

Abstract
Transcriptional upregulation of Kcc2b, the gene variant encoding the major isoform of the KCC2 chloride transporter, underlies a rapid perinatal decrease in intraneuronal chloride concentration (chloride shift), which is necessary for GABA to act inhibitory. Here we identify a novel repressor element-1 (RE-1) site in the 5′ regulatory region of Kcc2b. In primary cortical neurons, which recapitulate the chloride shift in culture, the novel upstream RE-1 together with a known intronic RE-1 site function in concerted interaction to suppress Kcc2b transcription. With critical relevance for the chloride shift, only in the presence of the dual RE-1 site could inhibition of REST upregulate Kcc2b transcription. For this, we confirmed increased KCC2 protein expression and decreased intraneuronal chloride. Kcc2b developmental upregulation was potentiated by BDNF application, which was fully dependent on the presence of dual RE-1. In addition, the developmental chloride shift and GABA switch, from excitatory to inhibitory action, was accelerated by REST inhibition and slowed by REST overexpression. These results identify the REST–dual RE-1 interaction as a novel mechanism of transcriptional Kcc2b upregulation that significantly contributes to the ontogenetic shift in chloride concentration and GABA action in cortical neurons, which is fundamental for brain function in health and disease. Thus, we present here a new logic for the perinatal chloride shift, which is critical for establishment of GABAergic cortical inhibitory neurotransmission.
Introduction
Intraneuronal chloride concentration ([Cl−]i) determines the polarity and efficacy of synaptic transmission mediated by ligand-gated Cl− channels such as GABAA receptors (GABAARs). Potassium chloride cotransporter 2 (Slc12a5, Kcc2) is expressed almost exclusively in CNS neurons (Payne et al., 1996) and plays a major role in neuronal Cl− homeostasis by extruding Cl−, thereby maintaining a low neuronal [Cl−]i. The critical importance of KCC2 is indicated by the perinatal death of Kcc2−/− mice (Lu et al., 1999; Hübner et al., 2001; Delpire and Mount, 2002; Tornberg et al., 2005).
In developing neurons, suppression of KCC2 expression, along with robust expression of NKCC1, which accumulates intracellular Cl−, results in high [Cl−]i. As a result of the outward electrochemical Cl− gradient, GABAAR activation leads to depolarization. Developmentally, NKCC1 decreases and KCC2 increases, producing lower [Cl−]i that yields hyperpolarizing GABA responses (Lu et al., 1999; Ben-Ari, 2002; Fiumelli and Woodin, 2007; Galanopoulou, 2007), which coincides with the GABA switch (Sernagor et al., 2003). KCC2 expression is reduced in several pathological states, including chronic pain (Coull et al., 2003; Morales-Aza et al., 2004; Zhang et al., 2008), epilepsy (Huberfeld et al., 2007; Munakata et al., 2007; Muñoz et al., 2007), traumatic brain injury (Bonislawski et al., 2007), and even constraint stress (Hewitt et al., 2009).
The murine Kcc2 gene leads to transcription of two isoforms, with the major isoform (Kcc2b) contributing ∼90% expression in mature animals' cortex (Uvarov et al., 2007, 2009). It is not yet clear how expression of Kcc2 is regulated. One study (Ganguly et al., 2001) reports that perinatal Kcc2b increase depends on neuronal activity and the depolarizing actions of GABAAR, a result not fully confirmed by another study (Ludwig et al., 2003). Another study (Liu et al., 2006) reports that calcium influx through nicotinic acetylcholine receptors promotes the GABA switch. However, transcriptional mechanisms underlying regulation of Kcc2b are mostly elusive. To address this, reporter gene methodology has been used. A 21 bp repressor element-1 (RE-1) within the first intron of murine Kcc2b [position +378 relative to transcriptional start site (TSS)] can repress Kcc2 in a cell line (Karadsheh and Delpire, 2001). This finding prompted the generation of transgenic mice with reporter genes under the control of Kcc2b (Uvarov et al., 2005). In these animals, reporter activity was found almost exclusively in the CNS in a pattern similar to that of endogenous Kcc2b. This in vivo study did not address whether the intronic RE-1 plays a role in the perinatal chloride shift. Other studies reported that an Egr element (−226 relative to TSS) and an E-box element (−321 relative to TSS) can upregulate Kcc2 (Uvarov et al., 2006; Markkanen et al., 2008). However, it is not known whether these enhancer mechanisms are critical for the perinatal chloride shift.
The 21 bp RE-1 binds to the REST transcriptional repressor complex and silences a large network of genes by recruiting corepressor proteins (Lunyak and Rosenfeld, 2005; Yeo et al., 2005). Because RE-1 was initially found only in neuronal genes, it was assumed to be a master regulator of neurogenesis (Kraner et al., 1992; Mori et al., 1992; Chong et al., 1995; Schoenherr and Anderson, 1995) but subsequently has been implicated in more complex gene regulation (Ballas and Mandel, 2005; Lunyak and Rosenfeld, 2005; Ooi and Wood, 2007). REST expression also has been found to be altered in epilepsy and ischemia (Palm et al., 1998; Calderone et al., 2003; Garriga-Canut et al., 2006).
In this study, we describe a novel RE-1 located in the 5′ promoter region (−1702/−1682 for the murine gene) of Kcc2b, which acts in concert with intronic RE-1 to repress Kcc2b. In rat primary cortical neurons, only in the presence of “dual RE-1” does REST inhibition lead to upregulation of Kcc2b. Furthermore, developmental regulation of Kcc2b, including its enhancement by BDNF, neuronal [Cl−]i, and GABA action all depend on REST.
Materials and Methods
Additional description of methods is provided in the supplemental data (available at www.jneurosci.org as supplemental material).
Generation of mouse Kcc2 reporter gene constructs and bioinformatics analysis.
A 3.0 kb Kcc2 genomic DNA fragment containing 5′ regulatory region, exon 1 and part of intron 1 of Kcc2b, was PCR amplified from a bacterial artificial chromosome. The PCR product was cloned into pGlucBasic (New England Biolabs), sequenced, and used as a starting template for generating other deletion constructs, which were all verified by sequencing. Mouse, rat, human, and chimpanzee Kcc2 (alias Slc12a5) genomic sequences were aligned using the ClustalW program (Chenna et al., 2003). Putative transcription factor binding sites within 3.0 kb were identified by using MotifScanner in the Toucan platform version 3.0.2 (Aerts et al., 2003).
Primary cultures of embryonic rat cortical neurons.
Rat primary cortical neurons were prepared as described previously (Szatmari et al., 2007). Briefly, cortices were microdissected from Sprague Dawley rat embryos at 17–18 d of gestation. The tissue was dissociated using papain, followed by mechanical dissociation. Cytosine arabinoside (2.5 μm) was added to cultures on the second day after seeding [2 d in vitro (DIV)] to inhibit the proliferation of non-neuronal cells. Cell suspension was plated at a density of 1 × 106 cells/ml onto tissue-culture dishes coated with poly-d-lysine. Cortical neuronal culture prepared by this method yielded a majority population of neuronal cells, with negligible glia contamination, as evidenced by the absence of GFAP by Western blotting (data not shown). For imaging experiments, cortical neurons were plated onto 12-mm-diameter poly-d-lysine-coated glass coverslips. Neuronal viability and differentiation were ascertained microscopically before experiments. All procedures used in this study were performed following the guidelines of the Animal Care and Use Committee of Duke University and under a valid institutional animal protocol.
Transfection (using 0.4 μg of reporter plasmid and 50 ng of pSEAP plasmid per 24-well) of primary neurons was performed using Amaxa Nucleofactor kit according to the instructions of the manufacturer, leading to a 30–60% transfection efficiency, assessed by use of fluorescent reporters (data not shown).
Gaussia luciferase assay and normalization.
Promoter constructs were cloned into pGLuc Basic vector, coding for secreted Gaussia luciferase, which has been codon optimized for mammalian gene expression. Secreted luciferase activity was measured with a Gaussia Luciferase Assay kit (New England Biolabs) according to the instructions of the manufacturer. Veritas microplate luminometer was used to measure luminescence; typically 30 μl of cell culture medium (from 24-well tissue-culture dish) was used. Coelantarazine substrate (25 μl) was injected per well.
Secreted alkaline phosphatase plasmid was cotransfected in all experiments, and activity was used as normalization for transfection. Assays were conducted in triplicate, with three independent experiments per approach.
Immunocytochemistry and morphometry of labeled cells.
Cells grown on coverslips were fixed in 4% paraformaldehyde, and unspecific antibody binding sites were blocked using 2.5% goat serum/Tris-buffered saline. Rabbit anti-NKCC1 (Alpha Diagnostics) and rabbit anti-KCC2 antibody (Abcam) were used at 1:600 dilution, followed by goat anti-rabbit IgG heavy and light chains conjugated to the fluorophore, Alexa Fluor 594 (diluted 1:800) (Invitrogen). Omission of primary antibodies was used as a negative control. Coverslips were viewed using an Olympus BX60 upright microscope, equipped with 1200 W xenon illumination and appropriate filter sets.
Image analysis was performed using NIH ImageJ freeware of captured frames with constant exposure parameters. Regions of interest were customized for each cell, and mean density was measured and background corrected.
Ratiometric chloride imaging using Clomeleon.
We essentially followed methodology described previously (Kuner and Augustine, 2000). In brief, a Clomeleon expression plasmid was transfected into primary cortical neurons by Amaxa electroporation (see above). Transfection efficiency was again at ≥30% efficiency, transfected neurons were verified by yellow fluorescent protein (YFP) fluorescence, and ratiometric images (excitation at λ = 434 nm, dual emission at λ = 485 and 535 nm; for resting chloride, six stable frames at a rate 12 of per minute were captured, which were averaged) were acquired, again using RATIOTOOL. Calibration of Clomeleon signals (535 nm/485 nm emission ratio) was performed by using tributyltin-nigericin to establish a standard curve (Pond et al., 2006), which was then normalized for measured intraneuronal pH to take into account the pH sensitivity of Clomeleon (supplemental Fig. S2A–C, available at www.jneurosci.org as supplemental material). All [Cl−]i are provided as mean ± SEM.
