

Abstract
Ubiquitin ligases play an important role in the regulation of the immune system. Absence of Itch E3 ubiquitin ligase in mice has been shown to cause severe autoimmune disease. Using autozygosity mapping in a large Amish kindred, we identified a linkage region on chromosome 20 and selected candidate genes for screening. We describe, in ten patients, identification of a mutation resulting in truncation of ITCH. These patients represent the first reported human phenotype associated with ITCH deficiency. These patients not only have multisystem autoimmune disease but also display morphologic and developmental abnormalities. This disorder underscores the importance of ITCH ubiquitin ligase in many cellular processes.
Main Text
We describe ten Old Order Amish children with organomegaly, failure to thrive, developmental delay, dysmorphic features, and autoimmune inflammatory cell infiltration of the lungs, liver, and gut. Extensive testing failed to reveal a diagnosis. We used single-nucleotide polymorphism (SNP) autozygosity mapping to localize the disease gene to chromosome 20q11 and subsequently found that all affected patients were homozygous for a truncating mutation in ITCH (MIM 606409), which codes for an E3 ubiquitin ligase. To our knowledge, we provide the first direct evidence connecting ITCH deficiency to human disease.
The primary function of ubiquitin is to target proteins for degradation in the protesome. Ubiquitination of target proteins is important for immune regulation.1–3 Ubiquitin tagging affects antigen processing by antigen-presenting cells and promotes immunological tolerance by changing signaling components to shift the balance away from activation and toward anergy.4–6 In mice, mutations of the E3 ligase Itch cause fatal autoimmune disease characterized by histiocyte and lymphocyte infiltration of lungs, liver, kidneys, and heart.7,8 Our findings have broad implications for the study of autoimmunity in humans and underscore the important role of ubiquitination in the development of other organ systems.
Three related Amish children were evaluated for poor growth, developmental delay, hepatosplenomegaly, diarrhea, and chronic lung disease. Seven additional relatives (Indiana = 5, Pennsylvania = 1, New York = 1) with a similar phenotype were identified during a field study and by personal correspondence. Institutional review boards at Indiana University/Clarian Hospitals and at Lancaster General Hospital approved the collection of clinical information and DNA for genetic mapping. All patients demonstrated multiple lines of common ancestry, and each was the product of a consanguineous marriage (Figure 1).
Figure 1.
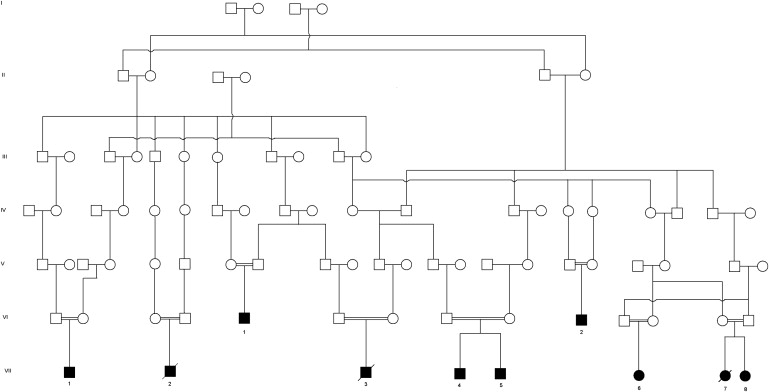
Pedigree Demonstrating Autosomal-Recessive Inheritance of Traits from Several Common Ancestors
Information obtained from a community genealogy book.22 Unaffected siblings (both carriers and noncarriers) are not displayed because of space constraints. Of the individuals tested, carriers include the parents of VII-1, the father of VII-3 (no record of testing the mother), the mother and three siblings of VII-4 and VII-5 (no record of testing the father), two brothers of VII-7 and VII-8, and one sister of VI-2.
