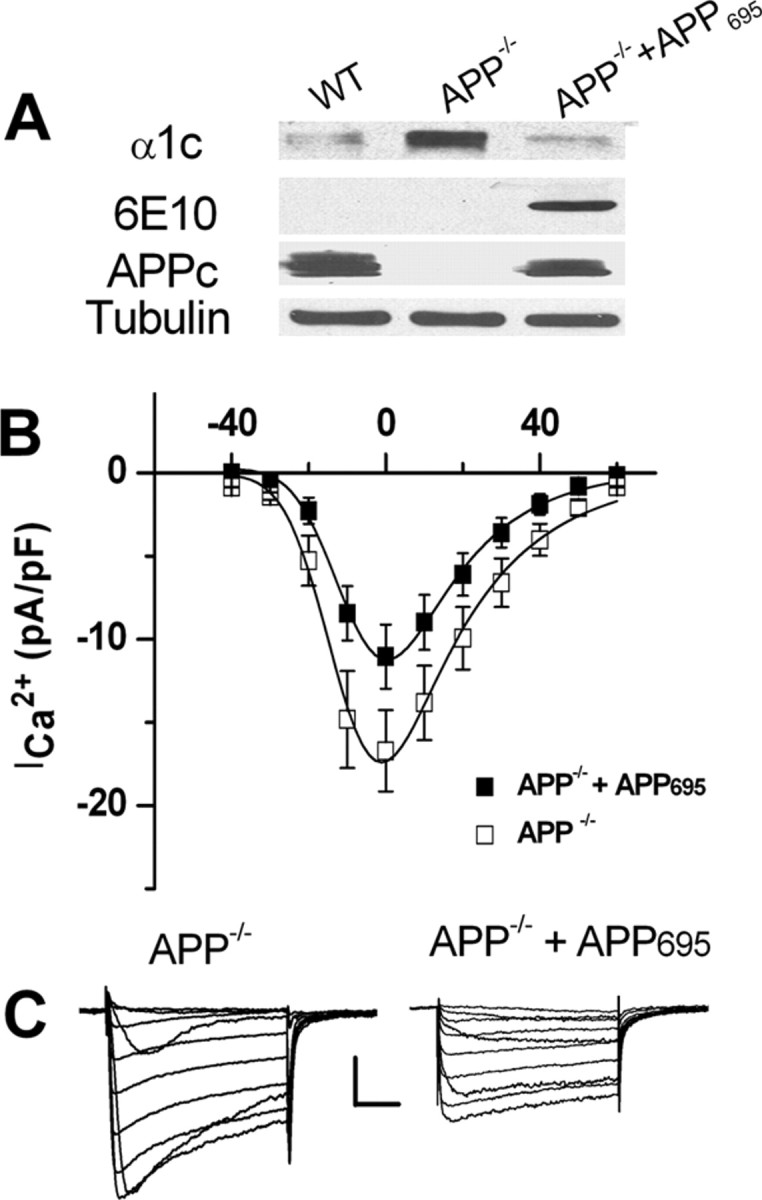
Lentiviral infection with APP695 rescued LTCC expression and ICa2+ in APP−/− striatal cultures. A, Immunoblots of striatal cultures of 12 DIV from WT, littermate APP−/− and APP−/−neurons infected with APP695. Lysates were blotted for LTCC (α1C), human APP (6E10) and mouse/human APP (APPc); α-tubulin was used as loading control. B, ICa2+ was reduced in APP−/− striatal cultures infected with APP695 (N = 15) compared with APP−/− alone (N = 12). Calibrations: 100 pA/50 ms. Data shown as mean ± SEM. C, Representative traces of whole-cell ICa2+ recorded from APP−/− and APP−/−+APP695 neurons, respectively.
Altered GABAergic short-term plasticity and rescue of GABAergic PPI and PTP by an LTCC blocker in APP−/− striatal and hippocampal cultures
Next, we looked at how increases in Cav1.2 in APP−/− mice might change GABAergic synaptic function. Because GABAergic PPI has been previously shown to be altered in hippocampal slices of APP−/− mice (Seabrook et al., 1999), we hypothesized that these changes might be due to alterations in Cav1.2 levels observed in APP−/− mice. To test this hypothesis, we examined two forms of GABAergic short-term plasticity (STP), PPI (a measure of presynaptic alteration) and PTP (a measure of presynaptic LTCC activity) of GABAergic IPSCs, in APP−/− and WT striatal cultures (Fig. 3). Utilizing 11–16 DIV striatal cultures in which GABA-mediated IPSCs can be isolated by local stimulation (Maximov et al., 2007) in the presence of glutamate receptor antagonists APV and CNQX, IPSCs were measured by paired-pulse (50 ms interpulse-intervals) stimulation at a rate of 0.05 Hz. The amplitude of the first IPSC (IPSC1) from each paired response was similar in WT and APP−/− mice (Fig. 3B), suggesting normal GABA-mediated inhibition under baseline conditions. However, the PPI, which provides a sensitive measure of changes in the regulation of GABA release (Schoch et al., 2002), was significantly reduced in APP−/− mice compared with WT controls. Specifically, the IPSC2/IPSC1 (P2/P1) ratio was significantly increased in APP−/− striatal neurons compared with WT controls (Fig. 3D, APP−/− Bef. vs WT Bef.). These changes in PPI suggest that APP deficiency results in altered presynaptic function in cultured striatal GABAergic synapses.
