

Abstract
Rationale
Phosphoinositide 3-kinase (PI3K)γ is expressed in hematopoietic cells, endothelial cells (ECs), and cardiomyocytes and regulates different cellular functions relevant to inflammation, tissue remodeling and cicatrization. Recently, PI3Kγ inhibitors have been indicated for the treatment of chronic inflammatory/autoimmune diseases and atherosclerosis.
Objective
We aimed to determine PI3Kγ contribution to the angiogenic capacity of ECs and the effect of PI3Kγ inhibition on healing of myocardial infarction (MI).
Methods and Results
Human umbilical ECs were treated with a selective PI3Kγ inhibitor, AS605240, or a pan-phosphoinositide 3-kinases inhibitor, LY294002. Both inhibitory treatments and small interfering RNA–mediated PI3Kγ knockdown strongly impaired ECs angiogenic capacity, because of suppression of the PI3K/Akt and mitogen-activated protein kinase pathways. Constitutive activation of Akt rescued the angiogenic defect. Reparative angiogenesis was studied in vivo in a model of MI. AS605240 did not affect MI-induced PI3Kγ upregulation, whereas it suppressed Akt activation and downstream signaling. AS605240 strongly reduced inflammation, enhanced cardiomyocyte apoptosis, and impaired survival and proliferation of ECs in peri-infarct zone, which resulted in defective reparative neovascularization. As a consequence, AS605240-treated MI hearts showed increased infarct size and impaired recovery of left ventricular function. Similarly, PI3Kγ-deficient mice showed impaired reparative neovascularization, enhanced cardiomyocyte apoptosis and marked deterioration of cardiac function following MI. Mice expressing catalytically inactive PI3Kγ also failed to mount a proper neovascularization, although cardiac dysfunction was similar to wild-type controls.
Conclusions
PI3Kγ expression and catalytic activity are involved at different levels in reparative neovascularization and healing of MI.
Keywords: PI3Kγ, Akt, angiogenesis, myocardial infarction
Phosphoinositide 3-kinases (PI3Ks) are a family of enzymes characterized by protein and lipid kinase activity. Class IA PI3Ks (PI3Kα, -β, and -δ) are mainly activated downstream tyrosine kinase receptors, whereas the single member of class I B PI3Ks, PI3Kγ, is activated on stimulation of G protein–coupled receptors (GPCRs) and is regulated by free Gβγ subunits of heterotrimeric G proteins. PI3Ks catalytic activity leads to the accumulation of phosphatidylinositol-3,4,5-tris-phosphate in the plasma membrane, which acts as docking site for pleckstrin homology domain containing effectors, including protein kinase B (PKB/Akt).1 The signaling pathway downstream of activated Akt controls cell-cycle progression, cell survival, growth, metabolism and movement.2
The contribution of class IA PI3K isoforms to angiogenic processes has been thoroughly dissected.3 In contrast, the involvement of PI3Kγ in reparative angiogenesis is not firmly established. Seminal studies showed that PI3Kγ is expressed not only in hematopoietic cells but also in endothelial cells (ECs) and cardiomyocytes,4 and acts as a modulator of leukocyte-EC interaction at inflammation sites, through the control of E-selectin–mediated adhesion.5 Moreover, PI3Kγ has been shown to be essential for Sphingosine-1-phosphate(S1P)-induced EC migration.6 Using PI3Kγ knockout (KO) mice with unilateral limb ischemia, we and others have recently demonstrated the contribution of PI3Kγ to reparative neovascularization and endothelial progenitor cell functions.7,8 Interestingly, mutant mice expressing catalytically inactive PI3Kγ (kinase dead [KD]) displayed normal angiogenesis following induction of limb ischemia.7 Of note, substantial differences were also denoted in the cardiac phenotype of PI3Kγ mutant animals. In fact, KO but not KD mice, showed a basal enhancement of cardiac contractility and developed cardiac damage following aortic constriction. These differential effects were attributed to the fact that PI3Kγ may exert distinct functions through its kinase activity and kinase-independent scaffolding action.9
Healing of the infarcted heart is accomplished through chemokine-mediated recruitment of inflammatory cells, differentiation of macrophages and myofibroblasts and formation of new vessels and scar tissue. We hypothesize that genetic or pharmacological inactivation of PI3Kγ might significantly interfere with this finely tuned process and thereby impact on functional recovery of the infarcted heart. To address this important question, we used AS605240 (AS), the most potent member of a new class of PI3Kγ-selective inhibitors recently introduced as powerful antiinflammatory agents for treatment of rheumatoid arthritis, systemic lupus and atherosclerosis,10-12 as well as genetically modified animal models with disrupted or inactivated PI3Kγ. Results newly demonstrate an unexpected complex contribution of PI3Kγ to reparative angiogenesis in myocardial infarction (MI).
Methods
An expanded Methods section is available in the Online Data Supplement at http://circres.ahajournals.org.
Cell Cultures
Human umbilical vein ECs (HUVECs) and adult mouse cardiomyocytes (HL-1 cells) were cultured according to manufacturer’s instruction and as described.13 In all in vitro experiments, culture media were supplemented with either 1 μmol/L AS, 15 μmol/L LY294002 (LY) (pan-class I PI3K inhibitor) in DMSO or an equal volume of DMSO alone (vehicle). AS is a potent and highly selective small-molecule PI3Kγ inhibitor that exhibits no notable activity against a wide panel of other protein kinases at 1 μmol/L.11
Adenoviral constructs have been described previously.14,15 Infections were performed overnight at 100 multiplicities of infection.
In Vitro Functional Assays
Cell Proliferation
5-Bromodeoxyuridine incorporation was detected using a colorimetric 5-bromodeoxyuridine assay kit according to the instructions of the manufacturer.
Scratch Assay
HUVEC migration was evaluated by measuring the distance between migrating fronts as described.16
In Vitro Angiogenesis Assay
HUVEC angiogenic capacity was assessed in a Matrigel-based assay. Number of branches and total length of ECs networks were calculated using the Image-Pro Plus software.
Apoptosis Assay
Hypoxia/starvation-induced activation of caspase-3/7 was assessed using a luminescent detection kit according to the instructions of the manufacturer.
Animal Procedures
Experiments involving mice were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals (the Institute of Laboratory Animal Resources, 1996) and with approval of the British Home Office and the University of Bristol. Nine-week-old male CD1 mice (Harlan) received AS (10 mg/kg, IP) or DMSO (vehicle) daily from 3 days before MI until euthanasia. KD and KO mice were generated as described9,17 and compared with wild-type (WT) littermates.
MI was induced by permanent ligation of left anterior descending artery using a 7 to 0 silk suture.18 Sham-operated animals underwent a similar procedure without ligation.
Cardiac function was evaluated using a mouse-dedicated echocardiography system with spatial resolution down to 30 μm (Vevo 770, VisualSonics) before and on day 14 after MI or sham operation.
On day 3 or 14 post-MI, hearts were stopped in diastole by intracardiac injection of cadmium chloride and perfused/fixed and collected for histological analyses. Alternatively, left ventricles were immediately frozen with liquid nitrogen for molecular analysis.
Immunoblot Analyses
Preparation of protein extracts and immunoblot analyses were performed as described.16
Assessment of Akt Kinase Activity in Heart Samples
Akt activity was assessed using the Akt/PKB Kinase Activity Assay kit according to the instructions of the manufacturer.
Immunohistochemical Detection of Leukocytes
To identify infiltrating leukocytes, paraffin-embedded sections (3 μm thick) were stained with an anti-CD45 monoclonal antibody. The average number of CD45-positive cells per square millimeter of tissue and per arteriole was calculated in periinfarct (PI) zone of sections from the midventricular area of AS- or DMSO-treated hearts 3 days post-MI.