Characteristic clinical features were dysmorphic facies, failure to thrive, hepatomegaly, splenomegaly, multisystem autoimmune disease, and delayed motor development (Table 1). The patients ranged from 5 months to 23 years of age; the mean age was 4.2 years, and the median age was 2 years. Growth was stunted, and patients displayed relative macrocephaly (typically at the 90th percentile or above), weight and height below the 3rd percentile (z-score less than −2), and body mass index below normal (z-score less than −1). Six of ten patients required gastrostomy tube placement within the first year of life after having failed to gain weight with oral feedings, although only two had overt symptoms of malabsorption. Distinctive craniofacial features included frontal bossing, dolichocephaly, orbital proptosis, flattened mid-face with a prominent occiput, small chin, and low, posteriorly rotated ears (Figure 2). The liver was typically 4–8 cm below the costal margin, and the spleen was 4–6 cm below the costal margin. The remainder of the physical examination was significant for global hypotonia and campto- or clinodactyly. All children were delayed in gross motor skills (they often did not walk until 3–4 years of age) and cognitive skills (they usually required special help in school). Several of the children were treated for recurrent infections. Complete blood counts, when available, did not demonstrate abnormal white blood cell counts, hematocrits, platelet counts, absolute neutrophil counts, or absolute lymphocyte counts except during acute infections.
Table 1.
Clinical and Autoimmune Features Seen in ITCH-Deficient Patients
| Clinical Features |
Patient Number |
Percent Affected | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| VII-4 | VII-5 | VI-2 | VII-3 | VII-6 | VII-8 | VII-1 | VI-1 | VII-2 | VII-7 | ||
| Hepatomegaly and/or splenomegaly | + | + | + | + | + | - | + | + | + | + | 90% |
| Failure to thrive | + | + | + | + | + | + | + | + | + | + | 100% |
| Developmental delay | + | + | + | + | + | + | + | + | + | + | 100% |
| Dysmorphic features | + | + | + | + | + | + | + | + | + | + | 100% |
| Relative macrocephaly | + | + | + | + | + | + | + | + | - | + | 90% |
| Chronic lung disease | + | + | - | + | + | + | + | + | + | + | 90% |
| Hypotonia | + | + | + | + | - | - | + | ? | + | ? | 60% |
| Autoimmune disease | + | - | + | - | + | - | + | + | + | - | 60% |
| −Hypothyroidism | + | - | + | - | - | - | + | - | + | - | 40% |
| −Hepatitis | - | - | - | - | + | - | + | - | + | - | 30% |
| −Enteropathy | + | - | - | - | - | - | + | - | - | - | 20% |
| −Diabetes mellitus | - | - | - | - | - | - | - | + | - | - | 10% |
| Current age or age at death (in years) |
12 | 2 | 3 | 3 | 9 | 4 | 6 | 23 | 0.6 | 1.2 | |
| Currently living? (yes or no) | Y | Y | Y | N | Y | Y | Y | Y | N | N | |
Figure 2.
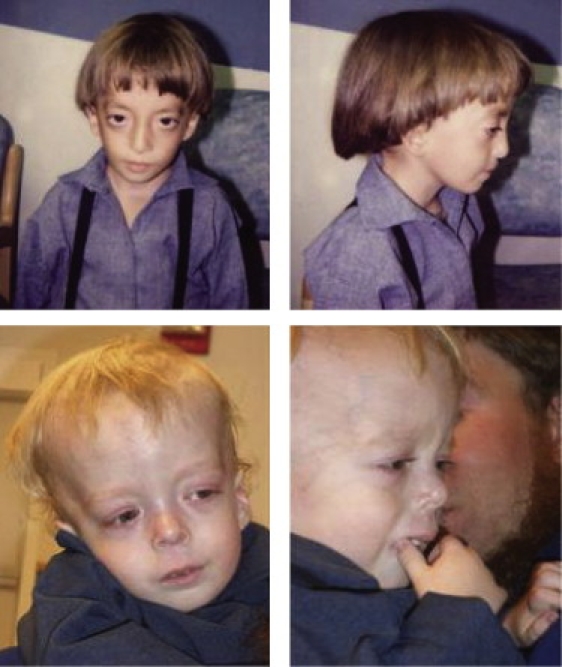
The Two Index Patients
Photos were used with the family's consent.