Figure 3.

Altered STP in APP−/− striatal GABAergic synapses and normalization of STP by LTCC blocker nifedipine. A, Representative traces of WT and APP−/− striatal neurons in response to paired-pulse stimulation (before tetanus), tetanus (20 Hz, 1 s) and after tetanus paired-pulse stimulation. B, Similar IPSC1 amplitude in the presence of CNQX and APV in APP−/− striatum (N = 20) compared with WT controls (N = 12). C, The ratio of IPSC1 amplitude after tetanus (IPSC1aft) vs before the tetanus (IPSC1bef) in APP−/− striatal (N = 18) and WT controls (N = 12). D, Paired-pulse ratio (P2/P1) before and after tetanic stimulation in WT [WT Bef., 0.62 ± 0.09 (N = 15); WT Aft., 0.82 ± 0.21 (N = 6)] and APP−/− [APP−/− Bef., 0.95 ± 0.11 (N = 17); APP−/− Aft., 0.6 ± 0.05 (N = 12)]. E, Representative traces showing paired-pulse response in the absence of nifedipine (Ctr.), in the presence of nifedipine before train stimulation (Bef. tetanus), during the train (20 at 20 Hz) and after the train stimulation (Aft. tetanus). ↓: indicating 15 min application of 10 μm nifedipine. F, Paired-pulse ratio (P2/P1) in APP−/− striatal cultures in the absence [Ctr., 0.74 ± 0.04 (N = 6)] and presence of nifedipine (before and after tetanus) (Bef., 0.57 ± 0.09; Aft., 0.49 ± 0.03 (N = 6)]. G, IPSC1 amplitude in the absence (Ctr) and presence of nifedipine in APP−/− hippocampal cultures (before and after tetanus). Data shown as mean ± SEM. Calibrations: 100 pA/50 ms. *p < 0.05.
Presynaptic LTCCs are known to be involved in PTP at GABAergic synapses (Jensen et al., 1999; Jensen and Mody, 2001); therefore, we examined GABAergic PTP, which provides a measure of presynaptic LTCC activity, in striatal cultures from APP−/− mice. PTP is not normally observed at GABAergic synapses using a tetanus of <40 pulses (Jensen et al., 1999; Jensen and Mody, 2001). However, when a tetanization of only 20 pulses at 20 Hz was applied to APP−/− and WT striatal neurons between measurements of paired IPSCs (Fig. 3A), we found that a 20-pulse tetanus induced GABAergic PTP in striatal neurons of APP−/− but not WT mice (Fig. 3A,C). Specifically, the first IPSC amplitude in APP−/− neurons after the tetanus (IPSC1aft.) was 1.8-fold compared with that before the tetanus (IPSC1bef.) (i.e., IPSC1aft./IPSC1bef. = 1.8). In contrast, the amplitude of IPSC1aft. and IPSC1bef. was similar in WT striatal cultures as the IPSCaft/IPSCbef is close to 1 (Fig. 3C). Furthermore, after the tetanus, the P2/P1 ratio was significantly reduced compared with APP−/− striatal cultures before tetanic stimulation (Fig. 3D, APP−/−Aft. vs APP−/− Bef., p < 0.05). Again, in WT striatal neurons in which no PTP was induced using the same protocol, P2/P1 ratios before and after the tetanus did not differ significantly (Fig. 3D, WT Aft. vs WT Bef.). Significant reduction of P2/P1 ratio in APP−/− cells after induction of PTP compared with before-tetanus controls suggests that APP deficiency increases GABAergic PTP, which is most likely due to changes at the presynaptic site.
To test whether increased Cav1.2 levels contribute to the observed reduction in PPI and GABAergic PTP seen in APP−/− striatal cultures, the LTCC blocker, nifedipine, was applied to the bath solution while measurements of presynaptic function were made of PPI and PTP (Fig. 3E–G). We found that the P2/P1 ratio was significantly reduced in nifedipine-treated APP−/− neurons before tetanus compared with untreated APP−/− controls (Fig. 3F, APP−/− Bef. vs APP−/− Ctr.). Additionally, PPI in APP−/− GABAergic synapses in the presence of nifedipine were returned to similar levels observed in WT synapses (Fig. 3F, APP−/− Bef. vs 3D WT Bef.). When we performed measurements of GABAergic PTP, we found that while we could elicit PTP after a tetanus of 20 pulses in the untreated APP−/− cultures (Fig. 3C), after application of 10 μm nifedepine, we no longer observed PTP since the IPSC1 amplitudes were identical in nifedepine-treated APP−/− neurons before and after the tetanus (Fig. 3G, APP−/− Bef. vs APP−/− Aft.), similar to what is observed in WT cultures (Fig. 3D, WT Bef. vs WT Aft.). The P2/P1 ratio remained similar in APP−/− cultures before and after the tetanus when GABAergic PTP was blocked by 10 μm nifedipine (Fig. 3F, APP−/− Bef. vs APP−/− Aft.). These experiments together provide evidence that the alteration of GABAergic synaptic function observed in APP−/− animals is due to increased levels of Cav1.2 in the neurons tested.
