

Abstract
We performed genome-wide homozygosity mapping in a large consanguineous family from Morocco and mapped the autosomal-recessive nonsyndromic hearing loss (ARNSHL) in this family to the DFNB79 locus on chromosome 9q34. By sequencing of 62 positional candidate genes of the critical region, we identified a causative homozygous 11 bp deletion, c.42_52del, in the TPRN gene in all seven affected individuals. The deletion is located in exon 1 and results in a frameshift and premature protein truncation (p.Gly15AlafsX150). Interestingly, the deleted sequence is part of a repetitive and CG-rich motive predicted to be prone to structural aberrations during crossover formation. We identified another family with progressive ARNSHL linked to this locus, whose affected members were shown to carry a causative 1 bp deletion (c.1347delG) in exon 1 of TPRN. The function of the encoded protein, taperin, is unknown; yet, partial homology to the actin-caping protein phostensin suggests a role in actin dynamics.
Main Text
Understanding the various mutational mechanisms and pathophysiological processes underlying inherited forms of hearing loss has an important impact on clinical patient management, genetic counseling, molecular diagnosis, and development of advanced therapeutic strategies. The approximate prevalence of hearing loss caused by genetic factors has been estimated to be at least 1 per 2000,1–3 and the most common hereditary form is autosomal-recessive nonsyndromic hearing loss (ARNSHL), accounting for > 70% of the cases.1 More than 60 loci for ARNSHL (DFNB) are known, and, to date, 32 of the corresponding genes have been identified (Hereditary Hearing Loss Homepage). Functional analysis of the encoded proteins provided intriguing insights into the complex structure of the inner ear as well as the mechanisms of hearing.4–6
Our strategy for identifying deafness genes is based mainly on both a collection of large consanguineous families with ARNSHL and the use of a homozygosity mapping approach. All subjects or their legal representatives gave written informed consent to the study, which was performed in accordance with the Declaration of Helsinki protocols and approved by the local institutional review boards. We ascertained 50 ARNSHL families from different regions of Morocco7 and 39 small ARNSHL families from The Netherlands. Participating members of all families underwent general otological examinations and pure-tone audiometry with air and bone conduction at 250 Hz, 500 Hz, 1000 Hz, 2000 Hz, 4000 Hz, and 8000 Hz (under sound-treated booth conditions). In family SF40 from Morocco (Figure 1A), all seven affected individuals were born to consanguineous parents and presented with severe to profound ARNSHL (Figure 1B). In affected individuals, hearing loss was reported to be congenital, with a progressive course leading to profound hearing loss. Developmental milestones were normal, and Romberg test did not reveal any symptoms of vestibular dysfunction in all patients.
Figure 1.
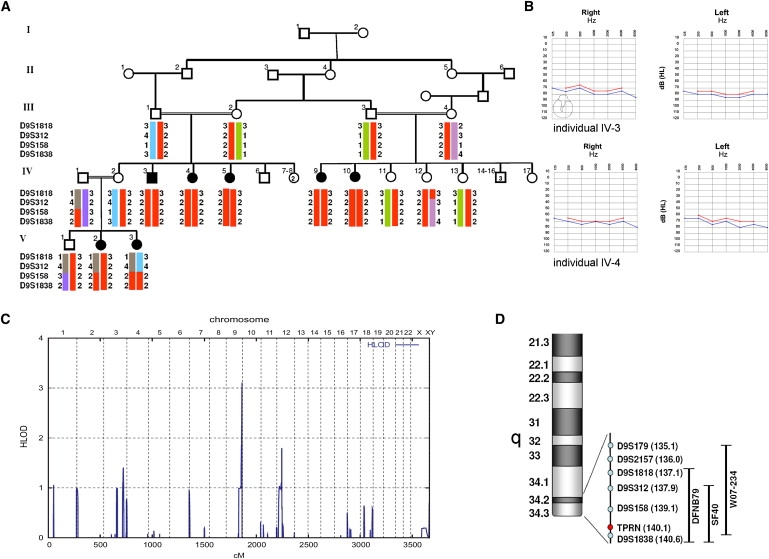
Mapping of the SF40 Family to the DFNB79 Locus
(A) Haplotype analysis in family SF40 showing homozygous haplotypes of the DFNB79 locus on chromosome 9q34.