Three patients had autoimmune hepatitis resulting in alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels that were 3–30 times the upper limit of normal. One had isolated anti-liver/kidney microsomal (LKM) antibodies, one had both LKM and antinuclear antibodies (ANA), and a third had anti-neutrophil cytoplasmic antibodies (pANCA). Liver biopsies on two children showed dense periportal mixed inflammatory infiltrate with interface activity consistent with autoimmune hepatitis (Figures 3A and 3B). In one of these children, there was substantial bridging fibrosis associated with profound loss of hepatocytes. One child had improvement in transaminases in response to corticosteroids, one died of lung disease before autoimmune hepatitis was treated, and a third is currently under evaluation.
Figure 3.
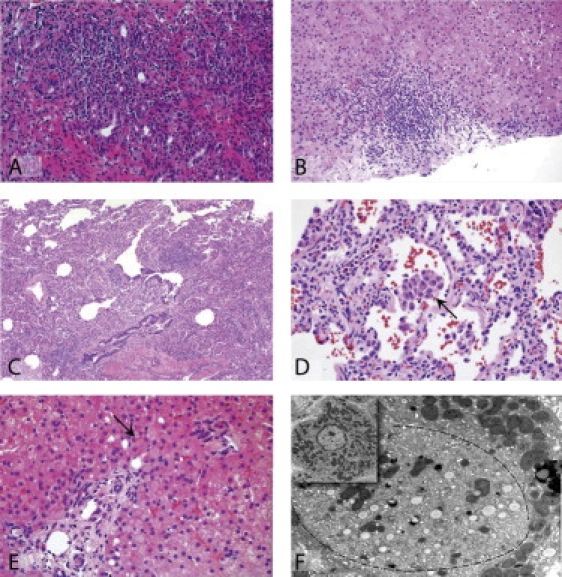
Histology Showing the Effects of ITCH Deficiency in Multiple Tissues
(A) A mid-power view of the liver in a subject with autoimmune hepatitis shows a dense mixed inflammatory infiltrate throughout the periportal region and an accompanying cholangitis and reactive proliferation of bile ducts.
(B) Mid-power view of the liver in another subject with autoimmune hepatitis shows a mixed inflammatory cell infiltrate in the periportal region.
(C) Lung showing a patchy expansion of the interstitium by lymphocytes and clusters of macrophages in the alveolar airspaces. There is no fibrosis.
(D) Higher magnification of the lung exhibiting a mild interstitial lymphocytic infiltrate. The central alveolar airspace contains a collection of macrophages (arrow).
(E) Although liver architecture is intact in a subject with normal liver enzymes but hepatomegaly, some multinucleated hepatocytes are present, and periportal hepatocytes contain large Lafora-body-like inclusions (arrow); there is no inflammation.
(F) An electron micrograph of the same specimen as in 3E reveals heterogeneous cytoplasmic material (bounded by a dotted line): small droplets of fat and endoplasmic reticulum displace mitochondria eccentrically (inset shows a normal hepatocyte for comparison).16
Two children had autoimmune enteropathy that resulted in chronic diarrhea. One of these children had anti-enterocyte antibodies, pANCA, and anti-smooth-muscle antibodies (SMA), whereas another had anti-enterocyte antibodies only. Both children had intestinal biopsies that showed lymphocytic inflammation of the lamina propria; one had severe villous blunting (not shown). The child with mild inflammation improved on low-dose steroids, whereas the second improved after being treated with an array of immunosuppressives, including corticosteroids, rapamycin, azathioprine, and tacrolimus.