We have thus far shown that GABAergic synapses are disrupted in striatal neurons of APP−/− animals due to increased levels of LTCCs. An important question is whether this is also the case in other brain regions, especially the hippocampus, where an alteration of GABAergic PPI has been implicated (Seabrook et al., 1999). Western blot analysis showed that levels of Cav1.2 were higher in striatum than hippocampus (supplemental Fig. S1, available at www.jneurosci.org as supplemental material), suggesting higher LTCCs in GABAergic neurons compared with excitatory neurons. However, we failed to detect significant differences in LTCC expression in the hippocampus between WT and APP−/− mice (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). We reasoned that it could be attributed to the low percentage of GABAergic neurons in this region. We thus performed double immunostaining of hippocampal slices using anti-α1C and anti-GABA antibodies to identify Cav1.2 and GABA double-positive neurons (Fig. 4), and quantified the levels of Cav1.2 fluorescent intensity in GABAergic interneurons of APP−/− mice and WT controls, which revealed significant increases in APP−/− neurons (120.2 ± 5.5) when normalized to WT controls (100.0 ± 6.2) (average ± SEM, N = 18/genotype; *p < 0.05). Our data suggest that upregulation of Cav1.2 as a result of APP deletion may be a general property of GABAergic neurons.
Figure 4.

Representative immunofluorescence staining of hippocampal sections of 2-month-old APP−/− and WT controls with anti-α1C (green) and anti-GABA (red) antibodies, showing increased α1C labeling in GABAergic interneurons in APP−/− hippocampus compared with the controls (Merge). Blue, DAPI showing CA1 pyramidal cells. Representative α1C- and GABA-double positive cells are labeled by arrows. Scale bars, 10 μm.
Having established an elevated LTCC expression in GABAergic interneurons of the APP−/− hippocampus, we assessed GABAergic synaptic function in cultured hippocampal neurons (Fig. 5). We measured local stimulation-induced GABAergic PPI and PTP in 11–16 DIV hippocampal neuronal cultures as we did in striatal cultures. In the presence of 50 μm APV and 20 μm CNQX to block excitatory responses, we found similar changes in APP−/− hippocampal GABAergic STP as seen in APP−/− striatal cultures, including reduced PPI and induction of GABAergic PTP by sub-threshold tetanic stimulation of APP−/− neurons (Fig. 5A–D). Consistent with previous reports (Jensen et al., 1999; Jensen and Mody, 2001), only high-frequency tetanus (1 s at 80 Hz) induced GABAergic PTP in WT controls (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). These changes in APP−/− hippocampal GABAergic STP were reversed by nifedepine (Fig. 5E–G). The fact that altered GABAergic STP, including PPI and PTP, which require intact presynaptic function, is normalized by an LTCC blocker suggests that changes in GABAergic PPI and PTP in APP−/− hippocampal cultures involves altered Cav1.2 LTCC activity in GABAergic neurons.
Figure 5.

Altered STP in APP−/− hippocampal GABAergic synapses and normalized STP by LTCC blocker nifedipine. A, Representative traces of WT and APP−/− hippocampal neurons in response to paired-pulse (Bef. tetanus), tetanic (20 pulses at 20 Hz) and after tetanus (Aft. tetanus) paired-pulse stimulation. B, Identical IPSC1 amplitude in the presence of CNQX and APV in hippocampal cultures of APP−/− and WT mice (WT, N = 13; APP−/− N = 26). C, IPSCaft./IPSCbef. ratio in APP−/− hippocampal neurons (N = 22) compared with WT controls (N = 12). D, P2/P1 ratio before and after tetanic stimulation in WT [WT Bef., 0.48 ± 0.06; WT Aft.0.48 ± 0.12 (N = 7)] and APP−/− neurons [APP−/− Bef., 0.72 ± 0.09; APP−/− Aft., 0.48 ± 0.04 (N = 13)]. E, Representative traces showing paired-pulse response in the absence of nifedipine (Ctr.), in the presence of nifedipine before tetanic stimulation (Bef. tetanus), during the train (20 at 20 Hz) and after the tetanic stimulation (Aft. tetanus) in APP−/− cultures. ↓ indicates 15 min application of 10 μm nifedipine. F, Paired-pulse ratio (P2/P1) in APP−/− hippocampal cultures in the absence [Ctr., 0.69 ± 0.02 (N = 5)] and presence of nifedipine (before and after tetanus) (Bef., 0.56 ± 0.048; Aft., 0.61 ± 0.06 (N = 5)]. G, IPSC1 amplitude in the absence (Ctr.) and presence of nifedipine (Bef. and Aft.) in APP−/− hippocampal cultures. Data shown as mean ± SEM. Calibrations: 100 pA/50 ms. *p < 0.05.
APP modulates Cav1.2 expression at the posttranscriptional level, possibly through physical interaction
APP processing is known to liberate various cleavage products, in particular the APP extracellular fragments, Aβ peptides and a cytoplasmic fragment, the APP intracellular domain (AICD), which has been shown to translocate to the nucleus and regulate gene transcription (Cao and Südhof, 2001). We have shown that APP deficiency causes a significant increase in the levels and function of Cav1.2 LTCCs. Thus, one possibility is that APP regulates Cav1.2 expression via the AICD. We looked for differences in the levels of Cav1.2 mRNA in APP−/− and WT striatum by quantitative RT-PCR. However, we failed to see any significant differences in Cav1.2 mRNA expression in the APP−/− samples compared with WT controls (supplemental Fig. S4A, available at www.jneurosci.org as supplemental material). Therefore, it is unlikely that APP modulates Cav1.2 levels via a transcriptional mechanism.