Ratiometric Ca2+ imaging using fura-2.
We followed previously described procedures. In brief, to load the dye, primary neurons plated on coverslips were incubated for 30 min at room temperature with 5 μm fura-2 AM in external solution containing the following (in mm): 134 NaCl, 6 KCl, 1.2 MgCl2, 2.5 CaCl2, 10 glucose, and 10 HEPES, pH 7.4. After dye loading, neurons were washed and allowed to de-esterify for 30 min. Coverslips with fura-2-loaded neurons were then placed in a perfusion chamber that was mounted on the stage of the Olympus BX60 upright microscope. Loaded neurons were imaged via a 40×/0.8 numerical aperture water-immersion objective (Olympus). Fluorescence of individual neurons, identified by cotransfection with fluorescent reporters [copepod-green fluorescent protein (GFP)], was determined at 340/380 nm excitation and 510 nm emission, and the time course of the ratio 340/380 was sampled at 0.33 Hz using RATIOTOOL software (ISee Imaging). Ca2+ imaging experiments were performed at room temperature (22°C). Neurons were pretreated by blocking glutamatergic receptors using CNQX (20 μm) and d-AP-5 (25 μm) before exposure to GABA (100 μm) or muscimol (10 μm). For controls, GABAARs were blocked with bicuculline/picrotoxin (10 μm/50 μm), and L-type voltage-gated Ca2+ channels were blocked with nifedipine (20 μm). A positive Ca2+ response to GABAAR stimulation was defined as ΔR ≥ 20% (within four frames) of the stable prestimulus ratio (R0).
Chromatin immunoprecipitation.
Chromatin immunoprecipitation (ChIP) was performed according to previously described methods (Spencer et al., 2003). Approximately 0.7 × 106 N2A neuronal cells were used for each ChIP experiment. Cells were crosslinked with 1% formaldehyde for 30 min, washed twice with cold PBS, resuspended in lysis buffer [1%SDS, 10 mm EDTA, and 50 mm Tris-HCl, pH 8.0, with protease inhibitor cocktail (Roche)] and sonicated for 15 s pulses. The lysates were clarified by centrifugation at 10,000 rpm for 10 min at 4°C in a microcentrifuge. One-tenth of the total lysate was used as input control of genomic DNA. Supernatants were collected and diluted in buffer (1% Triton X-100, 2 mm EDTA, 150 mm NaCl, 20 mm Tris-HCl, pH 8.0, and protease inhibitor cocktail), followed by immunoclearing with 1 mg of salmon sperm DNA, 10 ml of rabbit IgG, and 20 ml of protein A/G-Sepharose (Santa Cruz Biotechnology) for 1 h at 4°C. Immunoprecipitation was performed overnight at 4°C with 2 mg of each specific antibody. Precipitates were washed sequentially for 10 min each in TSE1 buffer (0.1% SDS, 1% Triton X-100, 2 mm EDTA, 150 mm NaCl, and 20 mm Tris-HCl, pH 8.0), TSE2 (TSE1 with 500 mm NaCl), and TSE3 (0.25 m LiCl, 1% NP-40, 1% deoxycholate, 1 mm EDTA, and 10 mm Tris-HCl, pH 8.0). Precipitates were then washed twice with 10 mm Tris/0.1 mm EDTA, pH 7.8 and extracted with 1% SDS containing 0.1 m NaHCO3. Eluates were pooled and heated at 65°C for 6 h to reverse formaldehyde crosslinking. DNA fragments were purified with Qiagen Qiaquick spin kit. For ChIP PCR, 1 μl of a 25 μl DNA extraction was used.
Data analysis.
Data are presented as mean ± SEM, and statistical significance was tested with the two-tailed t test, by ANOVA with post hoc Dunnett's test or by χ2 test.
Reagents.
The compounds were as follows: BDNF (25 ng/ml; R & D Systems), bicuculline (10 μm; tocris Bioscience), bumetanide (30 μm; Sigma), CNQX (20 μm; Tocris Bioscience), d-AP-5 (50 μm; Tocris Bioscience), fura-2 (5 μm; Invitrogen), furosemide (100 μm; Sigma), GABA (100 μm; Sigma), muscimol (10 μm; Sigma), nifedipine (20 μm; Sigma), picrotoxin (50 μm; Tocris Bioscience), SNARF-5F (2 μm; Invitrogen), and trichostatin-A (10 ng/ml; Sigma).
Sequences and ENSEMBL gene ID numbers.
The sequences used were as follows: rat Kcc2, ENSRNOG00000018111; mouse Kcc2, ENSMUSG00000017740; human KCC2, ENSG00000124140; chimpanzee Kcc2, ENSPTRG0000001 3578; pufferfish Kcc2, ENSTRUG00000018315.
Results
A novel RE-1 is located upstream of the mammalian Kcc2b transcriptional start site
Most active promoters are located within 2.5 kb of their respective TSS (Kim et al., 2005). To better understand transcriptional regulation of Kcc2b, we first used a cis-regulatory element discovery platform (Toucan) to analyze the 2.5 kb fragment surrounding the TSS of mouse Kcc2b. The predicted transcription factor binding sites are depicted in Figure 1. Our analysis identified three previously identified regulatory elements—an Egr binding site at position −226/−203, an E-box binding site at position −321/−314, and an intronic RE-1 at position +378/+398—validating our approach. In addition, a novel RE-1 site was identified at position −1702/−1682 upstream of the TSS (Fig. 1). Alignment of several mammalian genomic Kcc2 sequences revealed a high degree of conservation, with respect to upstream and intronic RE-1, in terms of position as well as sequence (Fig. 2). To determine whether this arrangement of Kcc2 RE-1 sites was similar in nonmammalian vertebrates, we also used Toucan to analyze the Kcc2 sequence of the pufferfish, Takifugu rubripes. The Takifugu genome is “compressed” and characterized by minimal-sized exons and regulatory sequences. As was the case in mammals, a Kcc2 intronic RE-1 was identified in the pufferfish. In addition, a noncanonical upstream RE-1 sequence was evident at position −964 (Fig. 2A), suggesting that the novel mammalian upstream RE-1 site we have identified is phylogenetically more recent than its intronic partner.
Figure 1.
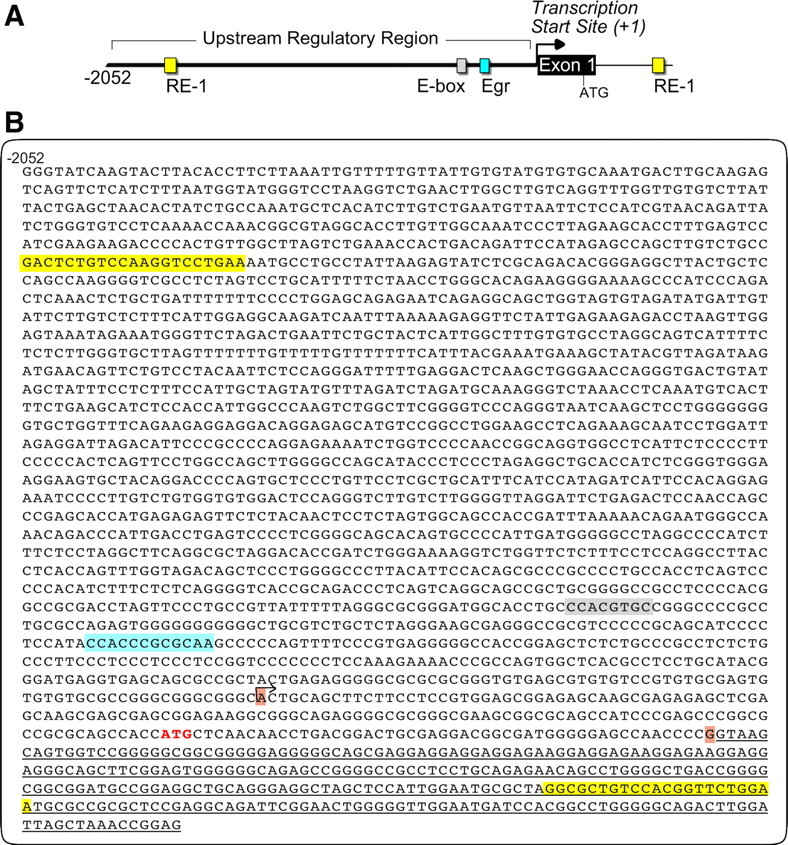
Regulatory DNA sequences surrounding the TSS of mouse Kcc2b (major isoform). A, Schematic of 2.5 kb mouse Kcc2b gene cloned 5′ of a secreted luciferase reporter construct. B, Single-stranded (forward orientation) DNA sequence of the mouse Kcc2b gene depicting upstream regulatory region, exon 1 and part of intron 1. The two RE-1 sites are highlighted yellow. Egr and E-box elements are highlighted gray and turquoise. Horizontal arrow indicates the first nucleotide (position +1, TSS). Nucleotides shaded brown delineate the first exon, and translation initiator ATG is colored in red. Underlined nucleotides denote partial first intron sequence.
Figure 2.