Four of ten children developed hypothyroidism. Autoantibodies were found in the three children who were tested: two tested positive for anti-thyroid peroxidase (TPO), and one tested positive for anti-thyroglobulin. Biochemical abnormalities resolved with appropriate thyroid hormone replacement. One subject was diagnosed with type 1 diabetes mellitus; autoantibodies were not measured.
Nine of ten children had chronic lung disease, often characterized as asthma. Five children had chronic lung opacities seen on chest radiography (n = 3) or chest computed tomography (n = 2). Three had a chronic oxygen requirement, and one child required tracheostomy placement and home ventilator support. Lung disease was treated with bronchodilators, antibiotics, and occasionally, corticosteroids with apparent response. A wedge lung biopsy of one severely affected child revealed mononuclear infiltrates expanding the interstitium and clusters of macrophages filling the alveolar airspaces (Figures 3C and 3D). These findings are somewhat difficult to classify but seem to fit best as a cellular, nonspecific interstitial pneumonitis. Three children died of respiratory failure.
Intermittent hepatomegaly was seen in several children in the absence of elevated serum transaminases. A liver biopsy in one such child was read as normal, but another demonstrated large pink inclusions in the cytoplasm of periportal hepatocytes after hematoxylin and eosin staining; electron microscopy of these cells, but not other hepatocytes, revealed cytoplamic accumulations of large amounts of smooth endoplasmic reticulum (Figures 3E and 3F). A third child with intermittent marked elevations in transaminases had steatohepatitis on one biopsy and giant cell hepatitis without typical autoimmune features on another.
Total genomic DNA from whole blood was isolated with the PureGene DNA Isolation Kit (QIAGEN, Valencia, CA, USA) according to the manufacturer's protocol. SNP genotyping was performed with the GeneChip Mapping 10K Assay Kit (Affymetrix, Santa Clara, CA, USA) as previously described.9 In brief, 250 ng of double-stranded genomic DNA was digested with XbaI, the restriction fragments were ligated to adapters, and the ligated products were amplified in quadruplicate with a generic primer. PCR products were purified, fragmented with DNase, labeled with terminal deoxynucleotidyl transferase, and finally hybridized to a GeneChip Human Mapping 10K Array (XbaI 142, 2.0). Microarrays were then washed with a fluidics station, incubated, and scanned with the GeneChip Scanner 3000 enabled by GeneChip GCOS software (Affymetrix).
Data analyses ascertained genomic regions identically homozygous among affected individuals and assumed mutation and locus homogeneity. We calculated cumulative two-point LOD scores (location scores) for each shared block of homozygous SNPs to determine the relative likelihood that the shared homozygous region harbored the disease gene. We used SNPs bounding a shared block to construct a candidate gene list by using the NCBI Genome Browser. For each gene, we assessed function and expression to generate a priority list for sequencing. Suitable candidates were subjected to polymerase chain reaction (PCR) amplification and sequencing of the coding regions and adjacent intron-exon boundaries. PCR primers were designed with Primer3. A 25 μl reaction was prepared with 1 U of Taq polymerase (QIAGEN, Valencia, CA), 200 mM each of dATP, dCTP, dGTP, and dTTP, 2.5 μl of 10× PCR buffer (QIAGEN), and 50 ng of genomic DNA. We used a Perkin-Elmer 480 or Applied Biosystems GeneAmp 9700 thermocycler to cycle the reaction for 30 cycles (30 s at 96°C, 10 s at 60°C, 30 s at 72°C), and this was followed by an 8 min incubation at 72°C. PCR products were sequenced with the BigDye Terminator cycle sequencing protocol (Applied Biosystems). Extension products were then size-fractionated on an ABI 310 Genetic Analyzer. Coding exons and adjacent intronic regions from candidate genes from patient sequences were compared to the human reference sequence and dbSNP (NCBI) so that pathogenic variants could be detected.