Another possibility is that Aβ peptides is involved in LTCC expression and it has been shown to induce calcium influx in neuronal cultures via LTCCs (Ekinci et al., 1999). To test this possibility, we blocked Aβ production by adding the γ-secretase inhibitor DAPT (1 μm) to WT striatal cultures. Cav1.2 expression was not upregulated in DAPT-treated WT cultures compared with untreated control cultures (supplemental Fig. S4B,C). This results suggests that APP γ-secretase cleavage products, and associated physiological functions of Aβ, are not responsible for altered Cav1.2 levels observed in APP−/− striatal neurons. This result is corroborated by the indistinguishable Cav1.2 protein levels in a strain of APP knock-in mice in which the human Aβ sequence and the Swedish/London FAD mutation have been introduced into the endogenous mouse APP locus (Hartmann et al., 2004; Köhler et al., 2005) (supplemental Fig. S5, available at www.jneurosci.org as supplemental material).
To further address the mechanism of how APP regulates levels of Cav1.2 LTCCs expression and function, we investigated whether APP can interact with Cav1.2. HEK293 cells were transfected with plasmids encoding Cav1.2 and APP695, then immunoprecipitated using an anti-Cav1.2 (anti-α1C) antibody. APP was detected in the anti-Cav1.2 immunoprecipitate but not that of the IgG control (Fig. 6A). Reverse IP using the anti-APP antibody 4G8 also detected Cav1.2 (Fig. 6B). Next, we tested whether endogenous Cav1.2 and APP interact in the brain. We performed immunoprecipitation of Cav1.2 (α1C) from striatal lysates and measured whether APP coimmunoprecipitated with Cav1.2. Indeed, when an anti-α1C antibody was used for the immunoprecipitation, we could detect the coimmunoprecipitation of APP (Fig. 6C). However, when we immunoprecipitated with antibodies directed against other VGCCs, including α1A of P/QTCC subunit and α1B of NTCC subunit, we failed to detect APP indicating that the interaction between APP and Cav1.2 is specific (Fig. 6D). The physical interaction between APP and Cav1.2 suggests a mechanism in which APP is involved in the regulation of functional Cav1.2 levels, and that loss of APP leads to an inappropriate accumulation of CaV1.2.
Figure 6.

Interaction of APP with Cav1.2. A, α1C interacts with full-length APP. HEK293 cells were transfected with full-length APP and α1C cDNA; cell lysates were immunoprecipitated with an anti-α1C antibody (IP: α1C) and probed with APP antibody 22C11. Rabbit IgG (IP: IgG) was used as a negative control. B, Conversely, cell lysates were immunoprecipitated with an anti-APP antibody (IP: 4G8) and probed with α1C antibody. Mouse IgG (IP: IgG) was used as a negative control. C, Brain striatal tissue lysates were immunoprecipitated with anti-α1C and anti-APP (APPc) antibodies and probed with APP 22C11 antibody. Rabbit IgG was used as a negative control (IP: IgG). D, Striatal lysates were immunoprecipitated with antibodies against pore-forming subunits of three different VGCCs, α1A, α1B, and α1C, and probed with the anti-APP antibody, 22C11. Only α1C antibody was able to immunoprecipitate APP.
Discussion
We have discovered a new function for APP in the regulation of appropriate Cav1.2 LTCC levels. Loss of APP leads to an increase in Cav1.2 levels in striatal GABAergic neurons that can be rescued by reintroduction of APP. Increased Cav1.2 levels in GABAergic neurons lead to altered GABAergic STP, including PPI and PTP, which can be reversed by the LTCC blocker nifedipine. These results suggest that one function of APP in neurons involves the regulation of appropriate Cav1.2 levels and implicates it as a direct calcium-related molecular target of APP. The precise mechanism for the regulation of Cav1.2 by APP is still unknown; however, we have detected a direct interaction between APP and CaV1.2, suggesting that a posttranslational mechanism may explain how APP regulates plasma membrane expression of Cav1.2. This regulation of calcium channel levels by APP represents a new understanding of how APP might affect synaptic efficacy. This link between APP and synaptic strength may account for certain aspects of synaptic loss and cognitive decline that occur when APP becomes misregulated.
Presynaptic GABAergic Cav1.2 LTCCs and synaptic plasticity
LTCCs have been observed mainly on the cell body and dendrites (Westenbroek et al., 1990; Hell et al., 1993); consequently, they are generally thought to function exclusively in the soma and proximal dendrites of neurons. This subcellular functional localization is well supported by findings that Cav1.2 LTCCs are positioned near but not at the presynaptic active zone of axonal terminals and that synaptic transmission is blocked by antagonists of P/QTCCs and NTCCs, but not LTCCs (Castillo et al., 1994; Wheeler et al., 1994). However, several lines of evidence strongly suggest the existence of LTCCs on presynaptic terminals: the ultrastructural discovery of a presynaptic distribution of LTCCs in all hippocampal subfields (Tippens et al., 2008), electrophysiological and biochemical identification of the presynaptic localization of LTCCs (Bonci et al., 1998; Tokunaga et al., 2004; Fourcaudot et al., 2009), and the functional determination that neuronal LTCCs open quickly and are inhibited slowly by dihydropyridine antagonists (Helton et al., 2005). As such, the contributions of LTCCs to presynaptic function may have been underestimated. We found that increased Cav1.2 in APP−/− animals led to an alteration of GABAergic PPI and PTP that could be rescued by a dihydropyridine antagonist, nifedipine, suggesting that Cav1.2 channels positioned at GABAergic axon terminals may indirectly promote neurotransmitter release. Our data are consistent with LTCCs functioning near the synapse, where they are known to serve a presynaptic function during repetitive stimulation (Bonci et al., 1998; Brosenitsch and Katz, 2001; Jensen and Mody, 2001).