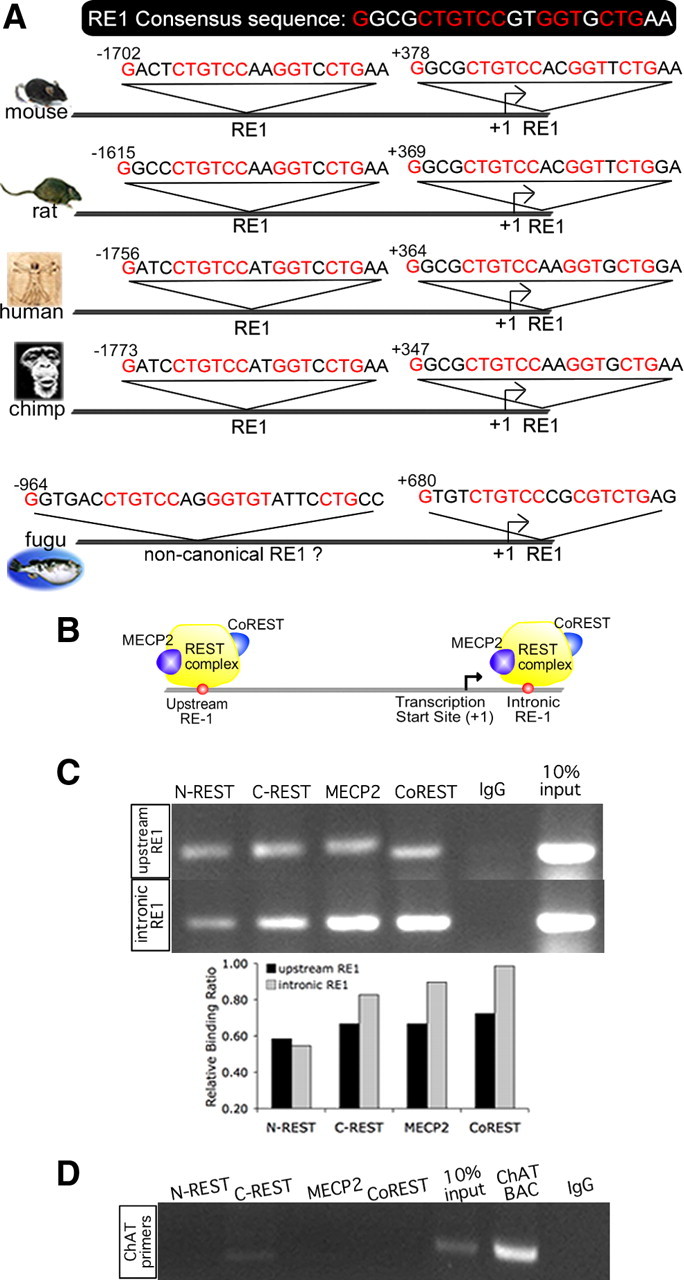
Conservation of sequence and position of a novel RE-1 site located upstream of the TSS of mouse Kcc2b; upstream and intronic RE-1 bind to REST complex. A, Upstream and intronic RE-1 sites of mammalian species (mouse, rat, human, and chimpanzee) are highly similar in sequence to consensus RE-1 (Schoenherr et al., 1996). Positions of the RE-1 sequences are indicated relative to TSS, showing a preservation of position for the dual RE-1 in mammals as well. Predicted RE-1 sites of a nonmammalian vertebrate, the pufferfish Takifugu rubripes, are also shown, indicating a canonical intronic RE-1 and a noncanonical upstream site. B, Schematic showing REST complex–RE-1 binding interaction at the Kcc2b promoter. C, ChIP was conducted using N2A neuronal cells and antibodies against N- and C-REST and corepressor proteins MECP2 and Co-REST. Immunoprecipitated DNA was PCR amplified using primers that flank the upstream and intronic RE-1 sites. Bar diagram depicts results of band densitometry. D, Results of the control ChIP PCR are depicted, using primer sequences specific for choline acetyltransferase, a gene without RE-1 sites.
Upstream and intronic RE-1 bind the REST complex
Based on these findings, we next addressed whether REST binds to both of the RE-1 elements within Kcc2. REST negatively regulates gene transcription primarily by recruiting corepressor proteins to two different repressor domains. One of these proteins, Co-REST, is associated with the C-terminus domain of REST, whereas another protein, MECP2, binds to a complex associated with the N terminus (Fig. 2B). To determine whether these proteins interact with the RE-1 elements surrounding the TSS of Kcc2b, we performed a series of ChIP assays in N2A neuronal cells, using specific antibodies against REST, MECP2, and Co-REST. We selected this cell line because the similarity of Kcc2b regulation by REST between N2A permanent neuronal cells and rat primary cortical neurons, demonstrated in the next section, validated our choice of N2A, which allows these assays to be conducted on a robust amount of starting material.
Both upstream and intronic RE-1 bind to N- and C-REST, as well as to the corepressors MECP2 and Co-REST (Fig. 2C). After normalizing the ChIP PCR signal for variable amounts of input, the relative binding of MECP2 and Co-REST to the intronic RE-1 appeared higher than that measured for the upstream RE-1. This suggests a tighter association of REST and corepressors with intronic RE-1. For control, we amplified choline acetyltransferase, which does not contain RE-1 sites, yielding a negative signal, indicating specific binding of REST and its corepressors to the RE-1 sites within Kcc2b (Fig. 2D). Thus, we conclude that the REST complex binds to both RE-1 sites of Kcc2b, with intronic RE-1 possibly binding REST with higher affinity.
Concerted interaction between both RE-1 elements and REST complex regulates Kcc2b promoter activity
Having demonstrated binding of REST and its corepressors to the Kcc2 RE-1 sites, we next addressed the role of the REST–RE-1 interaction in transcriptional regulation of Kcc2b. For this purpose, we generated six different luciferase reporter constructs to probe the impact of Kcc2b RE-1 sites and their flanking DNA sequence on Kcc2 transcriptional regulation (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Promoter gene constructs drove secreted Gaussia luciferase (Tannous et al., 2005). Measuring secreted luciferase is a feasible way to monitor increased levels of gene expression, because it avoids intracellular reporter accumulation.
Our data from permanent N2A neuronal cells—robust regulation of Kcc2b promoter via concerted repressive action of dual RE-1, functioning of the novel upstream RE-1 site as a strong repressor (more explicitly referred to in supplemental data, Fig. S1B, available at www.jneurosci.org as supplemental material)—prompted us to study RE-1 regulation of Kcc2b promoter activity in cortical neurons, in which the striking developmental upregulation of Kcc2b leads to a shift in chloride concentration and GABA action. Results from cultured rat embryonic day 17 cortical neurons were found in agreement. The findings suggest that the intronic RE-1 is sufficient to repress the Kcc2b promoter (supplemental Fig. S1C, lane 2, available at www.jneurosci.org as supplemental material), whereas the novel upstream RE-1 by itself can act as repressor site as well (supplemental Fig. S1C, lanes 3 and 4 vs 5, available at www.jneurosci.org as supplemental material); for more in-depth analysis, see also supplemental data file (available at www.jneurosci.org as supplemental material). In aggregate, results from both primary neurons and permanent cell lines indicate that the novel upstream RE-1 site functions in concerted interaction with its intronic partner to repress Kcc2b promoter activity.
Altering REST regulates Kcc2 promoter activity via dual RE-1 sites
Given that the REST complex binds both RE-1 sites and that the dual RE-1 sites repress Kcc2b promoter activity by concerted interaction, we next asked whether REST inhibition upregulated Kcc2b activity, which would imply this regulation as possibly contributory to the ontogenetic perinatal chloride shift in forebrain neurons. To address this question, we measured Kcc2b reporter gene activity while inhibiting REST genetically and chemically and, as a control, while increasing REST function by overexpression. For this purpose, we cotransfected primary cortical neurons with full-length REST (FL-REST), dominant-negative REST (dn-REST) (Chen et al., 1998), or control plasmid, together with reporter plasmids that contained no, one, or both RE-1 sites, and assessed reporter activity on DIV6. We also chemically inhibited REST with trichostatin-A, a potent pan-histone deacetylase (HDAC) inhibitor that disrupts the REST complex by inhibiting HDAC1 in primary cortical neurons (Huang et al., 1999).
Figure 3A shows that these changes in REST altered Kcc2b reporter activities of transfected primary cortical neurons. Activity measured with the dual RE-1 construct (−2052/+483) (Fig. 3A, lane 1, left bar), without REST modulation, was used as reference with a relative value of 1. Importantly, inhibition of REST, both genetically and chemically, caused striking upregulation of Kcc2b promoter activity only in the case when the dual RE-1 sites were included in the promoter (first four lanes, two right bars colored in green). In addition, REST overexpression caused additional robust repression of the dual RE-1 construct, although this construct already was repressed substantially by endogenous REST (first four lanes, red bar). For all other constructs, there was a complete lack of upregulation in response to any REST manipulation. Furthermore, the magnitude of responses to REST modulation was blunted in the absence of the novel upstream RE-1 (second four lanes). As expected, in the absence of both RE-1 sites, there was barely regulation, with generally derepressed reporter activity (fourth four lanes). Thus, dual RE-1 is absolutely necessary for Kccb2 promoter activity to be derepressed in response to inhibition of REST. There was transcriptional derepression only in the presence of dual RE-1 together with inhibition of REST.
Figure 3.

Luciferase reporter activity and regulation of Kcc2b in primary cortical neurons by gain-of-function and loss-of-function of REST. A, Effects of FL-REST, dn-REST, and the HDAC inhibitor trichostatin-A on reporter gene expression of the dual (2 red dots), single (1 red dot), and no RE-1 constructs. Normalized secreted Gaussia luciferase activity was compared with the activity of strongly repressed dual RE-1 construct (lane 1) assigned a value of 1.0. Note the presence of significant upregulation only in the presence of dual RE-1. Triplicate measurements were conducted in at least 3 independent experiments, and error bars represent SEM. B, Schematic rendering of the effect of REST inhibition versus overexpression on transcriptional activation of Kcc2b.