Analysis of Neovascularization
Paraffin-embedded sections (3 μm thick) were stained for isolectin IB4 (lectin IB4) to identify ECs and α-smooth muscle actin to identify smooth muscle cells. Capillary and arteriole densities at 14 days post-MI were evaluated in remote (R) and PI zone of heart sections from the midventricular area.
Proliferating ECs (recognized by double staining with an antibody for proliferating-cell nuclear antigen and with lectin IB4) or apoptotic (TUNEL-positive) ECs at 3 days post-MI were evaluated in PI zone of heart sections (3 μm thick) from the midventricular area.
Infarct Size
Hearts were harvested at 14 days post-MI. Sections from 3 levels of each heart (apical, midventricular, and basal) were stained by Azan Mallory and analyzed by morphometry.19
Statistical Analysis
Results are presented as means±SEM. Differences between multiple groups were compared by analysis of variance (1- or 2-way ANOVA and Bonferroni post test), and differences between 2 groups were compared by paired or unpaired Student t test. P<0.05 was considered significant. Stated n values represent biological replicates.
Results
Inhibition or Silencing of PI3Kγ Impair Angiogenesis-Related Processes
In HUVECs, the PI3Kγ inhibitor AS dose-dependently inhibited serum-stimulated phosphorylation of Akt and its downstream substrates, glycogen synthase kinase (GSK)3β and endothelial nitric oxide synthase (eNOS) (Online Figure I, A). Overexpression of PI3Kγ by adenovirus-mediated gene transfer resulted in Akt phosphorylation, which was inhibited by AS (Online Figure I, B). At 1 μmol/L, AS showed no inhibitory activity toward vascular endothelial growth factor (VEGF)-induced activation of the PI3K/Akt pathway (Online Figure I, C). Altogether, these data confirm selectivity of the PI3Kγ inhibitor at the cellular level.
Serum-induced proliferation of HUVECs was strongly decreased by AS and, to a greater extent, by the unselective PI3K inhibitor LY (Figure 1A). Moreover, both AS and LY equally affected HUVEC migration in in vitro scratch assays (Figure 1B). Furthermore, PI3Kγ inhibition impaired the ability of HUVECs to form networks in a Matrigel-based angiogenesis assay, as indicated by the reduced number of branches and network total length (Figure 1C), and increased caspase-3/7 activities following exposure of HUVECs to hypoxia and serum starvation (Figure 1D). Similar effects were observed in HUVECs treated with LY.
Figure 1.
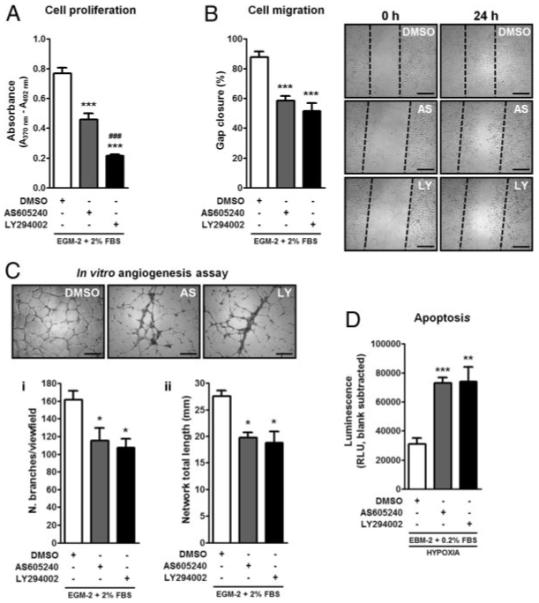
PI3Kγ inhibition impairs angiogenesis. A, Bar graph shows the effects of AS (1 μmol/L) or LY (15 μmol/L) on serum-induced EC proliferation as assessed by measurement of 5-bromodeoxyuridine incorporation. B, Bar graph and microphotographs illustrate gap closure (percentage) measured 24 hours after scratch in DMSO-, AS-, or LY-treated HUVECs. C, Endothelial network formation in Matrigel-based angiogenesis assay: both number of branches/view field (i) and network total length/view field (mm) (ii) were counted. D, Bar graph shows Caspase-3/7 activity assessed by measuring the luminescent signal generated by DEVD cleavage in extracts of HUVECs that were serum-starved and exposed to hypoxia (0.2% O2, 5% CO2) in the presence of DMSO, AS, or LY for 18 hour (n=3 for each assay). *P<0.05, **P<0.01, ***P<0.001 vs DMSO; ###P<0.001 vs AS. Scale bars: 500 μm.
To confirm the results obtained with AS, HUVECs were transduced with an adenoviral construct expressing a small interfering (si)RNA against p110γ (Ad.siRNAγ), PI3Kγ catalytic subunit, or scrambled control (Ad.scrambled). Ad.siRNAγ reduced PI3Kγ protein expression effectively (greater than 75%) and selectively (no inhibitory effect on other PI3K isoforms) (Figure 2A). Moreover, Ad.siRNAγ-transduced HUVECs showed a marked weakening in all angiogenic functions and increased apoptosis following hypoxia/starvation (Figure 2B through 2E). AS treatment of Ad.siRNAγ-transduced ECs did not induce any further functional falloff, thus underscoring the compound specificity (Online Figure II, A through D).
Figure 2.
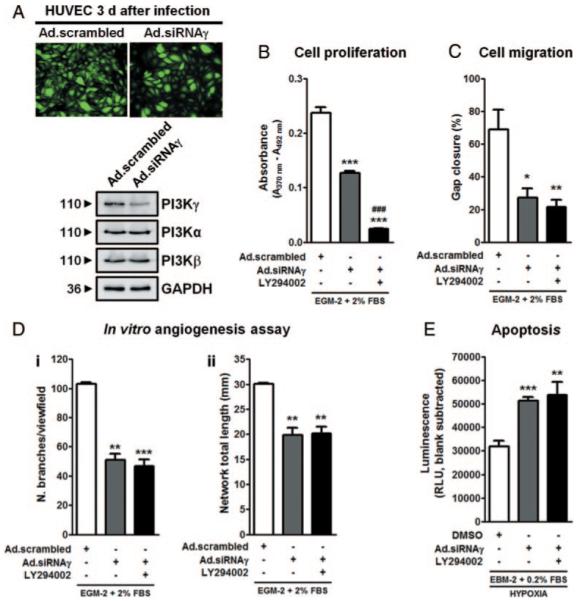
Silencing of PI3Kγ impairs angiogenesis. A, HUVECs were transduced with 100 multiplicities of infection of Ad.scrambled or Ad.siRNAγ overnight. Both adenoviral constructs carried GFP allowing straightforward assessment of infection efficiency. Top, GFP expression 3 days after infection. Bottom, Immunoblot analysis of PI3K isoforms expression in HUVECs harvested 3 days after infection. GAPDH is shown as loading control. The silencing approach does not affect PI3Kα or PI3Kβ expression. PI3Kγ is selectively and effectively downregulated. Assays were performed as in Figure 1, 3 days after adenoviral transduction. Three independent infections were performed for both Ad.siRNAγ and Ad.scrambled. Results indicate that PI3Kγ is essential to cell proliferation (B), migration (C), tube-like network formation (D), and survival (E) of ECs (n=3 for each experiment). *P<0.05, **P<0.01, ***P<0.001 vs scrambled; ###P<0.001 vs Ad.siRNAγ.