Genome-wide autozygosity mapping for five patients identified a large homozygous block in the pericentromeric region of chromosome 20 (Figure 4A). The 19 MB region was bounded by SNPs rs2038383 and rs2067084 and contained 258 known or hypothetical genes. Five of these candidate genes were sequenced on the basis of their predicted function. Prominent autoimmune disease in one patient directed candidate gene sequencing to ITCH because of similar autoimmune findings in Itch−/− mice. Sequence analysis of ITCH revealed a homozygous single basepair insertion in exon 6 (c.394_395insA) (Figure 4B). This frameshift mutation was predicted to truncate ITCH at amino acid position 139 (Figures 4C and 4D). All affected individuals were homozygous for this variant, and among those tested, their parents were heterozygous. None of the nine heterozygous siblings or parents were symptomatic or dysmorphic. Among 12 unaffected siblings tested from five families, none were homozygous for the mutation. We developed a SimpleProbe PCR assay for ITCH c.394_395insA to facilitate rapid carrier and diagnostic testing. Genotyping of 80 random Lancaster County Old Order Amish identified no carriers. This is not unexpected because all affected individuals descend from the Indiana Amish, a group genetically distinct from the Lancaster County Amish. We did not have banked DNA we could analyze from the Indiana Amish.
Figure 4.
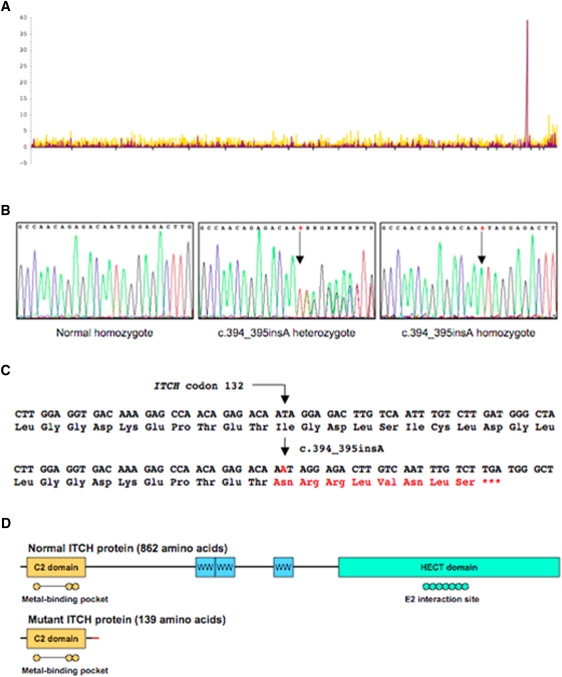
Genetic Mapping and Gene Mutation in ITCH-Deficient Patients
(A) Genetic mapping to chromosome 20p11.2-q12. A block of 37 contiguous SNPs on chromosome 20 provided overwhelming evidence for identity-by-descent in five affected Old Order Amish patients.
(B) The ITCH sequencing trace; sequencing of the 23 coding exons of ITCH identified a single base-pair insertion in exon 6. Panel 1 shows sequence from a control sample demonstrating normal exon 6 sequence. Panel 2 depicts sequence from an obligate heterozygote showing the single base-pair insertion. Panel 3 illustrates homozygosity for the c.394_395insA mutation (the red A denotes the mutation).
(C and D) The imputed effect of the insertion mutation on ITCH translation. The mutation causes a frameshift predicted to misincorporate eight amino acids in the peptide chain and cause a premature termination at codon 140. The truncated ITCH protein lacks the three WW domains as well as the HECT domain that contains the E2 interaction (active) site.17
Human ITCH deficiency results in a complex phenotype that affects physical growth, craniofacial morphology, muscle development, and immune function. The consequences of ITCH deficiency in humans appear to be similar to those in Itch−/− mice. Nine of the ten affected children had chronic lung disease, and respiratory failure caused death in three. This lung disease, similar to that seen in mice, was probably a result of immune dysregulation. Pathology results revealed a nonspecific interstitial pneumonitis that has been linked previously to autoimmune disease.10,11 Five other patients had autoimmune disease in other organ systems.