Thus, in rat primary cortical neurons, only the dual RE-1 promoter produced an upregulation of Kcc2 gene expression in response to REST inhibition. Because ontogenetic upregulation of Kcc2 is a critical element of the perinatal chloride shift, we propose that the dual RE-1 repressor sites in Kcc2 regulatory DNA sequences underlie the developmental chloride shift by mediating derepression in response to decreasing levels of REST (for conceptualization, see Fig. 3B).
Regulation of neuronal chloride depends on REST
We next asked whether transcriptional regulation of Kcc2b by the REST–RE-1 interaction translates into corresponding regulation of KCC2 protein and [Cl−]i. We inhibited REST–RE-1 binding in primary cortical neurons by expressing the dn-REST transgene and increased it by overexpressing FL-REST. KCC2 levels were determined by immunocytochemistry, whereas the fluorescent chloride indicator Clomeleon (Kuner and Augustine, 2000) was used to image [Cl−]i within cotransfected neurons.
Primary neurons were cotransfected with copepod-GFP, together with either FL-REST, dn-REST, or vector control, and subsequently immunolabeled for KCC2 at DIV7. KCC2 protein was most abundant in neurons expressing dn-REST, intermediate in controls, and lowest in FL-REST (Fig. 4). Figure 4 also shows that KCC2 is localized to the periphery of the soma, suggesting a plasma membrane location conducive to KCC2 chloride transporter function. Quantification of relative KCC2 abundance in cotransfected neurons indicated that these changes between groups were highly significant (Fig. 5A) (*p < 0.01, ANOVA with post hoc Dunnett's test).
Figure 4.

Functional KCC2 expression is regulated by REST: immunocytochemistry. Primary cortical neurons were cotransfected with copepod-GFP and FL-REST, dn-REST, or vector control, followed by immunostaining at DIV7. KCC2 immunoreactivity appears in red (left column), copepod-GFP in green (middle column), and merged (right column). Inset is a magnified view of KCC2 immunoreactive neurons, showing an increase of labeling intensity toward the edge of the soma, indicating transmembrane staining.
Figure 5.

Functional KCC2 expression is regulated by REST: quantitative immunocytochemistry, chloride measurements, and developmental chloride shift. A, Morphometry of copepod-GFP-positive neurons confirms robust upregulation of KCC2 by dn-REST and downregulation by FL-REST. *p < 0.01 versus control (ANOVA). Number of neurons analyzed: n = 95 (dn-REST), n = 64 (control), and n = 72 (FL-REST). B, Clomeleon-transfected neurons are shown: fluorescent micrograph in the bottom row and ratiometric pseudo images in the top row. Note the difference between high Clomeleon emission ratio (= low [Cl−]i) in dn-REST cotransfected neurons (pink pseudo image; see color scale on the right) versus the significantly lower ratio (= high [Cl−]i) in control and FL-REST cotransfected neurons (orange and yellow pseudo images). C, Measurement of neuronal [Cl−]i by cotransfection of the ratiometric chloride indicator Clomeleon together with dn-REST, FL-REST, and vector control are shown, indicating the robust downregulation of chloride by dn-REST and upregulation by FL-REST. Three independent Clomeleon experiments (DIV6) were conducted, n ≥ 18 per group per experiment; data were corrected for pH (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). *p < 0.01 (ANOVA). D, The effect of 100 μm furosemide on primary cortical neurons (DIV5), transfected with dn-REST versus control is shown, indicating the reversal to high [Cl−]i for both conditions. Note the steep change in dn-REST cotransfected neurons versus a modest increase in controls. Right-hand y-axis depicts p values of the difference between dn-REST- and control-transfected neurons; green line, p < 0.05. Both conditions finally equilibrate to identical levels of high [Cl−]i at the 12 min time point; n = 10 neurons per condition.
We next addressed whether these REST-induced changes in KCC2 abundance affected [Cl−]i. To address this question, we took advantage of the fluorescent chloride indicator Clomeleon, which is based on fluorescence resonance energy transfer between the chloride-insensitive cyan fluorescent protein (emission at λ = 485 nm) and the chloride-sensitive YFP (emission at λ = 535 nm), to ratiometrically measure [Cl−]i (supplemental Fig. S2A, available at www.jneurosci.org as supplemental material). Because Clomeleon is sensitive to pH (Kuner and Augustine, 2000) and gene regulation by REST and/or [Cl−]i may affect pH, we determined whether upregulation or downregulation of REST influenced intracellular pH. By imaging primary neurons loaded with the pH-sensitive ratiometric dye SNARF-5F (supplemental Fig. S2B, available at www.jneurosci.org as supplemental material), we observed a mild alkalinization at DIV3 and a subtle acidification for FL-REST and dn-REST at DIV6 compared with control vector (supplemental Fig. S2C, available at www.jneurosci.org as supplemental material). This result indicates regulation of intracellular pH by REST, a subject worthy of future analysis. These values of pH were taken into account when converting Clomeleon signals (E535 nm/E485 nm) into [Cl−]i.
Fluorescence emission ratios were measured in neurons cotransfected with Clomeleon and FL-REST or dn-REST at DIV7 (Fig. 5B). We noted a robust reduction of [Cl−]i from 25 to 18 mm for dn-REST and increase of [Cl−]i to 36 mm for FL-REST (Fig. 5C) (*p < 0.01, ANOVA post hoc Dunnett's test). Figure 5, A and C, suggests an inverse relationship between [Cl−]i and KCC2 protein expression. Thus, genetically encoded gain-of-function and loss-of-function of REST evoke significant upregulation and downregulation of intraneuronal chloride. To confirm that downregulation of [Cl−]i in the presence of dn-REST was caused by upregulation of KCC2, we treated neurons (DIV5) with 100 μm furosemide to chemically inhibit KCC2 transporter function, assuming a modest impact of NKCC1 at that developmental time point. Furosemide treatment caused a strong increase in [Cl−]i in the dn-REST-expressing neurons, in contrast to a significantly blunted response in neurons transfected with a control construct (Fig. 5D). The final [Cl−]i in both treatment groups did not differ, as it did approximate [Cl−]o. This disproportionate influence of furosemide strongly suggests that lower resting [Cl−]i in dn-REST-transfected neurons is caused by increased KCC2 chloride transporter expression.
We also used similar methodology to ask whether NKCC1 protein expression and function were altered as a result of REST regulation. NKCC1 does not harbor RE-1 sites in its regulatory DNA sequences and would be expected to be unaffected, at least directly, by our REST manipulations. In keeping with this reasoning, there was no significant difference in NKCC1 expression levels between groups, with NKCC1 expression seemingly low as judged by its modest immunoreactivity and lack of [Cl−]i response to 30 μm bumetanide (supplemental Fig. S3A,B, available at www.jneurosci.org as supplemental material). Thus, alteration of REST by gain-of-function or loss-of-function constructs led to significant downregulation or upregulation, respectively, of KCC2 protein in cultured neurons at DIV7. These effects occurred in the absence of changes in NKCC1.
In summary, genetically encoded inhibition of REST led to an increase of KCC2 protein abundance in primary neurons at approximately DIV7, which in turn led to a decrease of neuronal [Cl−]i. Opposite effects were observed for expression of FL-REST.
Developmental chloride shift and its acceleration by BDNF depend on REST
If the dual RE-1 repressor sites in Kcc2 regulatory DNA sequence underlie the developmental chloride shift, then Kcc2b expression should be derepressed in response to decreasing levels of REST–RE-1 binding over development. We confirmed increasing levels of KCC2 and decreasing levels of REST in primary cortical neurons (Fig. 6A,B) (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). This finding prompted us to look at Kcc2b transcriptional regulation as a function of maturation. For this purpose, cultured primary cortical neurons were transfected with either the dual RE-1 (−2052/+483) reporter gene construct, which conferred maximum repression by REST, or the construct (−1649/+49) lacking both RE-1 sites, which showed maximum derepression and complete lack of regulation by REST. The fact that the secreted reporter protein Gaussia luciferase provided sensitive assessment of reporter gene expression without the need for cell lysis allowed us to measure reporter expression over multiple time points (Fig. 6C). For both reporter promoters, the known ontogenetic upregulation of Kcc2 could be reflected in luciferase activity, reaching a plateau at approximately DIV8. However, the construct without RE-1 sites yielded 10-fold higher reporter levels compared with the dual-RE-1 construct, with reporter activity significantly different between these two groups at all time points (p < 0.001, t test). In summary, these findings indicate a powerful repressive effect mediated by the dual RE-1 site on developmental upregulation of Kcc2b, although developmental upregulation of Kcc2b transcription and subsequent plateauing does not depend on dual RE-1.
Figure 6.

Developmental regulation of Kcc2 gene expression depends on RE-1. A, Morphometry for REST immunoreactivity at different developmental time points. Number of neurons measured: n = 48 for each dn-REST, control, and FL-REST. B, Morphometry for KCC2 immunoreactivity at different developmental time points illustrates upregulation of KCC2 and downregulation of REST. Differences between subsequent time points were statistically highly significantly different (p < 0.001, ANOVA with post hoc Dunnett's test). n as given in A for each time point; the same neurons were recruited for REST and KCC2 densitometry. Note the inverse, nonlinear relation between time courses in A versus B. C, Cultured neurons were cotransfected with Kcc2b promoter constructs containing either dual RE-1 or no RE-1. Secreted Gaussia luciferase was monitored daily. Note the steady increase up to DIV8 for both constructs, also the striking 10-fold derepression in the absence of RE-1 sites (−1649/+49 construct). Reporter activity for all time points was statistically highly significantly different (p < 0.001, t test). D, Clomeleon-based chloride measurements in cultured primary cortical neurons at different developmental time points (DIV3, DIV6, DIV10); cultures were cotransfected with dn-REST, FL-REST, and vector control. Number of examined neurons were as follows: n = 30/44/65 (DIV3; control, FL-REST, dn-REST), n = 17/18/48 (DIV6), and n = 24/36/11 (DIV10). Note the decrease of neuronal [Cl−]i with maturation, also known as chloride shift, and its deceleration by increased expression of REST (FL-REST) versus acceleration by inhibition of REST (dn-REST). E, Neurons were transfected with constructs containing either dual RE-1 or no RE-1 and treated with 25 ng/ml BDNF. Secreted Gaussia luciferase activity was measured at DIV2 and DIV7.