(B) Audiograms of pure-tone audiometry with air (blue) and bone (red) conduction of individuals IV-3 (at 24 yrs of age) and IV-4 (at 21 yrs of age).
(C) Schematic representation of genome-wide LOD score calculations after 10K array SNP genotyping. LOD scores, calculated with ALLEGRO, are given along the y axis relative to genomic position in cM on the x axis. Note the highest peak in the telomeric region on chromosome 9q.
(D) Genomic overview of the chromosome 9q34 region and genomic localization of the used microsatellite markers. The black bars indicate the size and location of the described DFNB79 locus in comparison with the critical regions identified in the families SF40 and W07-234.
After exclusion of the GJB2 gene (MIM 121011), we genotyped DNAs from available SF40 family members by using the Affymetrix GeneChip Human Mapping 10K SNP Array (version 2.0). Genotypes were called by the GeneChip DNA Analysis Software (GDAS v2.0, Affymetrix). We verified sample genders by counting heterozygous SNPs on the X chromosome. Relationship errors were evaluated with the help of the Graphical Relationship Representation program.8 The PedCheck program was applied to detect Mendelian errors,9 and data for SNPs with such errors were removed from the data set. Nonparametric linkage analysis using all genotypes of a chromosome was simultaneously carried out with MERLIN. Parametric linkage analysis was performed by a modified version of the GENEHUNTER 2.1 program10,11 through stepwise use of a sliding window with sets of 110 or 300 SNPs and by the program ALLEGRO, assuming autosomal-recessive inheritance with full penetrance and a disease allele frequency of 0.0001.
We obtained a maximum LOD score of 3.11 for the telomeric region on chromosome 9q34.3 (Figure 1C). Subsequent fine mapping using microsatellite markers and including additional family members confirmed shared homozygosity of a 3.2 Mb region between D9S312 and the telomere in all affected individuals (Figures 1A and 1D). This region overlaps with the recently described DFNB79 locus (Figure 1D).12 A two-point LOD score of 4.62 for D9S158 was obtained (Table 1), under the assumption of an autosomal-recessive mode of inheritance with complete penetrance and a disease allele frequency of 0.001, with the use of LINKAGE software (version 5.2). A total of 88 protein-coding genes were located within the critical region (UCSC Genome Bioinformatics), and all genes were analyzed by their expression pattern and presumed functional properties (OMIM and Unigene). We sequenced the coding exons and adjacent splice sites of 62 of these positional candidate genes (Table S1, available online).
Table 1.
Two-Point LOD Score Results between the Disease in SF40 and Markers on Chromosome 9qter
| Marker | Physical Position |
Recombination Fraction (θ) |
||||||
|---|---|---|---|---|---|---|---|---|
| 0.0 | 0.01 | 0.05 | 0.1 | 0.2 | 0.3 | 0.4 | ||
| D9S1818 | 137135350–137135709 | 0.83 | 0.81 | 0.71 | 0.59 | 0.36 | 0.18 | 0.06 |
| D9S312 | 137919496–137919616 | −∞ | 2.48 | 2.80 | 2.62 | 1.97 | 1.24 | 0.53 |
| D9S158 | 139099048–139099430 | 4.62 | 4.52 | 4.08 | 3.53 | 2.41 | 1.31 | 0.38 |
| D9S1838 | 140636699–140636978 | 4.58 | 4.48 | 4.09 | 3.59 | 2.57 | 1.54 | 0.61 |
Highest LOD scores are shown in bold.