Rescue of AS-Inhibited Angiogenesis by Constitutively Active Akt
The activated Akt pathway has been shown to promote vascular cell survival and angiogenesis.20-22 Western blot analyses confirmed the inhibitory effect of AS and LY on serum-stimulated phosphorylation of Akt, GSK3β and eNOS (Figure 3A). Extracellular signal-regulated kinases 1/2 (Erk1/2) are considered downstream and ultimate effectors of the mitogen-activated protein kinase (MAPK) pathway, triggered also by PI3Kγ.23,24 Interestingly, both AS and LY caused a significant decrease of Erk1/2 phosphorylation levels (Figure 3A, iv). Similarly, on PI3Kγ knockdown, both the Akt and the MAPKs pathways were strongly downmodulated (Figure 3B).
Figure 3.

PI3K/Akt signaling suppression on PI3Kγ inhibition accounts for defective angiogenesis. A and B, HUVECs were incubated in EC basal medium with neither growth factors nor serum for 3 hours. During the last 30 minutes before stimulation, cells were pre-treated with DMSO, AS (1 μmol/L), or LY (15 μmol/L), followed by 10 minutes stimulation with complete medium containing growth factors and serum. A similar procedure was carried out on Ad.scrambled and Ad.siRNAγ HUVECs 3 days after transduction. Immunoblot analysis and averaged densitometric data show the effects of PI3K inhibition on phosphorylated (p)Akt (i), pGSK3β (ii), peNOS (iii), and pErk1/2 (iv) levels. C, Immunoblot analysis confirming the activation of Akt and downstream effectors achieved by infection with Ad.MyrAkt (hemagglutinin [HA]-tagged, top immunoblot image). D, i and ii, Bar graphs show the effects of the restoration of Akt activity by Ad.MyrAkt in Matrigel-based angiogenesis assay. In C and D, Ad.Null-HUVECs are shown for comparison. n=4. *P<0.05, **P<0.01, ***P<0.001 vs DMSO (A) or Ad.scrambled (B); #P<0.05, ##P<0.01, ###P<0.001 vs AS (A), Ad.siRNAγ (B), or Ad.Null AS/LY (D).
We next asked whether restoration of the Akt pathway could rescue the angiogenic defect resulting from PI3Kγ inhibition. To this aim, HUVECs were infected with a constitutively active form of Akt (hemagglutinin-tagged myr-istoylated Akt; Ad.MyrAkt), thus resulting in phosphorylation of the downstream targets (Figure 3C). Control HUVECs were infected with an empty adenovirus (Ad.Null). In vitro endothelial network formation was impaired in Null-transduced HUVECs treated with either AS or LY, whereas restoration of Akt activity completely rescued this defect (Figure 3D).
Effects of PI3Kγ Inhibition on Cell Signaling in Infarcted Hearts
We then evaluated the activation status of the PI3Kγ/Akt pathway in left ventricular (LV) samples collected from AS- or DMSO-treated mice 3 days post-MI or sham operation. MI induced a marked upregulation of PI3Kγ in the LV of both treatment groups, as assessed by immunoblot analysis (Figure 4A). By immunohistochemistry, we found that cardiomyocytes and ECs overexpressed PI3Kγ in the PI zone, whereas in sham-operated mice, PI3Kγ expression appeared rather diffuse and weak (Online Figure III). Akt phosphorylation levels and relative catalytic activity did not differ between DMSO- and AS-treated sham-operated mice, whereas MI-induced activation of Akt was not only completely abrogated by AS but also strongly decreased below the levels of AS-treated sham-operated mice (Figure 4B and 4C). Phosphorylation levels of GSK3β and eNOS followed the same trend observed for Akt (Figure 4D and 4E). Furthermore, Pim-1, a recently reported mediator of cardiomyocyte survival, downstream of Akt,25 was found strongly downmodulated in infarcted hearts of AS-treated mice (Figure 4F).
Figure 4.
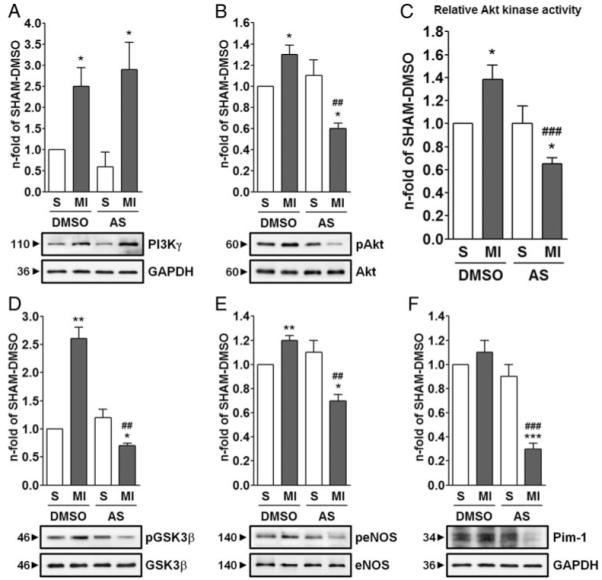
Effects of PI3Kγ inhibition on cell signaling in the infarcted heart. Immunoblot analyses (A, B, D, and F) of PI3Kγ/Akt signaling pathway and Akt relative kinase activity (C) in protein extracts from LV of DMSO- or AS-treated mice 3 days post-MI or sham (S). Bar graphs show fold changes calculated from the ratio between either phosphorylated and total protein content (B, D, and E) or total protein content and GAPDH, used as loading control (A and F). In all graphs, fold changes are calculated toward DMSO-treated Sham. n=4 hearts in each group. *P<0.05, **P<0.01, ***P<0.001 vs DMSO- or AS-treated sham; ##P<0.01, ###P<0.001 vs DMSO-treated MI.
PI3Kγ Inhibition by AS or PI3Kγ Knockout Exacerbates MI-Induced Cardiac Dysfunction
In sham-operated mice, AS did not cause any significant alteration in LV function, as assessed by echocardiography (Online Table I). However, AS significantly worsened MI-induced cardiac dysfunction compared to DMSO (Table).
Table. Echocardiographic Parameters.
| Echocardiography (Pre-MI) |
Echocardiography (14 Days Post-MI) |
|||
|---|---|---|---|---|
| Parameter | DMSO (n=9) |
AS605240 (n=15) |
DMSO (n=9) |
AS605240 (n=15) |
| LVAWs (mm) | 1.38±0.06 | 1.29±0.05 | 0.50±0.09 | 0.32±0.01* |
| LVAWd (mm) | 0.92±0.04 | 0.85±0.03 | 0.46±0.08 | 0.31±0.02* |
| LVPWs (mm) | 1.49±0.07 | 1.69±0.09 | 1.32±0.04 | 0.96±0.06*** |
| LVPWd (mm) | 1.04±0.04 | 1.10±0.06 | 0.98±0.03 | 0.81±0.05* |
| LVESD (mm) | 2.68±0.23 | 2.44±0.13 | 4.59±0.25 | 4.74±0.24 |
| LVEDD (mm) | 4.14±0.25 | 4.02±0.13 | 5.27±0.23 | 5.19±0.24 |
| LVESV (μL) | 25.7±5.1 | 22.1±2.8 | 99.8±12.7 | 109.4±12.8 |
| LVEDV (μL) | 71.8±9.8 | 71.3±5.0 | 136.6±14.2 | 133.3±14.5 |
| LVSV (μL) | 49.5±5.3 | 48.0±2.8 | 36.7±4.1 | 24.0±2.9* |
| LVEf (%) | 62.5±4.9 | 69.0±2.4 | 27.7±2.9 | 18.4±1.9* |
| LVFS (%) | 36.2±2.0 | 38.6±1.8 | 13.1±1.5 | 9.0±1.0* |
| HR (bpm) | 499.6±8.6 | 502.2±4.5 | 498.9±5.7 | 486.5±5.6 |
| Q (mL/min) | 24.7±2.7 | 24.1±1.4 | 18.3±2.0 | 12.1±1.4* |
Data are presented as means±SEM. LVAWs indicates LV anterior wall, end systole; LVAWd, LV anterior wall, end diastole; LVPWs, LV posterior wall, end systole; LVPWd, LV posterior wall, end diastole; LVESD, LV end-systolic diameter; LVEDD, LV end-diastolic diameter; LVESV, LV end-systolic volume; LVEDV, LV end-diastolic volume; LVSV, LV stroke volume; LVEf, LV ejection fraction; LVFS, LV fractional shortening; HR, heart rate; Q, cardiac output.