A role for APP in the modulation of Cav1.2 LTCC activity and synaptic plasticity
Earlier studies found impaired hippocampal LTP in APP−/− animals and suggested that it was a result of altered GABAergic interneuron activity (Seabrook et al., 1999). However, these studies did not address a mechanism by which APP deletion causes these functional changes at GABAergic synapses. Here, we show that GABAergic STP, including PPI and PTP of IPSCs, is disrupted in APP-deficient animals, and that we can normalize the responses with a dihydropyridine antagonist, nifedipine. These results suggest that increased Cav1.2 levels account for GABAergic synaptic alterations observed in APP-deficient mice. The data suggest that one endogenous function of APP is to regulate an appropriate neuronal complement of Cav1.2 in GABAergic neurons and thereby modulate STP. When APP function is compromised, Cav1.2 levels become misregulated and lead to changes in STP, which might ultimately lead to cognitive decline.
APP and its derivatives have been shown to play an important role in excitatory, glutamatergic, synaptic function (Kamenetz et al., 2003; Priller et al., 2006). A recent study reported that acute expression of human APP in cortical neurons increases LTCC-mediated ICa2+ through enhanced LTCC activation in glutamatergic neurons (Santos et al., 2009). While we were unable to detect significant changes in Cav1.2 levels in hippocampal lysates of APP-deficient mice, whether the LTCC-mediated synaptic property is altered in excitatory neurons resulting from APP loss of function was not explored. Overall, evidence from glutamatergic (Santos et al., 2009) and GABAergic systems (this study) support the notion that APP may affect LTCC function in multiple neurons and by multiple mechanisms.
Probing the mechanism of APP regulation of Cav1.2
We have tested a number of hypotheses to address the mechanism by which APP regulates levels of Cav1.2 to alter synaptic properties. The first hypothesis is that APP regulates expression of Cav1.2 in the nucleus. APP cleavage is known to release the AICD, which can transit to the nucleus and function as a transcription factor (Cao and Südhof, 2001). However, we observed no changes in Cav1.2 mRNA expression in APP−/− mice compared with WT controls, suggesting that APP most likely does not regulate Cav1.2 at the transcriptional level. The second hypothesis we tested was whether Aβ, generated by γ-secretase cleavage of APP, may serve to regulate Cav1.2 levels. Again, we failed to see any changes in Cav1.2 levels after prolonged exposure to the γ-secretase blocker DAPT, indicating that γ-secretase cleavage of APP, in particular Aβ generation, is not required for its regulation of CaV1.2. Finally, we tested whether APP might regulate Cav1.2 through a direct protein-protein interaction. Indeed, we did detect an interaction between these two proteins, but not between APP and other VGCC subunits. Cav1.2 has been shown to be regulated in neurons by endocytosis and intracellular trafficking in response to electrical activity (Green et al., 2007). Additionally, APP has been shown to actively traffic to and away from the membrane (Hill et al., 2003; Pietrzik et al., 2004). Given that we can detect an interaction between APP and Cav1.2, it is possible that APP is involved in the intracellular trafficking of Cav1.2 by pulling Cav1.2 into an intracellular compartment away from the plasma membrane, where it might be transported to endosomal and lysosomal compartments and subsequently degraded. When APP is genetically removed, Cav1.2 might accumulate at the plasma membrane because this trafficking mechanism has been eliminated, leading to higher levels of Cav1.2 at the plasma membrane, greater calcium currents, and defects in GABAergic synaptic plasticity, as we have observed in APP-deficient animals.
APP, Cav1.2 LTCC and calcium homeostasis in AD
We reveal here a previously unrecognized role of APP in the maintenance of LTCC levels and activity in selected neurons, and we provide the first causal link between APP loss-of-function and failed calcium homeostasis, which is a well recognized pathological feature of AD (Bezprozvanny and Mattson, 2008; Dreses-Werringloer et al., 2008; Green and LaFerla, 2008; Kuchibhotla et al., 2008;). Although the APP-LTCC regulation we studied here is specific to GABAergic inhibitory neurons in APP-deficient mice and the involvement of APP loss-of-function or misfunction in AD has not been established, in light of the central role of APP in AD pathogenesis, we believe our findings may provide novel pathogenic insights. In particular, evidence have shown that LTCCs are elevated during aging and in individuals with AD (Thibault and Landfield, 1996; Thibault et al., 2001), and this is correlated with reduced soluble APP extracellular protein levels in the same population (Palmert et al., 1990). Therefore, it is conceivable that impaired APP processing and other regulatory pathways may directly contribute to aberrant LTCC expression, leading to altered calcium homeostasis, synaptic dysfunction and AD pathogenesis.