Sequencing of the four exons of the C9orf75 gene (named TPRN) revealed a homozygous 11 bp deletion in exon 1, c.42_52del, which cosegregated with the disease in all affected family members, was heterozygous in the parents, and was not found in 140 healthy control individuals from Morocco (Figure 2A). Screening of 35 index cases of additional Moroccan ARNSHL families did not reveal any other family carrying this mutation. The c.42_52del deletion is predicted to cause a frameshift and premature protein truncation (p.Gly15AlafsX150). Interestingly, the deletion is part of an 11 bp repetitive and CG-rich motive (Figure 2A). Also, the neighborhood of the c.42_52del mutation is very GC rich. It is well known that both (a) repetitive nucleotide stretches and (b) an open chromatin structure, which is found in GC-rich regions, can strongly contribute to the occurrence of structural alterations such as deletions and duplications.13 These repetitive sequences might increase the instability of DNA to induce either deletions or duplications by the molecular mechanisms of simple replication slippage, sister-chromosome slipped misalignment, and single-strand annealing.14 Therefore, this part of exon 1 is predicted to be prone to small structural alterations. Our hypothesis that the c.42_52del mutation might represent a mutational hotspot is further supported by the finding of Rehman et al.,15 who describe the same c.42_52del mutation in a large ARNSHL pedigree from Pakistan, as well as another Pakistani family with a duplication within this repeat, c.44_54 dup. Taken together, the data from both studies suggest that mutations in TPRN could be a more frequent cause of ARNSHL, resulting from the mutational mechanisms described above.
Figure 2.
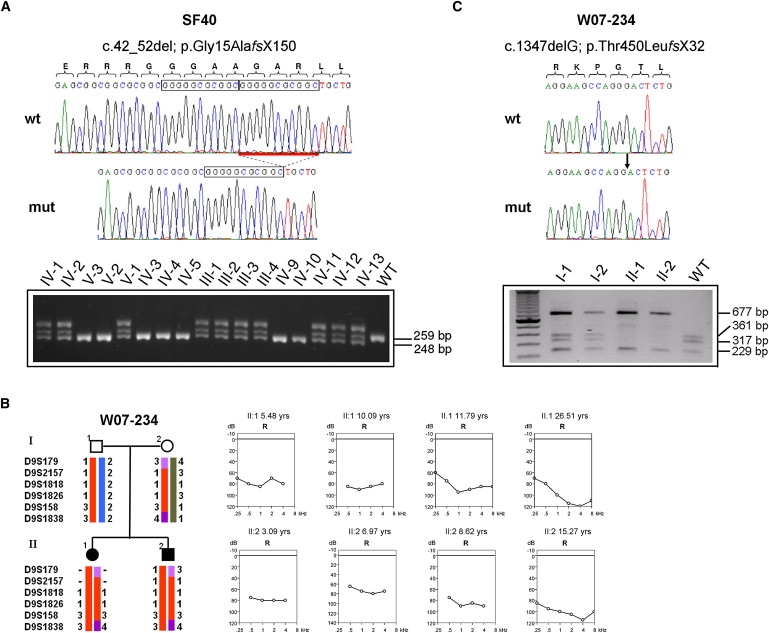
Identification of Mutations in TPRN in Two ARNSHL Families
(A and C) Sequence chromatograms showing the identified mutations in the TPRN gene. (A) The homozygous c.42_52del mutation affects a repetitive motive, as indicated by the boxes. Genotypes of the c.42_52del mutation in 17 family members of the SF40 family were analyzed by PCR amplification and separation on a 4% agarose gel. Fragment sizes are indicated. The upper band represents a heteroduplex fragment. (B) Haplotypes of microsatellite markers of the DFNB79 region and audiograms of both affected individuals in the Dutch family W07-234 at different time points, showing the progression of hearing impairment. (C) Chromatogram of the homozygous c.1347delG mutation identified in the W07-234 family in comparison with a normal wild-type sequence. The mutation removes a restriction site for BsmFI. Genotypes of parents and mutation-affected siblings were analyzed with the use of a BsmFI digestion after PCR amplification. Restriction fragments were separated on a 1% agarose gel, resulting in four fragments for the wild-type (361 bp, 317 bp, 229 bp, and 80 bp in size) and three fragments for the homozygous mutant (677 bp, 229 bp, and 80 bp in size).