Having demonstrated developmental regulation of Kcc2b and strong modulatory impact of the dual RE-1 site in primary cortical neurons, we next addressed whether this would translate into changes in neuronal chloride levels by modulating the chloride shift via inhibition of REST–RE-1 binding, by expression of dn-REST construct, or via boosting REST–RE-1 binding, by overexpression of REST. We examined three developmental time points between DIV3 and DIV10 to evaluate the influence of REST regulation on the chloride shift. Over the time course of development of these neurons, there is upregulation of KCC2 and downregulation of REST (Fig. 6A,B) (supplemental Fig. S4, available at www.jneurosci.org as supplemental material), with highly significant differences between developmental time points (p < 0.001, ANOVA with post hoc Dunnett's test). If our hypothesis is correct, then overexpressing REST should slow the developmental shift in [Cl−]i, whereas inhibiting REST should have the opposite effect. These predictions were borne out by Clomeleon measurements: dn-REST strikingly downregulated [Cl−]i from DIV3 until DIV10, whereas overexpressing FL-REST produced the opposite effect (Fig. 6D) (p < 0.001, ANOVA with post hoc Dunnett's test). Thus, the known developmental regulation pattern of Kcc2b together with reporter gene findings indicative of REST–RE-1 influencing developmental Kcc2 regulation (Fig. 6A–C) suggest a transcriptional logic for the observed effects of REST modulation on [Cl−]i (Fig. 6D).
Based on known trophic effects of BDNF–TrkB signaling on Kcc2b transcriptional regulation in development (Rivera et al., 2002, 2004), we wanted to find out whether BDNF signaling is affected by the presence of RE-1 sites. BDNF is not only a neural-activity-induced gene (West et al., 2001) but is also known to regulate spontaneous activity and increase KCC2 expression in developing hippocampal neurons (Aguado et al., 2003). Our data (Fig. 6E) show that, for the dual RE-1 promoter construct, BDNF led to a 10-fold increase from DIV2 to DIV7 (2.5%, 25%) versus nontreated controls. In contrast, for the −1649/+49 promoter construct devoid of RE-1 sites, there were negligible changes (+2.5% to −2%, from DIV2 to DIV7). These striking differences indicate that the upstream and intronic-RE-1 sites are necessary for BDNF regulation of Kcc2b transcription in development.
Influence of REST on the developmental change of action of GABA, “GABA switch”
GABA-stimulated upregulation of KCC2 expression constitutes an important mechanism underlying the developmental change in GABA signaling during postnatal neuronal development (Kriegstein and Owens, 2001). In view of the robust effects of REST on Kcc2 expression, [Cl−]i, and hence the chloride reversal potential, we next addressed possible consequences for the excitatory actions of GABA in primary cortical neurons.
For this purpose, we stimulated primary cortical neurons at different developmental time points with GABA receptor agonists, GABA (100 μm) and the GABAAR-specific agonist muscimol (10 μm). It has been demonstrated previously that activation of GABAAR in immature CNS neurons with high [Cl−]i leads to depolarization and subsequent activation of voltage-gated calcium channels (Ganguly et al., 2001; Liu et al., 2006). As [Cl−]i decreases with maturation, so does the percentage of neurons that respond with calcium influx in response to GABAAR stimulation. We were able to recapitulate these findings in our primary cortical cultures, using ratiometric calcium imaging: GABA excited 90% of neurons on DIV3 but only 25% on DIV12 (Fig. 7A,B). These responses were completely blocked by treatment with GABAAR antagonists (10 μm bicuculline plus 50 μm picrotoxin; data not shown). Furthermore, similar results were observed in response to muscimol (supplemental Fig. S5A,B, available at www.jneurosci.org as supplemental material). This confirms the role of GABAAR in the excitatory response to GABA. As observed by others, the calcium response depended on L-type voltage-dependent calcium channels because it was eliminated by nifedipine (20 μm), a specific blocker (data not shown).
Figure 7.
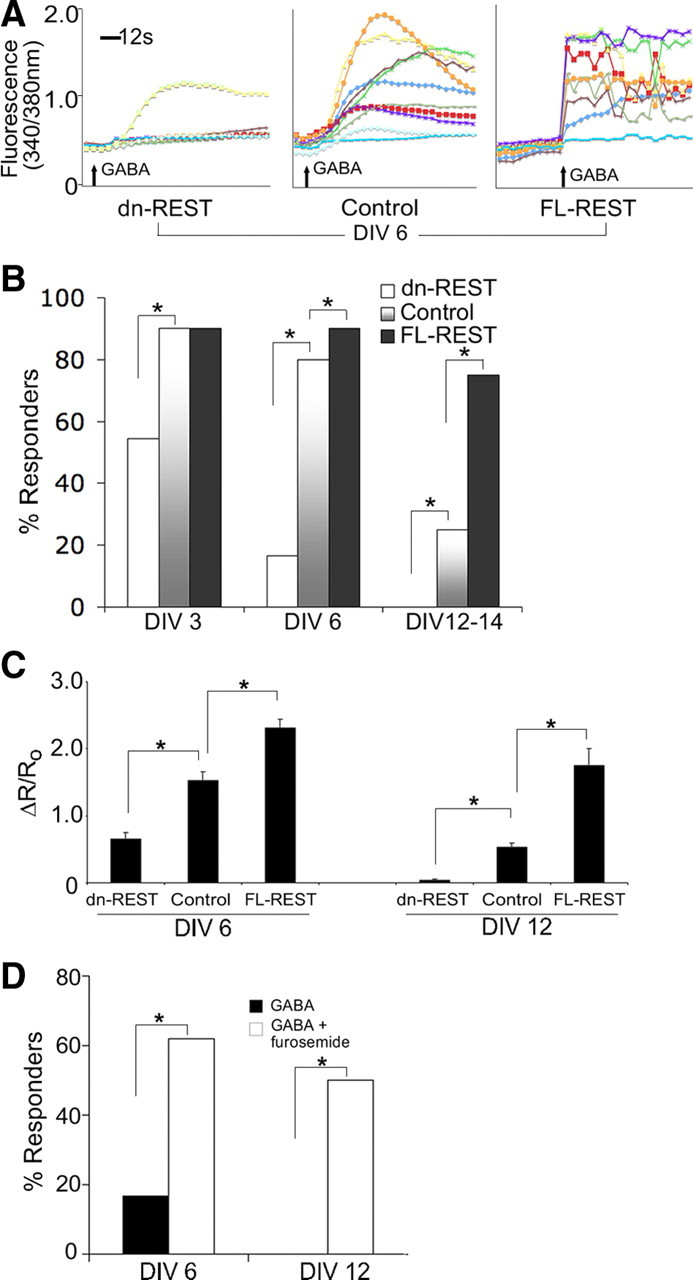
GABA switch depends on REST. A, Representative traces of the ratiometric calcium signal in individual neurons, imaged on DIV6, for dn-REST versus control versus FL-REST transfection. B, Bar diagrams depict percentage of cultured primary cortical neurons responding with a calcium signal, as a measure of excitation, in response to 100 μm GABA. Cultures were cotransfected with FL-REST, dn-REST, and vector control and examined on DIV3, DIV6, and DIV12–DIV14. A striking developmental decrease in the percentage of responding neurons is apparent, which can be accelerated robustly by inhibition of REST (dn-REST; n = 15, 12, 24 for DIV3, DIV6, and DIV12) and decelerated by REST overexpression (FL-REST, n = 18, 24, 24; for vector control, n = 30, 20, 25). Differences between REST-modulated and control group were statistically significantly different (p < 0.05, χ2 test). C, Quantitative measure of the calcium response to GABA: ΔR/R0 is the change in ratio after stimulation divided by the prestimulation ratio. Calcium ratios were recorded at DIV6 and DIV12. First, note that ΔR/R0 decreases with maturation (DIV6 vs DIV12) and, second, that it is increased by increasing REST (FL-REST) and decreased by inhibiting REST (dn-REST); n as given in B. *p < 0.01, ANOVA and post hoc Dunnett's test. D, Reversal of the effects of dn-REST on the GABA switch by inhibition of KCC2 chloride transporter function. This bar diagram indicates the percentage of dn-REST-transfected primary cortical neurons responding to 100 μm GABA with a calcium signal, on DIV6 and DIV12 (n = 12 and n = 8). After washout, the neurons were exposed to 100 μm furosemide for 15 min and restimulated with 100 μm GABA. Note the robust increase of responder neurons after furosemide-mediated inhibition of KCC2 to levels comparable with vector control-transfected neurons on DIV6 and DIV12-14 (see B).