Footnotes
This work was supported by grants from the National Institutes of Health (AG032051 and AG033467), the Alzheimer's Association (IIRG-06-25779), and the American Health and Assistance Foundation (A2008-052). We thank Dr. F. A. Pereira for a critical reading of the manuscript and Dr. J. A. Dani for valuable discussions. We thank D. Shim, E. Peethumnongsin, M. Wiese, and other members of the Zheng laboratory for constructive suggestions. We are grateful to N. Aithmitti and X. Chen for technical assistance. We thank Dr. D. Lipscombe for providing the Cav1.2 cDNA.
References
- Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer's disease. Trends Neurosci. 2008;31:454–463. doi: 10.1016/j.tins.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonci A, Grillner P, Mercuri NB, Bernardi G. L-type calcium channels mediate a slow excitatory synaptic transmission in rat midbrain dopaminergic neurons. J Neurosci. 1998;18:6693–6703. doi: 10.1523/JNEUROSCI.18-17-06693.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosenitsch TA, Katz DM. Physiological patterns of electrical stimulation can induce neuronal gene expression by activating N-type calcium channels. J Neurosci. 2001;21:2571–2579. doi: 10.1523/JNEUROSCI.21-08-02571.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calin-Jageman I, Lee A. Ca(v)1 L-type Ca2+ channel signaling complexes in neurons. J Neurochem. 2008;105:573–583. doi: 10.1111/j.1471-4159.2008.05286.x. [DOI] [PubMed] [Google Scholar]
- Cao X, Südhof TC. A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science. 2001;293:115–120. doi: 10.1126/science.1058783. [DOI] [PubMed] [Google Scholar]
- Castillo PE, Weisskopf MG, Nicoll RA. The role of Ca2+ channels in hippocampal mossy fiber synaptic transmission and long-term potentiation. Neuron. 1994;12:261–269. doi: 10.1016/0896-6273(94)90269-0. [DOI] [PubMed] [Google Scholar]
- Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol. 2000;16:521–555. doi: 10.1146/annurev.cellbio.16.1.521. [DOI] [PubMed] [Google Scholar]
- Chavis P, Fagni L, Lansman JB, Bockaert J. Functional coupling between ryanodine receptors and L-type calcium channels in neurons. Nature. 1996;382:719–722. doi: 10.1038/382719a0. [DOI] [PubMed] [Google Scholar]
- Dawson GR, Seabrook GR, Zheng H, Smith DW, Graham S, O'Dowd G, Bowery BJ, Boyce S, Trumbauer ME, Chen HY, Van der Ploeg LH, Sirinathsinghji DJ. Age-related cognitive deficits, impaired long-term potentiation and reduction in synaptic marker density in mice lacking the beta-amyloid precursor protein. Neuroscience. 1999;90:1–13. doi: 10.1016/s0306-4522(98)00410-2. [DOI] [PubMed] [Google Scholar]
- Dreses-Werringloer U, Lambert JC, Vingtdeux V, Zhao H, Vais H, Siebert A, Jain A, Koppel J, Rovelet-Lecrux A, Hannequin D, Pasquier F, Galimberti D, Scarpini E, Mann D, Lendon C, Campion D, Amouyel P, Davies P, Foskett JK, Campagne F, et al. A polymorphism in CALHM1 influences Ca2+ homeostasis, Abeta levels, and Alzheimer's disease risk. Cell. 2008;133:1149–1161. doi: 10.1016/j.cell.2008.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekinci FJ, Malik KU, Shea TB. Activation of the L voltage-sensitive calcium channel by mitogen-activated protein (MAP) kinase following exposure of neuronal cells to beta-amyloid. MAP kinase mediates beta-amyloid-induced neurodegeneration. J Biol Chem. 1999;274:30322–30327. doi: 10.1074/jbc.274.42.30322. [DOI] [PubMed] [Google Scholar]
- Fourcaudot E, Gambino F, Casassus G, Poulain B, Humeau Y, Lüthi A. L-type voltage-dependent Ca(2+) channels mediate expression of presynaptic LTP in amygdala. Nat Neurosci. 2009;12:1093–1095. doi: 10.1038/nn.2378. [DOI] [PubMed] [Google Scholar]
- Green EM, Barrett CF, Bultynck G, Shamah SM, Dolmetsch RE. The tumor suppressor eIF3e mediates calcium-dependent internalization of the L-type calcium channel CaV1.2. Neuron. 2007;55:615–632. doi: 10.1016/j.neuron.2007.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green KN, LaFerla FM. Linking calcium to Abeta and Alzheimer's disease. Neuron. 2008;59:190–194. doi: 10.1016/j.neuron.2008.07.013. [DOI] [PubMed] [Google Scholar]
- Hardy J. A hundred years of Alzheimer's disease research. Neuron. 2006;52:3–13. doi: 10.1016/j.neuron.2006.09.016. [DOI] [PubMed] [Google Scholar]
- Hartmann J, Erb C, Ebert U, Baumann KH, Popp A, König G, Klein J. Central cholinergic functions in human amyloid precursor protein knock-in/presenilin-1 transgenic mice. Neuroscience. 2004;125:1009–1017. doi: 10.1016/j.neuroscience.2004.02.038. [DOI] [PubMed] [Google Scholar]