P<0.05
P<0.001 vs DMSO.
We then evaluated the impact of MI on PI3Kγ genetically modified mice. Before MI, PI3Kγ KO mice showed higher LV ejection fraction (LVEf) and fractional shortening (LVFS) compared with KD or WT littermates (Online Table II), in line with previous reports indicating hyper-contractility of KO hearts under basal conditions.9,26 However, following MI, the function of KO hearts was more severely compromised as compared with WT and KD (Online Table II).
Consistent with hemodynamic data, AS-treated mice and PI3Kγ KO mice showed larger infarct sizes compared to DMSO-treated (Online Figure IV, A and B) or KD and WT (Online Figure IV, C and D), respectively.
Targeting of PI3Kγ Suppresses Post-MI Reparative Neovascularization
In DMSO-treated or WT mice, capillary density of the PI zone was higher than that of R zone. Importantly, reparative capillarization was abrogated by pharmacological inhibition (Figure 5A and B [i]) or genetic deletion (KO) or inactivation (KD) of PI3Kγ (Figure 5B, ii).
Figure 5.
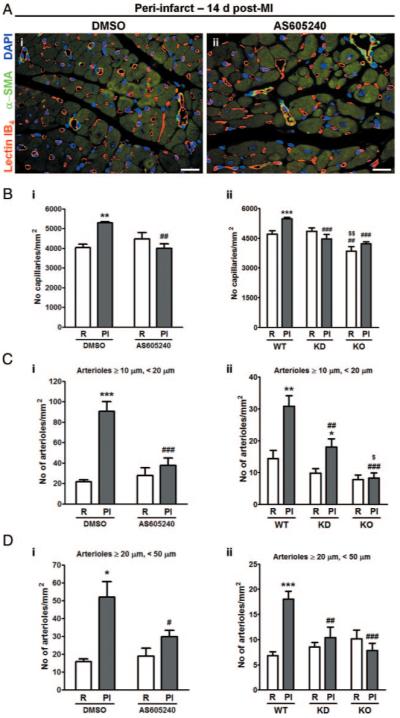
PI3Kγ is crucial for reparative neovascularization. A, Immunofluorescence images show the vascularization of PI zone of DMSO- or AS-treated hearts 14 days post-MI. ECs are stained by lectin IB4 (red), α-smooth muscle actin (α-SMA, green) identifies arterioles, and nuclei are stained by DAPI (blue). Scale bars: 20 μm. Bar graphs show the capillary density (B) or arteriole density (C and D) of R and PI zone of DMSO- or AS-treated hearts (i) and of WT, KD, or KO hearts (ii). n=4 to 8 hearts per group. *P<0.05, **P<0.01, ***P<0.001 vs DMSO-treated R or WT R; #P<0.05, ##P<0.01, ###P<0.001 vs DMSO-treated PI or WT PI; $P<0.05 vs KD PI; $$P<0.01 vs WT/KD R.
Analysis of arteriole density unraveled a significant increase of small arterioles (diameter <50 μm) in the PI zone of DMSO-treated hearts, which was abrogated by AS (Figure 5A, 5C [i], and 5D [i]). Similarly, arteriole density was reduced in KD and KO hearts as compared to WT (Figure 5C [ii] and 5D [ii]), with the arteriole defect being more prominent in KO. We did not find any difference in the number of arterioles with diameter ≥50 μm among groups, regardless the treatment received or genotype (data not shown).
Differential Effects of AS and PI3Kγ Genetic Inactivation on EC and Cardiomyocyte Biology
The finely tuned balance between EC proliferation and apoptosis early after an ischemic injury determines the extent of the neovascularization response.7,16 Therefore, we analyzed hearts at 3 days post-MI to quantify the fraction of proliferating/apoptotic ECs in PI zone. Interestingly, AS-treated hearts exhibited a 2-fold reduction in the percentage of proliferating ECs as compared to DMSO (Figure 6A and 6B [i]). Surprisingly, only KO hearts exhibited a similar phenotype, whereas KD hearts showed no impairment in EC proliferation (Figure 6B, ii). Of note, EC apoptosis was remarkably increased in the PI zone of AS-treated as well as of KD and KO hearts (Figure 7A and B [i and ii]).
Figure 6.
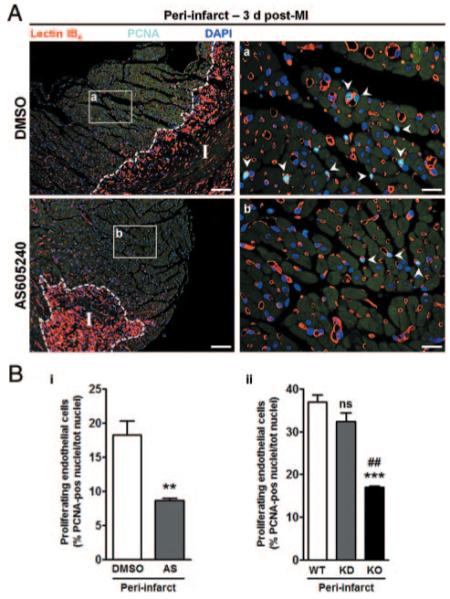
Targeting of PI3Kγ inhibits EC proliferation in infarcted hearts. Representative immunofluorescence images (A) show the fraction of proliferating ECs in PI zone of DMSO- or AS-treated hearts (a and b, respectively) 3 days post-MI. Proliferating ECs (arrowheads) are identified by lectin IB4 (red) and positivity for proliferating-cell nuclear antigen (PCNA) (light blue). Nuclei are stained by DAPI (blue). Scale bars: left, 100 μm; right, 20 μm. I indicates infarct; unspecific lectin IB4 binding delimiting infarcted area. B, Bar graph illustrates the number of proliferating ECs in PI zone of DMSO- or AS-treated mice (i) or of WT, KD and KO mice (ii). n=4 to 6 mice in each group. **P<0.01 vs DMSO; ***P<0.001 vs WT; ##P<0.01 vs KD; ns, not significant vs WT.
Figure 7.

PI3Kγ is crucial for EC and cardiomyocyte survival in vivo. Representative immunofluorescence images (A) and bar graphs (B) show the fraction of apoptotic ECs (i) and cardiomyocytes (iii) in PI zone of DMSO- and AS-treated hearts 3 days after MI. Apoptotic ECs and cardiomyocytes are positive to TUNEL reaction (light magenta). ECs are stained by lectin IB4 (green) and nuclei by DAPI (blue). n=6 DMSO- and 4 AS-treated hearts. Scale bars: left, 100 μm, right, 20 μm. I indicates infarct; unspecific lectin IB4 binding delimiting infarcted area. The number of apoptotic ECs (B, ii) and cardiomyocytes (B, iv) was also evaluated in PI zone of WT, KD, and KO hearts. n=4 for each group. C, HL-1 were serum starved and exposed to hypoxia (0.2% O2, 5% CO2) in the presence of DMSO or AS for 18 hours. Bar graph shows caspase-3/7 activity assessed as in Figure 1D; n=3. *P<0.05, **P<0.01, ***P<0.001 vs DMSO or WT; #P<0.05, ###P<0.001 vs KD.