ITCH attaches ubiquitin to substrate proteins.12 Ubiquitination of the T cell receptor mediates its downregulation,7 and downstream signaling events are altered by ubiquitination of proteins such as JUNB, which may inhibit IL-2 production and thus T cell proliferation.8 Dysfunction of E3 ligases, which catalyze the final step of ubiquitin attachment, can lead to indiscriminate T cell activation and loss of tolerance to self-antigens.1,13 In mice, mutations of the E3 ligase Itch cause fatal autoimmune disease characterized by histiocyte and lymphocyte infiltration of the lungs, liver, kidneys, and heart.7,8 “Itchy” mice have a skewed Th2 phenotype and dysfunctional B cell responses.14 The mice are small, dysmorphic, and pruritic and have elevated levels of circulating lymphocytes, immunoglobulins, and anti-nuclear antibodies. Both antigen processing and T cell anergy are abnormal in affected animals,8,15 and multiple organs are infiltrated with lymphocytes, particularly autoreactive B cells, that lead to fatal lung disease early in life.7,8,14
Human ITCH deficiency causes disease beyond the immune system, consistent with the role of protein ubiquitination in cell trafficking, functional activation, proteosomal degradation, and programmed cell death.16 In humans, ITCH is strongly expressed in the GI tract, pancreas, neuronal cells, and lymphoid tissue but is present in many other tissues (Protein Atlas; see Web Resources). The craniofacial abnormalities that lead to the distinctive facies in all of these children suggest a developmental role for ITCH in nonhematopoietic cells. The organomegaly and occasional cytoplasmic inclusions suggest possible storage of nonubiquitinated protein in the cells. Ubiquitin-related protein accumulation in the brain has been associated with some CNS diseases17 and may provide an explanation for psychomotor delays seen in affected children. A recent report suggests that ITCH might regulate NOD2 signaling, which could explain the bowel inflammation in two of these patients.18
The link between ITCH deficiency and immune dysregulation has implications for treatment. Screening for autoimmune disease is important in these children, and autoimmune manifestations in liver, intestine, and lung sometimes respond to immunosuppressive treatment. In patients who can be diagnosed early in life, such therapy could halt or slow progression of the disease. Moreover, exciting oncology research in recent years has focused on drugs that might affect ubiquitin pathways and thus allow more specific molecular therapies for the disorder.19 The severe phenotype in a few of the patients even raises consideration of bone marrow transplantation as a potential therapy.20
The discovery of involvement of a ubiquitin ligase in autoimmune disease has significance beyond this isolated Amish community. The disease demonstrates the vital importance of ubiquitin pathways in the cell signaling that controls T cell responsiveness in humans. The fact that only some of the ITCH-deficient patients had severe autoimmune disease suggests that, not surprisingly, other genetic modifiers or environmental factors contribute to T cell anergy. On the other hand, it is notable that other ubiquitin ligases in the cell fail to complement the defect enough to restore a normal phenotype.
Further studies will be needed to clarify the pathophysiology of ITCH deficiency. Morphologic examination of the affected tissues might further clarify the site of substrate accumulation or the specific subcellular site of toxicity. Functional assays will be necessary for determining the fate of ubiquitinated substrates such as JUN in the cell and for studying downstream signaling pathways such as those involving NOTCH. Detailed studies of immunological function, T cell regulation, and autoimmunity are underway.
Our report describes an approach to the elucidation of a novel genetic disorder within an isolated population. Prior to our investigations, several of these children had undergone extensive medical evaluations. The cause of their illness had not been found. The molecular diagnosis that relates the disease process to a mutation in the immune regulatory gene ITCH provides a new biological framework within which clinical observations and selected laboratory data can be used for studying the natural history of this disorder and evaluating therapies. As in our previous reports about genetic mapping in the Old Order Amish and Mennonite populations,21 this approach represents a cost-efficient way to discover the genetic basis of disease.