- Hell JW, Westenbroek RE, Warner C, Ahlijanian MK, Prystay W, Gilbert MM, Snutch TP, Catterall WA. Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel alpha 1 subunits. J Cell Biol. 1993;123:949–962. doi: 10.1083/jcb.123.4.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helton TD, Xu W, Lipscombe D. Neuronal L-type calcium channels open quickly and are inhibited slowly. J Neurosci. 2005;25:10247–10251. doi: 10.1523/JNEUROSCI.1089-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill K, Li Y, Bennett M, McKay M, Zhu X, Shern J, Torre E, Lah JJ, Levey AI, Kahn RA. Munc18 interacting proteins: ADP-ribosylation factor-dependent coat proteins that regulate the traffic of beta-Alzheimer's precursor protein. J Biol Chem. 2003;278:36032–36040. doi: 10.1074/jbc.M301632200. [DOI] [PubMed] [Google Scholar]
- Hoe HS, Fu Z, Makarova A, Lee JY, Lu C, Feng L, Pajoohesh-Ganji A, Matsuoka Y, Hyman BT, Ehlers MD, Vicini S, Pak DT, Rebeck GW. The effects of amyloid precursor protein on postsynaptic composition and activity. J Biol Chem. 2009;284:8495–8506. doi: 10.1074/jbc.M900141200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen K, Mody I. L-type Ca2+ channel-mediated short-term plasticity of GABAergic synapses. Nat Neurosci. 2001;4:975–976. doi: 10.1038/nn722. [DOI] [PubMed] [Google Scholar]
- Jensen K, Jensen MS, Lambert JD. Post-tetanic potentiation of GABAergic IPSCs in cultured rat hippocampal neurones. J Physiol. 1999;519:71–84. doi: 10.1111/j.1469-7793.1999.0071o.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- Köhler C, Ebert U, Baumann K, Schröder H. Alzheimer's disease-like neuropathology of gene-targeted APP-SLxPS1mut mice expressing the amyloid precursor protein at endogenous levels. Neurobiol Dis. 2005;20:528–540. doi: 10.1016/j.nbd.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Koo EH, Sisodia SS, Archer DR, Martin LJ, Weidemann A, Beyreuther K, Fischer P, Masters CL, Price DL. Precursor of amyloid protein in Alzheimer disease undergoes fast anterograde axonal transport. Proc Natl Acad Sci U S A. 1990;87:1561–1565. doi: 10.1073/pnas.87.4.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchibhotla KV, Goldman ST, Lattarulo CR, Wu HY, Hyman BT, Bacskai BJ. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron. 2008;59:214–225. doi: 10.1016/j.neuron.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximov A, Pang ZP, Tervo DG, Südhof TC. Monitoring synaptic transmission in primary neuronal cultures using local extracellular stimulation. J Neurosci Methods. 2007;161:75–87. doi: 10.1016/j.jneumeth.2006.10.009. [DOI] [PubMed] [Google Scholar]
- Ouardouz M, Nikolaeva MA, Coderre E, Zamponi GW, McRory JE, Trapp BD, Yin X, Wang W, Woulfe J, Stys PK. Depolarization-induced Ca2+ release in ischemic spinal cord white matter involves L-type Ca2+ channel activation of ryanodine receptors. Neuron. 2003;40:53–63. doi: 10.1016/j.neuron.2003.08.016. [DOI] [PubMed] [Google Scholar]
- Palmert MR, Usiak M, Mayeux R, Raskind M, Tourtellotte WW, Younkin SG. Soluble derivatives of the beta amyloid protein precursor in cerebrospinal fluid: alterations in normal aging and in Alzheimer's disease. Neurology. 1990;40:1028–1034. doi: 10.1212/wnl.40.7.1028. [DOI] [PubMed] [Google Scholar]
- Phinney AL, Calhoun ME, Wolfer DP, Lipp HP, Zheng H, Jucker M. No hippocampal neuron or synaptic bouton loss in learning-impaired aged beta-amyloid precursor protein-null mice. Neuroscience. 1999;90:1207–1216. doi: 10.1016/s0306-4522(98)00645-9. [DOI] [PubMed] [Google Scholar]
- Pietrzik CU, Yoon IS, Jaeger S, Busse T, Weggen S, Koo EH. FE65 constitutes the functional link between the low-density lipoprotein receptor-related protein and the amyloid precursor protein. J Neurosci. 2004;24:4259–4265. doi: 10.1523/JNEUROSCI.5451-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priller C, Bauer T, Mitteregger G, Krebs B, Kretzschmar HA, Herms J. Synapse formation and function is modulated by the amyloid precursor protein. J Neurosci. 2006;26:7212–7221. doi: 10.1523/JNEUROSCI.1450-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovelet-Lecrux A, Hannequin D, Raux G, Le Meur N, Laquerrière A, Vital A, Dumanchin C, Feuillette S, Brice A, Vercelletto M, Dubas F, Frebourg T, Campion D. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet. 2006;38:24–26. doi: 10.1038/ng1718. [DOI] [PubMed] [Google Scholar]
- Santos SF, Pierrot N, Morel N, Gailly P, Sindic C, Octave JN. Expression of human amyloid precursor protein in rat cortical neurons inhibits calcium oscillations. J Neurosci. 2009;29:4708–4718. doi: 10.1523/JNEUROSCI.4917-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoch S, Castillo PE, Jo T, Mukherjee K, Geppert M, Wang Y, Schmitz F, Malenka RC, Südhof TC. RIM1alpha forms a protein scaffold for regulating neurotransmitter release at the active zone. Nature. 2002;415:321–326. doi: 10.1038/415321a. [DOI] [PubMed] [Google Scholar]