PI3Kγ is also crucial for cardiomyocyte survival. In fact, apoptosis was enhanced by 2- and 2.3-fold in the PI zone of AS-treated and KD hearts (Figure 7A and 7B [iii and iv]). Of note, cardiomyocyte apoptosis was even more activated in KO hearts (Figure 7B, iv). To verify the direct effect of PI3Kγ inhibition on cardiomyocytes survival, serum-starved adult mouse cardiomyocytes (HL-1) were treated with DMSO or AS and exposed to hypoxia. As shown by measurement of Caspase-3/7 activation, AS treatment enhanced cardiomyocyte apoptosis (Figure 7C).
PI3Kγ Is Crucial for Leukocyte Homing
Leukocytes recruited in the infarct and PI zone are of central importance for myocardial and vascular remodeling.27-30 AS strikingly reduced the number of CD45pos leukocytes in the PI zone at 3 days post-MI as compared to DMSO (Online Figure V, A and B [i]). Further immunohistochemical studies showed that neutrophils and monocytes/macrophages are the most affected populations, although a less marked decrease in T lymphocytes recruitment was also observed (Online Figure VI). Importantly, leukocytes adhering to the arteriolar endothelial lining or infiltrating the arteriole wall were 13.3-fold less frequent in AS-treated hearts as compared to DMSO-treated hearts (Online Figure V, A and C [i]). Molecular analysis on protein extracts of primary monocytes/macrophages and lymphocytes isolated from the peripheral blood of infarcted mice 2 days post-MI showed a strong inhibition of the Akt pathway on AS treatment (Online Figure VII, A and B). Moreover, AS-treated bone marrow mononuclear cells from the same infarcted mice displayed impaired directional migratory capacity in vitro (Online Figure VII, C).
In line with the above results, analyses performed in hearts from genetically modified mice revealed a markedly reduced leukocyte infiltration in PI zone of KD and, to a bigger extent, KO mice as compared to WT (Online Figure V, B [ii] and C [ii]).
Discussion
The high degree of functional specialization among members of the PI3K family combined with the recent understanding of their involvement in several human diseases has fostered the development of isoform-specific inhibitors.31 In this context, a recent study identified PI3Kα as a major regulator of developmental angiogenesis and VEGF-dependent EC migration.3
Here, we present novel data supporting the concept that PI3Kγ, which is primarily activated on stimulation of GPCRs, plays a crucial role in reparative angiogenesis. Inhibition of PI3Kγ catalytic activity, achieved by either a highly selective PI3Kγ inhibitor, AS, or siRNA-mediated knockdown of PI3Kγ catalytic subunit, exerts detrimental effects on EC proliferation, migration, network formation, and survival in vitro. The decisive role of PI3Kγ in controlling angiogenesis-related processes is underscored in that LY, a pan PI3K inhibitor, did not affect further any EC function but cell proliferation. Because PI3Kα is not involved in the regulation of EC proliferation and PI3Kδ is scarcely expressed in HUVECs,3 PI3Kβ might be responsible for the modulation of EC proliferation, together with PI3Kγ. We also demonstrate that Akt is fundamental for PI3Kγ-driven angiogenesis. Indeed, PI3Kγ inhibition and PI3Kγ knockdown resulted in reduced activation of Akt and eNOS, accompanied by the release of Akt inhibitory action on GSK3β, which can therefore inhibit downstream targets required for cell cycle progression.32 Importantly, restoration of the Akt pathway led to recovery of EC angiogenic capacity. PI3Kγ inhibition also hampered the MAPK pathway, thus contributing to the observed proliferative defect.
MI remains one of the leading causes of morbidity and mortality worldwide despite improved management of risk factors and state-of-the-art treatments.33 In theory, inhibitors of PI3K do not represent the best candidate for the treatment of MI, considering the proangiogenic and prosurvival action exerted by the PI3K/Akt signaling pathway in myocardial ischemia.34,35 However, selective PI3Kγ inhibitors might have potential cardio-protective applications, especially for the treatment of atherosclerosis,12 and under conditions of increased workload, through the reduction of leukocyte infiltration and myocardial fibrosis.9 Whether inhibitors of PI3Kγ may beneficially impact on post-MI healing by constraining excessive inflammation and fibrosis without jeopardizing reparative angiogenesis is a matter of growing interest. The dual PI3Kγ/δ inhibitor TG100-115 has been recently tested in animal models of ischemia/reperfusion injury, where it reduced infarct size and improved myocardial function without affecting the number of inflammatory cells infiltrating the infarcted myocardium. This compound had no effect on VEGF-induced EC proliferation, angiogenesis and Erk phosphorylation, but it blocked VEGF-induced phosphorylation of Akt.36 A puzzling aspect of the above study is that only the angiogenesis driven by VEGF, which signals through tyrosine kinase receptors, was explored, thus leaving open the possibility that the inhibitor might or might not interfere with GPCR-dependent angiogenesis. Furthermore, PI3Kγ is known to play a pivotal role in chemokine-induced migration of neutrophils, monocytes/macrophages, and T lymphocytes to hypoxic or inflamed tissues17,37-39; therefore, the lack of effects of TG100-115 on myocardial inflammation casts doubts about complete abrogation of PI3Kγ signaling in leukocytes recruited to the infarcted heart.
AS is the most representative member of a new class of PI3Kγ-selective inhibitors which proved to exert therapeutic effects in murine models of chronic inflammatory/autoimmune diseases and atherosclerosis.10-12 In our experimental design, mice received AS before MI induction, mimicking the hypothetical clinical situation in which patients are already under treatment when MI occurs. One primary molecular hallmark of MI hearts was the striking upregulation of PI3Kγ associated to activation of Akt/eNOS and inhibition of GSK3β. AS treatment completely suppressed the MI-dependent activation of Akt and phosphorylation/expression of its downstream targets, including Pim1, an Akt-regulated enhancer of cardiomyocyte survival.25 These molecular findings anticipate that inhibition of PI3Kγ may interfere with different cellular functions relevant to cardiac recovery.
A balanced and coordinated inflammatory response is instrumental to degradation of extracellular matrix, clearance of dead cells, and replacement of necrotic areas by connective tissue.27,40 Furthermore, recruited monocytes contribute to the restoration of perfusion through direct and paracrine promotion of neovascularization.30,40 In line with the established antiinflammatory action of PI3Kγ inhibitors, AS-treated hearts displayed a highly reduced infiltration of leukocytes and essentially no leukocytes surrounding arterioles. Importantly, we verified that AS causes a striking downregulation of Akt phosphorylation in monocytes and lymphocytes from peripheral blood of infarcted mice and remarkably reduces the migratory activity of bone marrow mononuclear cells from the same animals. After acute ischemia, the circulating monocyte fraction becomes enriched with proangiogenic progenitor cells. We have recently demonstrated that PI3Kγ is constitutively expressed in human proangiogenic progenitor cells and recruited to the cell membrane in a polarized fashion on stimulation with the GPCR ligand bradykinin.41 Furthermore, PI3Kγ-silenced human progenitor cells, as well as PI3Kγ-deficient murine BM-derived proangiogenic cells exhibited remarkably depressed migratory activity, reduced Akt and eNOS phosphorylation, and decreased nitric oxide production, which jeopardize their regenerative potential.41,42
We then investigated the effect of AS on angiogenesis at the initial (3 days post-MI) and stabilization phase (14 days post-MI) of the healing process. Of note, AS-treated mice failed in mounting an adequate neovascularization response at capillary and arteriole level, with enhanced EC apoptosis and decreased EC proliferation accounting for such dysfunction. Similarly, cardiomyocytes were found more apoptotic in AS-treated hearts as compared to controls. A direct action of AS on cardiomyocyte survival was documented in vitro. Finally, AS-treated mice showed larger scars with thinner LV walls and significantly depressed cardiac contractility, indicating that, by interfering with multiple cellular events, the inhibitor detrimentally impinges on cardiac recovery.