Among the Dutch families, we also identified a family with a TPRN mutation. In both affected subjects of family W07-234 (Figure 2B), results of the Ewing hearing distraction test at 9 mo of age were normal. Sensorineural hearing loss was diagnosed at the ages of 4 yrs, 3 mo and 2 yrs, 9 mo in patients II-1 and II-2, respectively. At these ages, speech development was delayed in both individuals. In patient II-1, the hearing impairment progressed from severe to profound between age at diagnosis and 26 yrs of age. The hearing loss in patient II-2 was severe at diagnosis and profound at the age of 15 (Figure 2B). Individual longitudinal linear regression analysis of binaural mean air-conduction threshold values on age was performed for both affected individuals. The annual threshold deterioration (ATD) was calculated, and the progression was called significant if the 95% confidence interval did not include zero. The level of significance used in all tests was p = 0.05. For individual II-1, the ATD increased from 0.42 at 0.25 kHz to 2.0 at 4 kHz. The progression at 2 kHz (ATD 1.85) was found to be statistically significant. For individual II-2, the ATD was 1.90, 1.91, 1.76, and 2.70 at 0.5 kHz, 1 kHz, 2 kHz, and 4 kHz, respectively. Progression was not statistically significant for any of these frequencies. The lack of significance in progression is likely to be due to the relatively large variation in the measurements during childhood and the low number of measurements. No additional clinical abnormalities were seen in the patients in ear, nose, and throat (ENT) and pediatric examinations, and no retinal abnormalities were detected in funduscopy (individual II-2). GJB2 was initially excluded. Given the fact that with 16 million inhabitants, the Dutch population is rather small and that population mixture by migration and mating could be lower than expected, we used a homozygosity mapping strategy in Dutch ARNSHL families without obvious consanguinity. Only affected individuals from families were analyzed for homozygous stretches with the use of the Affymetrix GeneChip Human Mapping 250K NspI SNP Array. Genotype calling and detection of homozygous regions was performed with the Genotyping Console software from Affymetrix. In family W07-234, the largest overlapping homozygous region, of 9.59 Mb, was located on chromosome 9q22.32-q31.2 (flanking SNPs: SNP_A-2028693, SNP_A-2061426). The second largest overlapping homozygous region, of 5.21 Mb, was identified on chromosome 9q34.13-34.3 (flanking SNPs: SNP_A-1803621, SNP_A-2186204), which partly overlapped with the DFNB79 locus (Figure 1D). In total, 11 shared homozygous regions greater than 1 Mb were identified for family W07-234. Independent analysis using microsatellite markers confirmed the DFNB79-overlapping homozygous stretch in affected family members (Figure 2B). In both siblings, we found a 1 bp deletion, c.1347delG, in exon 1 of the TPRN gene (Figure 2C). The deletion is predicted to cause a frameshift and a premature protein truncation (p.Thr450LeufsX32). Parents are heterozygous carriers, and the mutation was not detected in 160 healthy Dutch control individuals, tested by PCR and restriction enzyme digestion using BsmFI (Figure 2C).
Our data provide evidence that loss-of-function mutations in TPRN cause ARNSHL. Interestingly, the hearing phenotype caused by TPRN mutations shows a progressive course in the Dutch family described here. Speech acquisition—although delayed—was possible in the Dutch family without the use of hearing aids, but hearing impairment progressed in affected individuals, becoming profound by young adulthood. Most of the recessively inherited forms of ARNSHL show a congenital onset and more severe affection. Progression of ARNSHL has as yet been associated with mutations in only a few other genes, namely MYO3A (MIM 606808), TMPRSS3 (MIM 605511), PJVK (MIM 610219), and LOXHD1 (MIM 613072).16–20 TPRN-related hearing loss represents another example of this characteristic phenotype.
TPRN isoform 1 encodes a protein 650 amino acids (aa) in length, named taperin, with yet unknown function. Different transcripts of the gene are described (Figure 3A). The expression of the TPRN mouse ortholog C430004E15Rik (NM_175286.4) was tested in postnatal day 2 (P2) mice via RT-PCR experiments. We detected expression of TPRN in all tissues investigated, including cochlea (Figure 3B). Furthermore, TPRN expression was detected in human fetal cochlea (Figure 3C). In an accompanying paper, Rehman et al. show that taperin is mainly expressed at the base of inner ear stereocilia in a region referred to as the taper.15 Future investigations of expression profiles of TPRN in different stages of inner ear development will determine the spatiotemporal expression in the cochlea and the central auditory pathway. Homology searches identified a 58 aa stretch of taperin (482–539 aa) with a 76% similarity to phostensin, a protein involved in actin dynamics through capping to pointed ends of actin filaments.21 Whether taperin has a similar functional role in actin dynamics remains to be elucidated in the future.