- Seabrook GR, Smith DW, Bowery BJ, Easter A, Reynolds T, Fitzjohn SM, Morton RA, Zheng H, Dawson GR, Sirinathsinghji DJ, Davies CH, Collingridge GL, Hill RG. Mechanisms contributing to the deficits in hippocampal synaptic plasticity in mice lacking amyloid precursor protein. Neuropharmacology. 1999;38:349–359. doi: 10.1016/s0028-3908(98)00204-4. [DOI] [PubMed] [Google Scholar]
- Sisodia SS, Koo EH, Hoffman PN, Perry G, Price DL. Identification and transport of full-length amyloid precursor proteins in rat peripheral nervous system. J Neurosci. 1993;13:3136–3142. doi: 10.1523/JNEUROSCI.13-07-03136.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibault O, Landfield PW. Increase in single L-type calcium channels in hippocampal neurons during aging. Science. 1996;272:1017–1020. doi: 10.1126/science.272.5264.1017. [DOI] [PubMed] [Google Scholar]
- Thibault O, Hadley R, Landfield PW. Elevated postsynaptic [Ca2+]i and L-type calcium channel activity in aged hippocampal neurons: relationship to impaired synaptic plasticity. J Neurosci. 2001;21:9744–9756. doi: 10.1523/JNEUROSCI.21-24-09744.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tippens AL, Pare JF, Langwieser N, Moosmang S, Milner TA, Smith Y, Lee A. Ultrastructural evidence for presynaptic and postsynaptic localization of Cav1.2 L-type Ca2+ channels in the rat hippocampus. J Comp Neurol. 2008;506:569–583. doi: 10.1002/cne.21567. [DOI] [PubMed] [Google Scholar]
- Tokunaga T, Miyazaki K, Koseki M, Mobarakeh JI, Ishizuka T, Yawo H. Pharmacological dissection of calcium channel subtype-related components of strontium inflow in large mossy fiber boutons of mouse hippocampus. Hippocampus. 2004;14:570–585. doi: 10.1002/hipo.10202. [DOI] [PubMed] [Google Scholar]
- Tsien RW, Tsien RY. Calcium channels, stores, and oscillations. Annu Rev Cell Biol. 1990;6:715–760. doi: 10.1146/annurev.cb.06.110190.003435. [DOI] [PubMed] [Google Scholar]
- Tsien RW, Lipscombe D, Madison D, Bley K, Fox A. Reflections on Ca(2+)-channel diversity, 1988–1994. Trends Neurosci. 1995;18:52–54. [PubMed] [Google Scholar]
- Ventimigilia R, Lindsay RM. Rat hippocampal neurons in low-density, serum-free culture. In: Banker G, Goslin K, editors. Culturing nerve cells. Ed 2. Cambridge, MA: MIT; 1998. pp. 371–394. [Google Scholar]
- Wang P, Yang G, Mosier DR, Chang P, Zaidi T, Gong YD, Zhao NM, Dominguez B, Lee KF, Gan WB, Zheng H. Defective neuromuscular synapses in mice lacking amyloid precursor protein (APP) and APP-like protein 2. J Neurosci. 2005;25:1219–1225. doi: 10.1523/JNEUROSCI.4660-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Wang B, Yang L, Guo Q, Aithmitti N, Songyang Z, Zheng H. Presynaptic and postsynaptic interaction of the amyloid precursor protein promotes peripheral and central synaptogenesis. J Neurosci. 2009;29:10788–10801. doi: 10.1523/JNEUROSCI.2132-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westenbroek RE, Ahlijanian MK, Catterall WA. Clustering of L-type Ca2+ channels at the base of major dendrites in hippocampal pyramidal neurons. Nature. 1990;347:281–284. doi: 10.1038/347281a0. [DOI] [PubMed] [Google Scholar]
- Wheeler DB, Randall A, Tsien RW. Roles of N-type and Q-type Ca2+ channels in supporting hippocampal synaptic transmission. Science. 1994;264:107–111. doi: 10.1126/science.7832825. [DOI] [PubMed] [Google Scholar]
- Yamazaki T, Selkoe DJ, Koo EH. Trafficking of cell surface beta-amyloid precursor protein: retrograde and transcytotic transport in cultured neurons. J Cell Biol. 1995;129:431–442. doi: 10.1083/jcb.129.2.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Wang B, Long C, Wu G, Zheng H. Increased asynchronous release and aberrant calcium channel activation in amyloid precursor protein deficient neuromuscular synapses. Neuroscience. 2007;149:768–778. doi: 10.1016/j.neuroscience.2007.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Koo EH. The amyloid precursor protein: beyond amyloid. Mol Neurodegener. 2006;1:5. doi: 10.1186/1750-1326-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Jiang M, Trumbauer ME, Sirinathsinghji DJ, Hopkins R, Smith DW, Heavens RP, Dawson GR, Boyce S, Conner MW, Stevens KA, Slunt HH, Sisoda SS, Chen HY, Van der Ploeg LH. beta-Amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell. 1995;81:525–531. doi: 10.1016/0092-8674(95)90073-x. [DOI] [PubMed] [Google Scholar]