To gain further insight into the relevance of PI3Kγ in reparative angiogenesis, we investigated the response of PI3Kγ KD and KO mice to MI. The results obtained in genetically modified animals overall confirm an important role of PI3Kγ in reparative neovascularization and healing of MI. Nonetheless, some intriguing differences were uncovered, with KO animals showing more remarkable activation of EC and cardiomyocyte apoptosis and inhibition of EC proliferation as compared to KD. This translated into larger scars and more profoundly compromised LV function in KO animals. These data are in line with susceptibility of PI3Kγ KO mice to cardiac damage, which was attributed to elevation of cAMP in KO hearts.9 Increased myocardial cAMP in settings of acute MI is detrimental, causing perfusion–contraction mismatching, increased myocardial energetic requirements, and an unfavorable flow redistribution away from the ischemic subendocardium.43 In KD mice, reparative angiogenesis was less severely impaired compared to KO, although MI-induced cardiac dysfunction was similar to WT controls.
Our genetic models show that, in reparative angiogenesis, the absence of PI3Kγ protein is more detrimental than the inactivation of its catalytic activity. This is in agreement with previous reports,7 but is in apparent contrast with our pharmacological studies. One caveat of our pharmacological approach is that some of the effects induced by AS might be attributable to partial inhibition of other PI3K isoforms (ie, PI3Kα), which is unlikely at the elected dosage, or to interference with unrelated enzymes. In human ECs, we have shown that AS specifically suppresses Akt phosphorylation induced by adenovirus-mediated PI3Kγ overexpression, while not inhibiting VEGF-induced Akt activation. Furthermore, both AS and PI3Kγ silencing inhibit angiogenesis in vitro, with no additional effect when AS is superimposed to PI3Kγ silencing. Although we cannot completely exclude that the AS compound is not selective enough to uniquely block PI3Kγ function in vivo, it is conceivable that the milder phenotype of infarcted KD mice compared AS-treated mice may be attributable to the development of compensatory/adaptive mechanisms in the genetically modified mice.44
Taken together, our findings clearly establish PI3Kγ as a key player in physiological and reparative angiogenesis, as well as healing of MI. Additionally, our results point out the need for new chemical structures with improved selectivity profiles and devoid of the harmful effects shown by AS.
Novelty and Significance.
Phosphoinositide-3 kinase (PI3K)γ is a pivotal mediator of leukocytes chemotaxis. Given the participation of inflammatory cells in reparative and pathological angiogenesis, PI3Kγ has become a hot topic for vascular and cancer researchers and pharmaceutical industry, which claims PI3Kγ inhibitors to be “the aspirin of the new millennium.” We and others concurrently reported that knockout of PI3Kγ impairs endothelial progenitor cells functions and homing to site of ischemic injury, thus alerting against the risk of PI3Kγ signaling perturbation. We now show that pharmacological targeting of PI3Kγ inhibits angiogenesis in a model of myocardial infarction, leading to enhanced apoptosis in the peri-infarct zone, enlarged scars and increased cardiac dysfunction. Akt-dependent signaling, which regulates migration and survival of endothelial cells, is remarkably hampered following PI3Kγ inhibition. We then verified whether genetically modified mice lacking PI3Kγ or carrying a kinase-dead mutation are phenocopies of mice treated with PI3Kγ inhibitor. Interestingly, knockout mice showed severely impaired reparative angiogenesis and large infarcts, whereas kinase-dead mice failed to mount a proper neovascularization, but showed no additional cardiac dysfunction compared to controls. These results reveal an unexpected complex contribution of PI3Kγ to vascular and cardiac repair processes and call for caution in the use of first-generation PI3Kγ inhibitors in myocardial ischemia.
What Is Known?
PI3Kγ, a kinase that links G protein–coupled receptors to the Akt pathway, plays a key role in inflammation by acting as a compass for the directional migration of leukocytes.
Pharmacological inhibitors of PI3Kγ may be useful as antiinflammatory agents.
Genetic disruption of PI3Kγ impairs neovascularization and integrin-dependent homing of endothelial progenitor cells in a model of peripheral ischemia.
What New Information Does This Article Contribute?
PI3Kγ plays a major role in reparative angiogenesis in a model of myocardial infarction.
Pharmacological inhibition of PI3Kγ to fight inflammation may jeopardize the ischemic heart.
Acknowledgments
Ad.siRNAγ and Ad.scrambled were kindly provided by Prof Patrick E. MacDonald and Dr Jocelyn E. Manning Fox (University of Alberta, Canada). We thank Dr Graciela Sala-Newby and Jill Tarlton for adenoviruses generation, Paul Savage for technical support, and Dr Nicolle Kränkel, Dr Andrea Caporali, Dr Paola Campagnolo, and Brunella Cristofaro for valuable advice.
Sources of Funding: This study was supported by the British Heart Foundation (PhD studentship FS/06/083/21828).
Non-standard Abbreviations and Acronyms
- AS
AS605240
- EC
endothelial cell
- eNOS
endothelial nitric oxide synthase
- Erk
extracellular signal-regulated
- GSK
glycogen synthase kinase
- GPCR
G protein-coupled receptor
- HUVEC
human umbilical vein endothelial cell
- KD
kinase dead
- KO
knockout
- LV
left ventricular
- LY
LY294002
- MAPK
mitogen-activated protein kinase
- MI
myocardial infarction
- p
phosphorylated
- PI
periinfarct
- PI3K
phosphoinositide-3 kinase
- R
remote
- siRNA
small interfering RNA
- VEGF
vascular endothelial growth factor
- WT
wild type
Footnotes
Disclosures: E.H. also operates as a consultant for Merck Serono and Cellzome.