Figure 3.
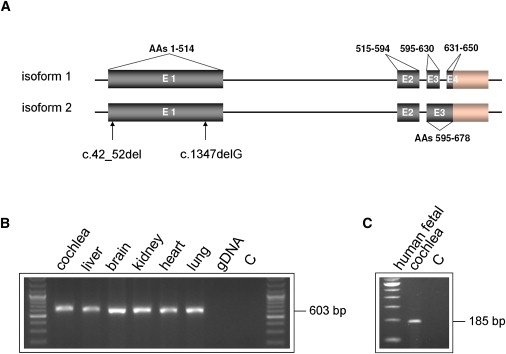
TPRN Structure and Expression
(A) Schematic representation of described TPRN transcripts (NM_001128228.1, NM_173691.3), encoded amino acids of the TPRN protein (NP_775962.2, NP_001121700.1), and location of identified mutations.
(B and C) Analysis of TPRN expression by RT-PCR in different tissues in P2 mouse and human fetal cochlea (gDNA, genomic DNA; C, water control). PCR primers for mouse RT-PCR were located in exon 1 (forward, 5′-TCTGACACAGACAAGTGTGTTAGG-3′) and exon 2 (reverse, 5′-CAGAG AGCTCTCAGAAGGGTACTC-3′) of TPRN. Primers for RT-PCR on human fetal cochlea were located in exon 3 (forward, 5′-CTCAGGCCTGTCCAGCTAC-3′) and exon 4 (reverse, 5′-AGTGCTGGGCTCAGAAATAC-3′).
In conclusion, we provide evidence that mutations in TPRN cause ARNSHL charcterized by a progressive course of hearing impairment. The identified c.42_52del deletion is a recurrent mutation possibly originating by recombination errors due to a GC-rich and repetitive nature of this region. Future screening of additional ARNSHL families of different origins will provide important information about the frequency of TPRN mutations.
Acknowledgments
We thank all family members who participated in this study. We also thank Michaela Thoenes, Esther Milz, and Nicole Weegerink for preparing the figures of the audiograms and the longitudinal linear regression analysis and Karin Boss for critically reading the manuscript. This work was supported by the European Commission FP6 Integrated Project EUROHEAR (LSHG-CT-20054-512063 to C.K., H.K., and B.W.), by a German Federal Ministry of Education and Research (BMBF) grant under the E-RARE network CRANIRARE consortium (01GM0801 to B.W.), by the RNID (GR36 to H.K.), by Fonds NutsOhra (project SNO-T-0702-102 to H.K.), and by grants from the Janivo Stichting (to H.K.), and the Heinsius Houbolt Foundation (to H.K.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Ensembl, http://www.ensembl.org/index.html
Hereditary Hearing Loss Homepage, http://webhost.ua.ac.be/hhh/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/omim/
UCSC Genome Bioinformatics, http://www.genome.ucsc.edu/
Unigene, http://www.ncbi.nlm.nih.gov/unigene
References
- 1.Morton N.E. Genetic epidemiology of hearing impairment. Ann. N Y Acad. Sci. 1991;630:16–31. doi: 10.1111/j.1749-6632.1991.tb19572.x. [DOI] [PubMed] [Google Scholar]
- 2.Mehl A.L., Thomson V. Newborn hearing screening: the great omission. Pediatrics. 1998;101:E4. doi: 10.1542/peds.101.1.e4. [DOI] [PubMed] [Google Scholar]
- 3.Mehl A.L., Thomson V. The Colorado newborn hearing screening project, 1992-1999: on the threshold of effective population-based universal newborn hearing screening. Pediatrics. 2002;109:E7. doi: 10.1542/peds.109.1.e7. [DOI] [PubMed] [Google Scholar]
- 4.Dror A.A., Avraham K.B. Hearing loss: mechanisms revealed by genetics and cell biology. Annu. Rev. Genet. 2009;43:411–437. doi: 10.1146/annurev-genet-102108-134135. [DOI] [PubMed] [Google Scholar]
- 5.Brown S.D., Hardisty-Hughes R.E., Mburu P. Quiet as a mouse: dissecting the molecular and genetic basis of hearing. Nat. Rev. Genet. 2008;9:277–290. doi: 10.1038/nrg2309. [DOI] [PubMed] [Google Scholar]