We next asked how the excitatory responses to GABA were affected by inhibition of REST–RE-1 binding or by REST overexpression. We found that the developmental switch in GABA action could be strikingly accelerated by REST inhibition and slowed by transfection of FL-REST (Fig. 7A,B) (muscimol response shown in supplemental Fig. S5A–C, available at www.jneurosci.org as supplemental material). These differences between the REST-modulated and control groups were highly significant (p < 0.01, χ2 test). REST manipulation not only altered the percentage of neurons that responded to GABA with calcium influx but also affected the amplitude of responses in these neurons. The amplitude of ratiometric calcium signals (ΔR), normalized for prestimulation (R0), were upregulated by FL-REST and downregulated with dn-REST (Fig. 7C) (supplemental Fig. S5C, available at www.jneurosci.org as supplemental material). The amplitudes of the excitatory GABA responses were appreciable with increased magnitude in earlier DIVs (Fig. 7C). Again, differences between the REST-modulated and control groups were highly significant (p < 0.01, ANOVA with post hoc Dunnett's test).
To examine the contribution of KCC2 to the acceleration of the developmental GABA switch produced by REST inhibition, we used furosemide (100 μm) to inhibit KCC2 chloride transporter function at DIV6 and DIV12, at which time points KCC2 is expressed appreciably. We found that dn-REST-transfected neurons that did not show a calcium signal in response to GABA reverted to a depolarizing response to GABA during exposure to furosemide at both DIV6 and DIV12–DIV14 (Fig. 7D). These differences between dn-REST and controls were significant (p < 0.01, χ2 test). This indicates that the effect of dn-REST on the acceleration of the GABA switch is associated with an increase in KCC2 function. Our findings described above indicate that this switch in GABA action is caused by transcriptional derepression of Kcc2b, which is critically dependent on the dual RE-1 in Kcc2b. This mechanism leads to increased KCC2 protein expression and decreased neuronal chloride. Our findings that dn-REST accelerated and FL-REST decelerated the developmental GABA switch is consistent with this conclusion.
Discussion
In this study, we have shown that the major isoform of Kcc2 is regulated at the transcriptional level by the REST complex and furthermore that REST functions as transcriptional repressor by binding to two RE-1 sites in Kcc2b regulatory DNA sequences. We found a novel RE-1 DNA binding site upstream (−1702 relative to TSS in mouse), in Kcc2b in addition to the known intronic RE-1 (+378 relative to TSS). These sites are located as “dual RE-1” site 2080 bases apart, “bracketing” the TSS. Strikingly, Kcc2b transcription was upregulated in response to inhibition of REST only when both RE-1 sites were present. We propose that the concerted interaction between the REST complex bound to the dual RE-1 site enables, on the one hand, tight transcriptional regulation of Kcc2b and, on the other hand, the robust and rapid increase in the perinatal expression of Kcc2b in the forebrain. We demonstrate this regulation in primary cortical neurons derived from embryonic rats and prove transcriptional regulation of Kcc2b to reflect KCC2 protein expression and chloride transporter function by using Clomeleon fluorescent chloride indicator. Moreover, we have observed the naturally occurring chloride shift in these neurons to be inversely related to changes in expression of Kcc2b, pertaining to every level of regulation of the Kcc2 gene, namely Kcc2 transcription, KCC2 protein expression, and chloride extrusion from neurons. When inhibiting REST–RE-1 DNA binding, we observed a robust derepression of Kcc2b expression and likewise an increased repression of Kcc2b in response to REST overexpression. These changes led to corresponding changes in [Cl−]i at different developmental time points. Acceleration of the developmental chloride shift could be accomplished by application of BDNF, a change in kinetics fully dependent on the dual RE-1 site in Kcc2b. In keeping with our ability to accelerate and decelerate the chloride shift by modulating REST, we could influence depolarization in response to activation of GABAAR in a similar manner. Thus, plasticity modeling of the GABA switch was accomplished by upregulation or downregulation of REST. These findings represent a novel transcriptional mechanism that underlies the enigmatic chloride and subsequent GABA shift partial to all vertebrate CNS at approximately the time of birth.
Our starting point was a comparison of Kcc2 sequences found in several mammalian species, then extending the alignment to a “lower” vertebrate. High evolutionary conservation of sequence and position of the mammalian Kcc2b RE-1 elements points toward the critical importance of these DNA binding sites for Kcc2 transcriptional regulation. When considering the alignment with pufferfish Kcc2 gene, the intronic RE-1 was conserved, whereas the upstream RE-1 was present in a noncanonical version, indicating that the upstream RE-1 was a more recent evolutionary development. Our findings are in keeping with a recently published evolutionary study that compared, at the genomic level, mammalian and vertebrate RE-1 sites, concluding that phylogenetically more conserved sites bind REST tighter, are located closer to the TSS, and repress more robustly (Johnson et al., 2009). Our present demonstration of REST complex binding both RE-1 sites in neuronal cells is essentially confirmed and complemented by recent findings describing genome-wide application of ChIP-Seq methodology in a non-neuronal cell line (Jurkat T cells) (Johnson et al., 2007). An attractive hypothesis emanating from our mammalian Kcc2 alignment is the significance of dual RE-1, exerting repression by binding to its cognate transcriptional repressor complex REST to meet the regulatory needs of Kcc2b: exquisitely fine regulation for plasticity adaptation of inhibitory transmission by GABA and glycine in mature neurons and the dramatic perinatal increase in KCC2. Our novel transcriptional mechanism presented here is in synchrony with the latter: in primary cortical neurons that recapitulate the chloride shift, only in the presence of the dual RE-1 could Kcc2b transcription be increased when REST–RE-1 binding was inhibited.
When considering the inhibition of Kcc2b transcription by REST–RE-1, one has to bear in mind that the REST complex is dynamic and can vary in composition depending on target gene, cellular context, or stimulation. It is conceivable that REST–RE-1 transcription factor–DNA interaction in the repression of Kcc2b promoter activity proceeds in a one-on-one manner for the two RE-1 yet that corepressor components of the REST complex may be shared between the two DNA-bound REST molecules, permitting the dynamic regulation of the Kcc2 gene as needed both in plasticity and perinatally. If this were the case, it implies that regulation of Kcc2 by REST–RE-1, involving shared corepressor components of the REST complex, depends on DNA folding of the Kcc2 gene (Dillon, 2006). This reasoning is conceptualized in Figure 8. For comparison, we identified a total of 1341 REST target genes (mouse) in a databank (http://www.broad.mit.edu/∼xhx/projects/NRSE/), 149 of which have more than one RE-1 site. Interestingly, apart from Kcc2, three other genes have a second RE-1 that is located within a distance of between 1000 and 3000 bp, namely the POU class 2 homeobox 2; cyclin-dependent kinase 5, regulatory subunit 2 (p39); and Hu antigen B. Only the dual RE-1 in Kcc2b is positioned both upstream and downstream of the respective TSS. Thus, KCC2 is not only the sole chloride transporter for extrusion of chloride from mature CNS neurons in mammals, but it is also unique for the following two features: (1) it is the only REST target gene that encodes an ion channel/transporter and at the same time harbors a dual RE-1 site, and (2) it is the only gene with dual RE-1 that bracket the TSS.
Figure 8.

Schematic rendering of REST complex binding to Kcc2b dual RE-1, leading to transcriptional repression or derepression in development.
Because the upstream RE-1 site has not been described in previous work, our findings cannot be readily compared with the literature. However, a recent study (Uvarov et al., 2005) using transgenic mice bearing the intronic RE-1 site as part of the transgenic promoter demonstrated a modest but appreciable degree of regulation of the transgene in vivo. At parturition and 2 weeks after birth, there appeared to be no difference in reporter gene expression in cortices of transgenic mice lacking the intronic RE-1 in the promoter of the transgene, whereas in the presence of the intronic RE-1 site, transgene expression was elevated at 2 weeks of age compared with at birth. This finding indicates a modest derepression of Kcc2b in vivo mediated by the intronic RE-1 site over 2 weeks. However, at 4 weeks of age, there were no differences between genotypes. Derepression mediated by the intronic RE-1 site was observed in our experiments, but upregulation of Kcc2 transcription by inhibiting REST–RE-1 binding was robust only when both upstream and intronic RE-1 sites were present. In addition, whereas the transgenic mouse study (Uvarov et al., 2005) provided strong evidence that a minimal Kcc2 promoter, devoid of RE-1, is sufficient to direct Kcc2 expression virtually exclusively to the CNS, a deleterious dysregulation of Kcc2 expression in the CNS or a critical subregion might have gone unnoticed by assaying reporter gene expression in whole-brain lysates because KCC2 function was maintained by the wild-type alleles. Dysregulation of Kcc2b transcription, as evident at the 2 week time point in the absence of the intronic RE-1 site (Uvarov et al., 2005), could suffice to exert dramatic consequences during this critical period of development. Particularly during the first postnatal week, which the referenced study does not examine (Uvarov et al., 2005), dysregulation of Kcc2b transcription could lead to deleterious interference with migration of cortical interneurons (Bortone and Polleux, 2009).
In this respect, it should be noted that KCC2 not only functions as the dominant chloride transporter that extrudes chloride from postnatal CNS neurons but it also functions in yet another role, independent of anion transport, namely that of a powerful morphogen and accelerator of dendritic spine maturation and function (Li et al., 2007). Therefore, small differences in expression of this critical regulator of neuronal synchrony are likely to exert a far-reaching, possibly dramatic effect. This concept, conversely, points toward the need for the organism to tightly and finely regulate Kcc2b transcription. However, regulation of Kcc2b transcription also needs to have the dynamic range necessary for the rapid and robust upregulation in forebrain neurons in the perinatal period. Our data are in agreement with this concept, and we propose that the novel upstream RE-1, in concerted interaction with its intronic RE-1 partner, is a key player in this exquisite regulation. Last but not least, our identification of a transcriptional mechanism via the REST complex binding the dual RE-1 site in the Kcc2b gene, causing its regulation, builds a bridge between more canonical transcription factor–DNA binding models of gene regulation and more recent concepts of chromatin and epigenetic regulation of genes. This conclusion is ratified by the striking effect of the HDAC inhibitor trichostatin-A, which upregulated dual RE-1 Kcc2b reporter gene activity with similar potency as genetically encoded inhibition of REST.