Acknowledgments
Naomi Lohr was funded by an American Academy of Pediatrics Resident Research Grant. The authors thank Vicki Haviland-Wilhite and Sharon McPheeters for expert secretarial assistance. They also thank the Amish families who shared their medical histories and allowed physical examinations and blood draws. Jean Molleston receives research funding support from Hoffman LaRoche.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance of Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Genome Browser, http://www.ncbi.nlm.nih.gov/
Single Nucleotide Polymorphism Database, http://www.ncbi.nlm.nih.gov/SNP/
Protein Atlas, http://www.proteinatlas.org/
References
- 1.Anandasabapathy N., Ford G.S., Bloom D., Holness C., Paragas V., Seroogy C., Skrenta H., Hollenhorst M., Fathman C.G., Soares L. GRAIL: An E3 ubiquitin ligase that inhibits cytokine gene transcription is expressed in anergic CD4+ T cells. Immunity. 2003;18:535–547. doi: 10.1016/s1074-7613(03)00084-0. [DOI] [PubMed] [Google Scholar]
- 2.Mueller D.L. E3 Ubiquitin ligases as T cell anergy factors. Nat. Immunol. 2004;5:883–890. doi: 10.1038/ni1106. [DOI] [PubMed] [Google Scholar]
- 3.Safford M., Collins S., Lutz M., Allen A., Huang C.T., Kowalski J., Blackford A., Horton M.R., Drake C., Schwartz R.H., Powell J.D. Egr-2 and Egr-3 are negative regulators of T cell activation. Nat. Immunol. 2005;6:472–480. doi: 10.1038/ni1193. [DOI] [PubMed] [Google Scholar]
- 4.Bilodeau P.S., Urbanowski J., Winistorfer S., Piper R. The Vps27p-Hse1p complex binds ubiquitin and mediates endosomal sorting. Nat. Cell Biol. 2002;4:534–539. doi: 10.1038/ncb815. [DOI] [PubMed] [Google Scholar]
- 5.Lu Q., Hope W., Brasch M., Reinhard C., Cohen S. TSG101 interaction with HRS mediates endosomal trafficking and receptor down-regulation. Proc. Natl. Acad. Sci. USA. 2003;100:7626–7631. doi: 10.1073/pnas.0932599100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jeon M.-S., Atfield A., Venuprasad K., Krawczyk C., Sarao R., Elly C., Yang C., Arya S., Bachmaier K., Su L. Essential role of the E3 ubiquitin ligase Cbl-b in T cell anergy induction. Immunity. 2004;21:167–177. doi: 10.1016/j.immuni.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 7.Matesic L., Copeland N., Jenkins N. Itchy mice: The identification of a new pathway for the development of autoimmunity. In: Buetler B., editor. Immunology, Phenotype First: How Mutations Have Established New Principles and Pathways in Immunology. Current Topics in Microbiology and Immunology. Springer-Verlag; Heidelberg: 2008. pp. 185–200. [DOI] [PubMed] [Google Scholar]
- 8.Matesic L.E., Haines D., Copeland N., Jenkins N. Itch genetically interacts with Notch1 in a mouse autoimmune disease model. Hum. Mol. Genet. 2006;15:3485–3497. doi: 10.1093/hmg/ddl425. [DOI] [PubMed] [Google Scholar]
- 9.Puffenberger E.G., Hu-Lince D., Parod J.M., Craig D.W., Dobrin S.E., Conway A.R., Donarum E.A., Strauss K.A., Dunckley T., Cardenas J.F. Mapping of sudden infant death with dysgenesis of the testes syndrome (SIDDT) by a SNP genome scan and identification of TSPYL loss of function. Proc. Natl. Acad. Sci. USA. 2004;101:11689–11694. doi: 10.1073/pnas.0401194101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Katzenstein A.L.A., Myers J.L. Nonspecific interstitial pneumonia and the other idiopathic interstitial pneumonias: Classification and diagnostic criteria. Am. J. Surg. Pathol. 2000;24:1–3. doi: 10.1097/00000478-200001000-00001. [DOI] [PubMed] [Google Scholar]