References
- 1.Vanhaesebroeck B, Leevers SJ, Ahmadi K, Timms J, Katso R, Driscoll PC, Woscholski R, Parker PJ, Waterfield MD. Synthesis and function of 3-phosphorylated inositol lipids. Annu Rev Biochem. 2001;70:535–602. doi: 10.1146/annurev.biochem.70.1.535. [DOI] [PubMed] [Google Scholar]
- 2.Hawkins PT, Anderson KE, Davidson K, Stephens LR. Signalling through Class I PI3Ks in mammalian cells. Biochem Soc Trans. 2006;34:647–662. doi: 10.1042/BST0340647. [DOI] [PubMed] [Google Scholar]
- 3.Graupera M, Guillermet-Guibert J, Foukas LC, Phng LK, Cain RJ, Salpekar A, Pearce W, Meek S, Millan J, Cutillas PR, Smith AJ, Ridley AJ, Ruhrberg C, Gerhardt H, Vanhaesebroeck B. Angiogenesis selectively requires the p110alpha isoform of PI3K to control endothelial cell migration. Nature. 2008;453:662–666. doi: 10.1038/nature06892. [DOI] [PubMed] [Google Scholar]
- 4.Hirsch E, Lembo G, Montrucchio G, Rommel C, Costa C, Barberis L. Signaling through PI3Kgamma: a common platform for leukocyte, platelet and cardiovascular stress sensing. Thromb Haemost. 2006;95:29–35. [PubMed] [Google Scholar]
- 5.Puri KD, Doggett TA, Huang CY, Douangpanya J, Hayflick JS, Turner M, Penninger J, Diacovo TG. The role of endothelial PI3Kgamma activity in neutrophil trafficking. Blood. 2005;106:150–157. doi: 10.1182/blood-2005-01-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heller R, Chang Q, Ehrlich G, Hsieh SN, Schoenwaelder SM, Kuhlencordt PJ, Preissner KT, Hirsch E, Wetzker R. Overlapping and distinct roles for PI3Kbeta and gamma isoforms in S1P-induced migration of human and mouse endothelial cells. Cardiovasc Res. 2008;80:96–105. doi: 10.1093/cvr/cvn159. [DOI] [PubMed] [Google Scholar]
- 7.Madeddu P, Kraenkel N, Barcelos LS, Siragusa M, Campagnolo P, Oikawa A, Caporali A, Herman A, Azzolino O, Barberis L, Perino A, Damilano F, Emanueli C, Hirsch E. Phosphoinositide 3-kinase gamma gene knockout impairs postischemic neovascularization and endothelial progenitor cell functions. Arterioscler Thromb Vasc Biol. 2008;28:68–76. doi: 10.1161/ATVBAHA.107.145573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chavakis E, Carmona G, Urbich C, Gottig S, Henschler R, Penninger JM, Zeiher AM, Chavakis T, Dimmeler S. Phosphatidylinositol-3-kinase-gamma is integral to homing functions of progenitor cells. Circulation research. 2008;102:942–949. doi: 10.1161/CIRCRESAHA.107.164376. [DOI] [PubMed] [Google Scholar]
- 9.Patrucco E, Notte A, Barberis L, Selvetella G, Maffei A, Brancaccio M, Marengo S, Russo G, Azzolino O, Rybalkin SD, Silengo L, Altruda F, Wetzker R, Wymann MP, Lembo G, Hirsch E. PI3Kgamma modulates the cardiac response to chronic pressure overload by distinct kinase-dependent and -independent effects. Cell. 2004;118:375–387. doi: 10.1016/j.cell.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 10.Barber DF, Bartolome A, Hernandez C, Flores JM, Redondo C, Fernandez-Arias C, Camps M, Ruckle T, Schwarz MK, Rodriguez S, Martinez AC, Balomenos D, Rommel C, Carrera AC. PI3Kgamma inhibition blocks glomerulonephritis and extends lifespan in a mouse model of systemic lupus. Nat Med. 2005;11:933–935. doi: 10.1038/nm1291. [DOI] [PubMed] [Google Scholar]
- 11.Camps M, Ruckle T, Ji H, Ardissone V, Rintelen F, Shaw J, Ferrandi C, Chabert C, Gillieron C, Francon B, Martin T, Gretener D, Perrin D, Leroy D, Vitte PA, Hirsch E, Wymann MP, Cirillo R, Schwarz MK, Rommel C. Blockade of PI3Kgamma suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat Med. 2005;11:936–943. doi: 10.1038/nm1284. [DOI] [PubMed] [Google Scholar]
- 12.Fougerat A, Gayral S, Gourdy P, Schambourg A, Ruckle T, Schwarz MK, Rommel C, Hirsch E, Arnal JF, Salles JP, Perret B, Breton-Douillon M, Wymann MP, Laffargue M. Genetic and pharmacological targeting of phosphoinositide 3-kinase-gamma reduces atherosclerosis and favors plaque stability by modulating inflammatory processes. Circulation. 2008;117:1310–1317. doi: 10.1161/CIRCULATIONAHA.107.720466. [DOI] [PubMed] [Google Scholar]
- 13.White SM, Constantin PE, Claycomb WC. Cardiac physiology at the cellular level: use of cultured HL-1 cardiomyocytes for studies of cardiac muscle cell structure and function. Am J Physiol Heart Circ Physiol. 2004;286:H823–H829. doi: 10.1152/ajpheart.00986.2003. [DOI] [PubMed] [Google Scholar]
- 14.Pigeau GM, Kolic J, Ball BJ, Hoppa MB, Wang YW, Ruckle T, Woo M, Manning Fox JE, Macdonald PE. Insulin granule recruitment and exocytosis is dependent on p110{gamma} in insulinoma and human {beta}-cells. Diabetes. 2009 doi: 10.2337/db08-1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Emanueli C, Caporali A, Krankel N, Cristofaro B, Van Linthout S, Madeddu P. Type-2 diabetic Lepr(db/db) mice show a defective microvascular phenotype under basal conditions and an impaired response to angiogenesis gene therapy in the setting of limb ischemia. Front Biosci. 2007;12:2003–2012. doi: 10.2741/2205. [DOI] [PubMed] [Google Scholar]
- 16.Barcelos LS, Duplaa C, Krankel N, Graiani G, Invernici G, Katare R, Siragusa M, Meloni M, Campesi I, Monica M, Simm A, Campagnolo P, Mangialardi G, Stevanato L, Alessandri G, Emanueli C, Madeddu P. Human CD133+ progenitor cells promote the healing of diabetic ischemic ulcers by paracrine stimulation of angiogenesis and activation of Wnt signaling. Circ Res. 2009;104:1095–1102. doi: 10.1161/CIRCRESAHA.108.192138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirsch E, Katanaev VL, Garlanda C, Azzolino O, Pirola L, Silengo L, Sozzani S, Mantovani A, Altruda F, Wymann MP. Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science. 2000;287:1049–1053. doi: 10.1126/science.287.5455.1049. [DOI] [PubMed] [Google Scholar]
- 18.Spillmann F, Graiani G, Van Linthout S, Meloni M, Campesi I, Lagrasta C, Westermann D, Tschope C, Quaini F, Emanueli C, Madeddu P. Regional and global protective effects of tissue kallikrein gene delivery to the peri-infarct myocardium. Regen Med. 2006;1:235–254. doi: 10.2217/17460751.1.2.235. [DOI] [PubMed] [Google Scholar]
- 19.Takagawa J, Zhang Y, Wong ML, Sievers RE, Kapasi NK, Wang Y, Yeghiazarians Y, Lee RJ, Grossman W, Springer ML. Myocardial infarct size measurement in the mouse chronic infarction model: comparison of area- and length-based approaches. J Appl Physiol. 2007;102:2104–2111. doi: 10.1152/japplphysiol.00033.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 21.Jiang BH, Zheng JZ, Aoki M, Vogt PK. Phosphatidylinositol 3-kinase signaling mediates angiogenesis and expression of vascular endothelial growth factor in endothelial cells. Proc Natl Acad Sci U S A. 2000;97:1749–1753. doi: 10.1073/pnas.040560897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shiojima I, Walsh K. Role of Akt signaling in vascular homeostasis and angiogenesis. Circ Res. 2002;90:1243–1250. doi: 10.1161/01.res.0000022200.71892.9f. [DOI] [PubMed] [Google Scholar]