Just like all other biological processes, be they physiological or pathological, regulation of neuronal chloride relies on the coordinated expression of genes, the products of which act together to mediate cellular function. Understanding of the processes that control gene expression underlying neuronal chloride homeostasis and its developmental dynamics is essential to our comprehension of development and disease. Based on our findings presented here, we conclude that the REST–dual RE-1 Kcc2b mechanism provides a partial explanation for the ontogenetic Kcc2b upregulation and subsequent chloride downregulation, which is critical for structure and function of forebrain inhibitory neurotransmission circuitry. This fine regulation of gene transcription of the major isoform of Kcc2 and tight control over a wide dynamic range are accomplished via concerted interaction of two RE-1 DNA-binding sites, conserved throughout mammals, possibly extending to lower vertebrates. Developmental chloride-shift acceleratory actions of BDNF as a pro-differentiation factor in the cerebral cortex, via upregulation of Kcc2b, were strictly dependent on the presence of dual RE-1. Together with previous work (Rivera et al., 2002, 2004), it is attractive to reason that BDNF, acting via TrkB and possibly the Shc intracellular signaling pathway, leads to inhibition of the REST complex binding the Kcc2b dual RE-1 during ontogeny [but not in circumstances of neuronal injury (Coull et al., 2003; Price et al., 2005)]. Our results open the possibility of targeted inhibition of REST as a new therapeutic rationale for resetting physiological levels of inhibitory neurotransmission. From a translational medical standpoint, this exciting concept applies in the first place to disease states associated with downregulation of Kcc2b and upregulation of chloride in injured neurons, such as epilepsy, traumatic brain injury, and chronic pathological pain.
Footnotes
This work was supported by National Institutes of Health/National Institute of Neurological Disorders and Stroke Grant R21NS066307, National Institutes of Health/National Institute on Aging Pilot Project Grant P30AG028377, grant support from the Mathers Foundation (Mount Kisco, NY), the Esther A. and Joseph Klingenstein Fund (New York, NY), and startup funds from Duke University (W.L.). dn-REST and FL-REST expression plasmids were generous gifts from Dr. David Anderson (California Institute of Technology, Pasadena, CA). Suk Hee Lee (Duke University) provided excellent technical assistance with Ca2+ imaging. Dr. Erszebet Szatmari (Duke University) contributed advice and guidance in the initial stages of rat cortical neuronal cultures, and Dr. Aurel Betz (The Rockefeller University, New York, NY) provided insightful comments on this manuscript.
References
- Aerts S, Thijs G, Coessens B, Staes M, Moreau Y, De Moor B. Toucan: deciphering the cis-regulatory logic of coregulated genes. Nucleic Acids Res. 2003;31:1753–1764. doi: 10.1093/nar/gkg268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguado F, Carmona MA, Pozas E, Aguiló A, Martínez-Guijarro FJ, Alcantara S, Borrell V, Yuste R, Ibañez CF, Soriano E. BDNF regulates spontaneous correlated activity at early developmental stages by increasing synaptogenesis and expression of the K+/Cl− co-transporter KCC2. Development. 2003;130:1267–1280. doi: 10.1242/dev.00351. [DOI] [PubMed] [Google Scholar]
- Ballas N, Mandel G. The many faces of REST oversee epigenetic programming of neuronal genes. Curr Opin Neurobiol. 2005;15:500–506. doi: 10.1016/j.conb.2005.08.015. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y. Excitatory actions of gaba during development: the nature of the nurture. Nat Rev Neurosci. 2002;3:728–739. doi: 10.1038/nrn920. [DOI] [PubMed] [Google Scholar]
- Bonislawski DP, Schwarzbach EP, Cohen AS. Brain injury impairs dentate gyrus inhibitory efficacy. Neurobiol Dis. 2007;25:163–169. doi: 10.1016/j.nbd.2006.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bortone D, Polleux F. KCC2 expression promotes the termination of cortical interneuron migration in a voltage-sensitive calcium-dependent manner. Neuron. 2009;62:53–71. doi: 10.1016/j.neuron.2009.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderone A, Jover T, Noh KM, Tanaka H, Yokota H, Lin Y, Grooms SY, Regis R, Bennett MV, Zukin RS. Ischemic insults derepress the gene silencer REST in neurons destined to die. J Neurosci. 2003;23:2112–2121. doi: 10.1523/JNEUROSCI.23-06-02112.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZF, Paquette AJ, Anderson DJ. NRSF/REST is required in vivo for repression of multiple neuronal target genes during embryogenesis. Nat Genet. 1998;20:136–142. doi: 10.1038/2431. [DOI] [PubMed] [Google Scholar]
- Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003;31:3497–3500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong JA, Tapia-Ramírez J, Kim S, Toledo-Aral JJ, Zheng Y, Boutros MC, Altshuller YM, Frohman MA, Kraner SD, Mandel G. REST: a mammalian silencer protein that restricts sodium channel gene expression to neurons. Cell. 1995;80:949–957. doi: 10.1016/0092-8674(95)90298-8. [DOI] [PubMed] [Google Scholar]
- Coull JA, Boudreau D, Bachand K, Prescott SA, Nault F, Sík A, De Koninck P, De Koninck Y. Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature. 2003;424:938–942. doi: 10.1038/nature01868. [DOI] [PubMed] [Google Scholar]
- Delpire E, Mount DB. Human and murine phenotypes associated with defects in cation-chloride cotransport. Annu Rev Physiol. 2002;64:803–843. doi: 10.1146/annurev.physiol.64.081501.155847. [DOI] [PubMed] [Google Scholar]
- Dillon N. Gene regulation and large-scale chromatin organization in the nucleus. Chromosome Res. 2006;14:117–126. doi: 10.1007/s10577-006-1027-8. [DOI] [PubMed] [Google Scholar]
- Fiumelli H, Woodin MA. Role of activity-dependent regulation of neuronal chloride homeostasis in development. Curr Opin Neurobiol. 2007;17:81–86. doi: 10.1016/j.conb.2007.01.002. [DOI] [PubMed] [Google Scholar]
- Galanopoulou AS. Developmental patterns in the regulation of chloride homeostasis and GABA(A) receptor signaling by seizures. Epilepsia. 2007;48(Suppl 5):14–18. doi: 10.1111/j.1528-1167.2007.01284.x. [DOI] [PubMed] [Google Scholar]
- Ganguly K, Schinder AF, Wong ST, Poo M. GABA itself promotes the developmental switch of neuronal GABAergic responses from excitation to inhibition. Cell. 2001;105:521–532. doi: 10.1016/s0092-8674(01)00341-5. [DOI] [PubMed] [Google Scholar]
- Garriga-Canut M, Schoenike B, Qazi R, Bergendahl K, Daley TJ, Pfender RM, Morrison JF, Ockuly J, Stafstrom C, Sutula T, Roopra A. 2-Deoxy-d-glucose reduces epilepsy progression by NRSF-CtBP-dependent metabolic regulation of chromatin structure. Nat Neurosci. 2006;9:1382–1387. doi: 10.1038/nn1791. [DOI] [PubMed] [Google Scholar]
- Hewitt SA, Wamsteeker JI, Kurz EU, Bains JS. Altered chloride homeostasis removes synaptic inhibitory constraint of the stress axis. Nat Neurosci. 2009;12:438–443. doi: 10.1038/nn.2274. [DOI] [PubMed] [Google Scholar]
- Huang Y, Myers SJ, Dingledine R. Transcriptional repression by REST: recruitment of Sin3A and histone deacetylase to neuronal genes. Nat Neurosci. 1999;2:867–872. doi: 10.1038/13165. [DOI] [PubMed] [Google Scholar]
- Huberfeld G, Wittner L, Clemenceau S, Baulac M, Kaila K, Miles R, Rivera C. Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J Neurosci. 2007;27:9866–9873. doi: 10.1523/JNEUROSCI.2761-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hübner CA, Stein V, Hermans-Borgmeyer I, Meyer T, Ballanyi K, Jentsch TJ. Disruption of KCC2 reveals an essential role of K-Cl cotransport already in early synaptic inhibition. Neuron. 2001;30:515–524. doi: 10.1016/s0896-6273(01)00297-5. [DOI] [PubMed] [Google Scholar]
- Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- Johnson R, Samuel J, Ng CK, Jauch R, Stanton LW, Wood IC. Evolution of the vertebrate gene regulatory network controlled by the transcriptional repressor REST. Mol Biol Evol. 2009;26:1491–1507. doi: 10.1093/molbev/msp058. [DOI] [PubMed] [Google Scholar]
- Karadsheh MF, Delpire E. Neuronal restrictive silencing element is found in the KCC2 gene: molecular basis for KCC2-specific expression in neurons. J Neurophysiol. 2001;85:995–997. doi: 10.1152/jn.2001.85.2.995. [DOI] [PubMed] [Google Scholar]
- Kim TH, Barrera LO, Zheng M, Qu C, Singer MA, Richmond TA, Wu Y, Green RD, Ren B. A high-resolution map of active promoters in the human genome. Nature. 2005;436:876–880. doi: 10.1038/nature03877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraner SD, Chong JA, Tsay HJ, Mandel G. Silencing the type II sodium channel gene: a model for neural-specific gene regulation. Neuron. 1992;9:37–44. doi: 10.1016/0896-6273(92)90218-3. [DOI] [PubMed] [Google Scholar]