- 11.Kinder B.W., Collard H.R., Koth L., Daikh D.I., Wolters P.J., Elicker B., Jones K.D., King T.E., Jr. Idiopathic nonspecific interstitial pneumonia lung manifestation of undifferentiated connective tissue disease? Am. J. Respir. Crit. Care Med. 2007;176:691–697. doi: 10.1164/rccm.200702-220OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lorick K.L., Yang Y., Jensen J.P., Iwai K., Weissman A.M. Current Protocols in Cell Biology. Chapter 15, Unit 15.9. John Wiley and sons; Hoboken, NJ:: 2006. Studies of the ubiquitin proteasome system. [DOI] [PubMed] [Google Scholar]
- 13.Bachmaier K., Krawczyk C., Kozieradzki I., Kong Y.Y., Sasaki T., Oliveira-dos-Santos A., Mariathasan S., Bouchard D., Wakeham A., Itie A. Negative regulation of lymphocyte activation and autoimmunity by the molecular adaptor Cbl-b. Nature. 2000;403:211–216. doi: 10.1038/35003228. [DOI] [PubMed] [Google Scholar]
- 14.Parravicini V., Field A.-C., Tomlinson P.D., Basson M.A., Zamoyska R. Itch −/− αβ and γδ T cells independently contribute to autoimmunity in Itchy mice. Blood. 2008;111:4273–4282. doi: 10.1182/blood-2007-10-115667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang M., Veselits M., O'Neill S., Hou P., Reddi A.L., Berlin I., Ikeda M., Nash P.D., Longnecker R., Band H., Clark M.R. Ubiquitinylation of IgB dictates the endocytic fate of the B cell antigen receptor. J. Immunol. 2007;179:4435–4443. doi: 10.4049/jimmunol.179.7.4435. [DOI] [PubMed] [Google Scholar]
- 16.Hershko A., Ciechanover A., Varshavsky A. The ubiquitin system. Nat. Med. 2000;6:1073–1081. doi: 10.1038/80384. [DOI] [PubMed] [Google Scholar]
- 17.Martinez A., Potero-Otin M., Pamplona R., Ferrer I. Protein targets of oxidative damage in human neurodegenerative disease with abnormal protein aggregates. Brain Pathol. 2009 doi: 10.1111/j.1750-3639.2009.00326.x. Published online August 6, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tao M., Scacheri P.C., Marinis J.M., Harhaj E.W., Matesic L.E., Abbott D.W. ITCH K63-ubiquitinates the NOD2 binding protein, RIP2, to influence inflammatory signaling pathways. Curr. Biol. 2009;19:1255–1263. doi: 10.1016/j.cub.2009.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nalepa G., Rolfe M., Harper J.W. Drug discovery in the ubiquitin-proteasome system. Nat. Rev. Drug Discov. 2006;5:596–613. doi: 10.1038/nrd2056. [DOI] [PubMed] [Google Scholar]
- 20.Burt R.K., Slavin S., Burns W.H., Marmont A.M. Induction of tolerance in autoimmune disease by hematopoietic stem cell transplantation: getting closer to a cure? Blood. 2002;99:768–784. doi: 10.1182/blood.v99.3.768. [DOI] [PubMed] [Google Scholar]
- 21.Strauss K.A., Puffenberger E.G. Genetics, medicine, and the plain people. Annu. Rev. Genomics Hum. Genet. 2009;10:513–536. doi: 10.1146/annurev-genom-082908-150040. [DOI] [PubMed] [Google Scholar]
- 22.Eicher B.J., Schartz M.N. Print Shop; Gordonville, PA: 1990. Das Schwatz Buch Descendants of John J. Schwartz 1862–1990. pp. 17529. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.