- 23.Bondeva T, Pirola L, Bulgarelli-Leva G, Rubio I, Wetzker R, Wymann MP. Bifurcation of lipid and protein kinase signals of PI3Kgamma to the protein kinases PKB and MAPK. Science. 1998;282:293–296. doi: 10.1126/science.282.5387.293. [DOI] [PubMed] [Google Scholar]
- 24.Rubio I, Rodriguez-Viciana P, Downward J, Wetzker R. Interaction of Ras with phosphoinositide 3-kinase gamma. Biochem J. 1997;326(pt 3):891–895. doi: 10.1042/bj3260891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muraski JA, Rota M, Misao Y, Fransioli J, Cottage C, Gude N, Esposito G, Delucchi F, Arcarese M, Alvarez R, Siddiqi S, Emmanuel GN, Wu W, Fischer K, Martindale JJ, Glembotski CC, Leri A, Kajstura J, Magnuson N, Berns A, Beretta RM, Houser SR, Schaefer EM, Anversa P, Sussman MA. Pim-1 regulates cardiomyocyte survival downstream of Akt. Nat Med. 2007;13:1467–1475. doi: 10.1038/nm1671. [DOI] [PubMed] [Google Scholar]
- 26.Crackower MA, Oudit GY, Kozieradzki I, Sarao R, Sun H, Sasaki T, Hirsch E, Suzuki A, Shioi T, Irie-Sasaki J, Sah R, Cheng HY, Rybin VO, Lembo G, Fratta L, Oliveira-dos-Santos AJ, Benovic JL, Kahn CR, Izumo S, Steinberg SF, Wymann MP, Backx PH, Penninger JM. Regulation of myocardial contractility and cell size by distinct PI3K-PTEN signaling pathways. Cell. 2002;110:737–749. doi: 10.1016/s0092-8674(02)00969-8. [DOI] [PubMed] [Google Scholar]
- 27.Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, Libby P, Weissleder R, Pittet MJ. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204:3037–3047. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fraccarollo D, Galuppo P, Schraut S, Kneitz S, van Rooijen N, Ertl G, Bauersachs J. Immediate mineralocorticoid receptor blockade improves myocardial infarct healing by modulation of the inflammatory response. Hypertension. 2008;51:905–914. doi: 10.1161/HYPERTENSIONAHA.107.100941. [DOI] [PubMed] [Google Scholar]
- 29.Jujo K, Ii M, Losordo DW. Endothelial progenitor cells in neovascularization of infarcted myocardium. J Mol Cell Cardiol. 2008;45:530–544. doi: 10.1016/j.yjmcc.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ito WD, Arras M, Winkler B, Scholz D, Schaper J, Schaper W. Monocyte chemotactic protein-1 increases collateral and peripheral conductance after femoral artery occlusion. Circ Res. 1997;80:829–837. doi: 10.1161/01.res.80.6.829. [DOI] [PubMed] [Google Scholar]
- 31.Hirsch E, Ciraolo E, Ghigo A, Costa C. Taming the PI3K team to hold inflammation and cancer at bay. Pharmacol Ther. 2008;118:192–205. doi: 10.1016/j.pharmthera.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 32.Liang J, Slingerland JM. Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle. 2003;2:339–345. [PubMed] [Google Scholar]
- 33.Lloyd-Jones D, Adams R, Carnethon M, De Simone G, Ferguson TB, Flegal K, Ford E, Furie K, Go A, Greenlund K, Haase N, Hailpern S, Ho M, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott M, Meigs J, Mozaffarian D, Nichol G, O’Donnell C, Roger V, Rosamond W, Sacco R, Sorlie P, Stafford R, Steinberger J, Thom T, Wasserthiel-Smoller S, Wong N, Wylie-Rosett J, Hong Y. Heart disease and stroke statistics–2009 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2009;119:e21–181. doi: 10.1161/CIRCULATIONAHA.108.191261. [DOI] [PubMed] [Google Scholar]
- 34.Naga Prasad SV, Perrino C, Rockman HA. Role of phosphoinositide 3-kinase in cardiac function and heart failure. Trends Cardiovasc Med. 2003;13:206–212. doi: 10.1016/s1050-1738(03)00080-x. [DOI] [PubMed] [Google Scholar]
- 35.Tong H, Imahashi K, Steenbergen C, Murphy E. Phosphorylation of glycogen synthase kinase-3beta during preconditioning through a phosphatidylinositol-3-kinase-dependent pathway is cardioprotective. Circ Res. 2002;90:377–379. doi: 10.1161/01.res.0000012567.95445.55. [DOI] [PubMed] [Google Scholar]
- 36.Doukas J, Wrasidlo W, Noronha G, Dneprovskaia E, Fine R, Weis S, Hood J, Demaria A, Soll R, Cheresh D. Phosphoinositide 3-kinase gamma/delta inhibition limits infarct size after myocardial ischemia/reperfusion injury. Proc Natl Acad Sci U S A. 2006;103:19866–19871. doi: 10.1073/pnas.0606956103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sasaki T, Irie-Sasaki J, Jones RG, Oliveira-dos-Santos AJ, Stanford WL, Bolon B, Wakeham A, Itie A, Bouchard D, Kozieradzki I, Joza N, Mak TW, Ohashi PS, Suzuki A, Penninger JM. Function of PI3Kgamma in thymocyte development, T cell activation, and neutrophil migration. Science. 2000;287:1040–1046. doi: 10.1126/science.287.5455.1040. [DOI] [PubMed] [Google Scholar]
- 38.Jones GE, Prigmore E, Calvez R, Hogan C, Dunn GA, Hirsch E, Wymann MP, Ridley AJ. Requirement for PI 3-kinase gamma in macrophage migration to MCP-1 and CSF-1. Exp Cell Res. 2003;290:120–131. doi: 10.1016/s0014-4827(03)00318-5. [DOI] [PubMed] [Google Scholar]
- 39.Reif K, Okkenhaug K, Sasaki T, Penninger JM, Vanhaesebroeck B, Cyster JG. Cutting edge: differential roles for phosphoinositide 3-kinases, p110gamma and p110delta, in lymphocyte chemotaxis and homing. J Immunol. 2004;173:2236–2240. doi: 10.4049/jimmunol.173.4.2236. [DOI] [PubMed] [Google Scholar]
- 40.Frantz S, Bauersachs J, Ertl G. Post-infarct remodelling: contribution of wound healing and inflammation. Cardiovasc Res. 2009;81:474–481. doi: 10.1093/cvr/cvn292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krankel N, Katare RG, Siragusa M, Barcelos LS, Campagnolo P, Mangialardi G, Fortunato O, Spinetti G, Tran N, Zacharowski K, Wojakowski W, Mroz I, Herman A, Manning Fox JE, MacDonald PE, Schanstra JP, Bascands JL, Ascione R, Angelini G, Emanueli C, Madeddu P. Role of kinin B2 receptor signaling in the recruitment of circulating progenitor cells with neovascularization potential. Circ Res. 2008;103:1335–1343. doi: 10.1161/CIRCRESAHA.108.179952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stone OA, Richer C, Emanueli C, van Weel V, Quax PH, Katare R, Kraenkel N, Campagnolo P, Barcelos LS, Siragusa M, Sala-Newby GB, Baldessari D, Mione M, Vincent MP, Benest AV, Al Haj Zen A, Gonzalez J, Bates DO, Alhenc-Gelas F, Madeddu P. Critical role of tissue kallikrein in vessel formation and maturation: implications for therapeutic revascularization. Arterioscler Thromb Vasc Biol. 2009;29:657–664. doi: 10.1161/ATVBAHA.108.182139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leineweber K, Bohm M, Heusch G. Cyclic adenosine monophosphate in acute myocardial infarction with heart failure: slayer or savior? Circulation. 2006;114:365–367. doi: 10.1161/CIRCULATIONAHA.106.642132. [DOI] [PubMed] [Google Scholar]
- 44.Vanhaesebroeck B, Rohn JL, Waterfield MD. Gene targeting: attention to detail. Cell. 2004;118:274–276. doi: 10.1016/j.cell.2004.07.018. [DOI] [PubMed] [Google Scholar]