- Kriegstein AR, Owens DF. GABA may act as a self-limiting trophic factor at developing synapses. Sci STKE. 2001;2001:PE1. doi: 10.1126/stke.2001.95.pe1. [DOI] [PubMed] [Google Scholar]
- Kuner T, Augustine GJ. A genetically encoded ratiometric indicator for chloride: capturing chloride transients in cultured hippocampal neurons. Neuron. 2000;27:447–459. doi: 10.1016/s0896-6273(00)00056-8. [DOI] [PubMed] [Google Scholar]
- Li H, Khirug S, Cai C, Ludwig A, Blaesse P, Kolikova J, Afzalov R, Coleman SK, Lauri S, Airaksinen MS, Keinänen K, Khiroug L, Saarma M, Kaila K, Rivera C. KCC2 interacts with the dendritic cytoskeleton to promote spine development. Neuron. 2007;56:1019–1033. doi: 10.1016/j.neuron.2007.10.039. [DOI] [PubMed] [Google Scholar]
- Liu Z, Neff RA, Berg DK. Sequential interplay of nicotinic and GABAergic signaling guides neuronal development. Science. 2006;314:1610–1613. doi: 10.1126/science.1134246. [DOI] [PubMed] [Google Scholar]
- Lu J, Karadsheh M, Delpire E. Developmental regulation of the neuronal-specific isoform of K-Cl cotransporter KCC2 in postnatal rat brains. J Neurobiol. 1999;39:558–568. [PubMed] [Google Scholar]
- Ludwig A, Li H, Saarma M, Kaila K, Rivera C. Developmental up-regulation of KCC2 in the absence of GABAergic and glutamatergic transmission. Eur J Neurosci. 2003;18:3199–3206. doi: 10.1111/j.1460-9568.2003.03069.x. [DOI] [PubMed] [Google Scholar]
- Lunyak VV, Rosenfeld MG. No rest for REST: REST/NRSF regulation of neurogenesis. Cell. 2005;121:499–501. doi: 10.1016/j.cell.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Markkanen M, Uvarov P, Airaksinen MS. Role of upstream stimulating factors in the transcriptional regulation of the neuron-specific K-Cl cotransporter KCC2. Brain Res. 2008;1236:8–15. doi: 10.1016/j.brainres.2008.08.007. [DOI] [PubMed] [Google Scholar]
- Morales-Aza BM, Chillingworth NL, Payne JA, Donaldson LF. Inflammation alters cation chloride cotransporter expression in sensory neurons. Neurobiol Dis. 2004;17:62–69. doi: 10.1016/j.nbd.2004.05.010. [DOI] [PubMed] [Google Scholar]
- Mori N, Schoenherr C, Vandenbergh DJ, Anderson DJ. A common silencer element in the SCG10 and type II Na+ channel genes binds a factor present in nonneuronal cells but not in neuronal cells. Neuron. 1992;9:45–54. doi: 10.1016/0896-6273(92)90219-4. [DOI] [PubMed] [Google Scholar]
- Munakata M, Watanabe M, Otsuki T, Nakama H, Arima K, Itoh M, Nabekura J, Iinuma K, Tsuchiya S. Altered distribution of KCC2 in cortical dysplasia in patients with intractable epilepsy. Epilepsia. 2007;48:837–844. doi: 10.1111/j.1528-1167.2006.00954.x. [DOI] [PubMed] [Google Scholar]
- Muñoz A, Méndez P, DeFelipe J, Alvarez-Leefmans FJ. Cation-chloride cotransporters and GABA-ergic innervation in the human epileptic hippocampus. Epilepsia. 2007;48:663–673. doi: 10.1111/j.1528-1167.2007.00986.x. [DOI] [PubMed] [Google Scholar]
- Ooi L, Wood IC. Chromatin crosstalk in development and disease: lessons from REST. Nat Rev Genet. 2007;8:544–554. doi: 10.1038/nrg2100. [DOI] [PubMed] [Google Scholar]
- Palm K, Belluardo N, Metsis M, Timmusk T. Neuronal expression of zinc finger transcription factor REST/NRSF/XBR gene. J Neurosci. 1998;18:1280–1296. doi: 10.1523/JNEUROSCI.18-04-01280.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne JA, Stevenson TJ, Donaldson LF. Molecular characterization of a putative K-Cl cotransporter in rat brain. A neuronal-specific isoform. J Biol Chem. 1996;271:16245–16252. doi: 10.1074/jbc.271.27.16245. [DOI] [PubMed] [Google Scholar]
- Pond BB, Berglund K, Kuner T, Feng G, Augustine GJ, Schwartz-Bloom RD. The chloride transporter Na+-K+-Cl− cotransporter isoform-1 contributes to intracellular chloride increases after in vitro ischemia. J Neurosci. 2006;26:1396–1406. doi: 10.1523/JNEUROSCI.1421-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price TJ, Cervero F, de Koninck Y. Role of cation-chloride-cotransporters (CCC) in pain and hyperalgesia. Curr Top Med Chem. 2005;5:547–555. doi: 10.2174/1568026054367629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera C, Li H, Thomas-Crusells J, Lahtinen H, Viitanen T, Nanobashvili A, Kokaia Z, Airaksinen MS, Voipio J, Kaila K, Saarma M. BDNF-induced TrkB activation down-regulates the K+-Cl− cotransporter KCC2 and impairs neuronal Cl− extrusion. J Cell Biol. 2002;159:747–752. doi: 10.1083/jcb.200209011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Thomas-Crusells J, Li H, Emri Z, Sipilä S, Payne JA, Minichiello L, Saarma M, Kaila K. Mechanism of activity-dependent downregulation of the neuron-specific K-Cl cotransporter KCC2. J Neurosci. 2004;24:4683–4691. doi: 10.1523/JNEUROSCI.5265-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenherr CJ, Anderson DJ. Silencing is golden: negative regulation in the control of neuronal gene transcription. Curr Opin Neurobiol. 1995;5:566–571. doi: 10.1016/0959-4388(95)80060-3. [DOI] [PubMed] [Google Scholar]
- Schoenherr CJ, Paquette AJ, Anderson DJ. Identification of potential target genes for the neuron-restrictive silencer factor. Proc Natl Acad Sci USA. 1996;93:9881–9886. doi: 10.1073/pnas.93.18.9881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sernagor E, Young C, Eglen SJ. Developmental modulation of retinal wave dynamics: shedding light on the GABA saga. J Neurosci. 2003;23:7621–7629. doi: 10.1523/JNEUROSCI.23-20-07621.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer VA, Sun JM, Li L, Davie JR. Chromatin immunoprecipitation: a tool for studying histone acetylation and transcription factor binding. Methods. 2003;31:67–75. doi: 10.1016/s1046-2023(03)00089-6. [DOI] [PubMed] [Google Scholar]
- Szatmari E, Kalita KB, Kharebava G, Hetman M. Role of kinase suppressor of Ras-1 in neuronal survival signaling by extracellular signal-regulated kinase 1/2. J Neurosci. 2007;27:11389–11400. doi: 10.1523/JNEUROSCI.3473-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannous BA, Kim DE, Fernandez JL, Weissleder R, Breakefield XO. Codon-optimized Gaussia luciferase cDNA for mammalian gene expression in culture and in vivo. Mol Ther. 2005;11:435–443. doi: 10.1016/j.ymthe.2004.10.016. [DOI] [PubMed] [Google Scholar]
- Tornberg J, Voikar V, Savilahti H, Rauvala H, Airaksinen MS. Behavioural phenotypes of hypomorphic KCC2-deficient mice. Eur J Neurosci. 2005;21:1327–1337. doi: 10.1111/j.1460-9568.2005.03959.x. [DOI] [PubMed] [Google Scholar]
- Uvarov P, Pruunsild P, Timmusk T, Airaksinen MS. Neuronal K+/Cl− co-transporter (KCC2) transgenes lacking neurone restrictive silencer element recapitulate CNS neurone-specific expression and developmental up-regulation of endogenous KCC2 gene. J Neurochem. 2005;95:1144–1155. doi: 10.1111/j.1471-4159.2005.03434.x. [DOI] [PubMed] [Google Scholar]
- Uvarov P, Ludwig A, Markkanen M, Rivera C, Airaksinen MS. Upregulation of the neuron-specific K+/Cl− cotransporter expression by transcription factor early growth response 4. J Neurosci. 2006;26:13463–13473. doi: 10.1523/JNEUROSCI.4731-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uvarov P, Ludwig A, Markkanen M, Pruunsild P, Kaila K, Delpire E, Timmusk T, Rivera C, Airaksinen MS. A novel N-terminal isoform of the neuron-specific K-Cl cotransporter KCC2. J Biol Chem. 2007;282:30570–30576. doi: 10.1074/jbc.M705095200. [DOI] [PubMed] [Google Scholar]
- Uvarov P, Ludwig A, Markkanen M, Soni S, Hübner CA, Rivera C, Airaksinen MS. Coexpression and heteromerization of two neuronal K-Cl cotransporter isoforms in neonatal brain. J Biol Chem. 2009;284:13696–13704. doi: 10.1074/jbc.M807366200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AE, Chen WG, Dalva MB, Dolmetsch RE, Kornhauser JM, Shaywitz AJ, Takasu MA, Tao X, Greenberg ME. Calcium regulation of neuronal gene expression. Proc Natl Acad Sci USA. 2001;98:11024–11031. doi: 10.1073/pnas.191352298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo M, Lee SK, Lee B, Ruiz EC, Pfaff SL, Gill GN. Small CTD phosphatases function in silencing neuronal gene expression. Science. 2005;307:596–600. doi: 10.1126/science.1100801. [DOI] [PubMed] [Google Scholar]
- Zhang W, Liu LY, Xu TL. Reduced potassium-chloride co-transporter expression in spinal cord dorsal horn neurons contributes to inflammatory pain hypersensitivity in rats. Neuroscience. 2008;152:502–510. doi: 10.1016/j.neuroscience.2007.12.037. [DOI] [PubMed] [Google Scholar]