- 6.Gillespie P.G., Müller U. Mechanotransduction by hair cells: models, molecules, and mechanisms. Cell. 2009;139:33–44. doi: 10.1016/j.cell.2009.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boulouiz R., Li Y., Soualhine H., Abidi O., Chafik A., Nürnberg G., Becker C., Nürnberg P., Kubisch C., Wollnik B., Barakat A. A novel mutation in the Espin gene causes autosomal recessive nonsyndromic hearing loss but no apparent vestibular dysfunction in a Moroccan family. Am. J. Med. Genet. A. 2008;146A:3086–3089. doi: 10.1002/ajmg.a.32525. [DOI] [PubMed] [Google Scholar]
- 8.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. GRR: graphical representation of relationship errors. Bioinformatics. 2001;17:742–743. doi: 10.1093/bioinformatics/17.8.742. [DOI] [PubMed] [Google Scholar]
- 9.O'Connell J.R., Weeks D.E. PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am. J. Hum. Genet. 1998;63:259–266. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kruglyak L., Daly M.J., Reeve-Daly M.P., Lander E.S. Parametric and nonparametric linkage analysis: a unified multipoint approach. Am. J. Hum. Genet. 1996;58:1347–1363. [PMC free article] [PubMed] [Google Scholar]
- 11.Strauch K., Fimmers R., Kurz T., Deichmann K.A., Wienker T.F., Baur M.P. Parametric and nonparametric multipoint linkage analysis with imprinting and two-locus-trait models: application to mite sensitization. Am. J. Hum. Genet. 2000;66:1945–1957. doi: 10.1086/302911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khan S.Y., Riazuddin S., Shahzad M., Ahmed N., Zafar A.U., Rehman A.U., Morell R.J., Griffith A.J., Ahmed Z.M., Riazuddin S., Friedman T.B. DFNB79: reincarnation of a nonsyndromic deafness locus on chromosome 9q34.3. Eur. J. Hum. Genet. 2009;18:125–129. doi: 10.1038/ejhg.2009.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Costantini M., Bernardi G. Mapping insertions, deletions and SNPs on Venter's chromosomes. PLoS ONE. 2009;4:e5972. doi: 10.1371/journal.pone.0005972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bzymek M., Lovett S.T. Instability of repetitive DNA sequences: the role of replication in multiple mechanisms. Proc. Natl. Acad. Sci. USA. 2001;98:8319–8325. doi: 10.1073/pnas.111008398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rehman A.U., Morell R.J., Khan S.Y., Belyantseva I.A., Boger E.T., Shazad M., Ahmed Z.M., Riazuddin S., Khan S.N., Riazuddin S., Friedman T.B. Targeted capture and next-generation sequencing identifies C9orf75, encoding taperin, as the mutated gene in nonsyndromic deafness DFNB79. Am. J. Hum. Genet. 2010;86:378–388. doi: 10.1016/j.ajhg.2010.01.030. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walsh T., Walsh V., Vreugde S., Hertzano R., Shahin H., Haika S., Lee M.K., Kanaan M., King M.C., Avraham K.B. From flies' eyes to our ears: mutations in a human class III myosin cause progressive nonsyndromic hearing loss DFNB30. Proc. Natl. Acad. Sci. USA. 2002;99:7518–7523. doi: 10.1073/pnas.102091699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elbracht M., Senderek J., Eggermann T., Thürmer C., Park J., Westhofen M., Zerres K. Autosomal recessive postlingual hearing loss (DFNB8): compound heterozygosity for two novel TMPRSS3 mutations in German siblings. J. Med. Genet. 2007;44:e81. doi: 10.1136/jmg.2007.049122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scott H.S., Kudoh J., Wattenhofer M., Shibuya K., Berry A., Chrast R., Guipponi M., Wang J., Kawasaki K., Asakawa S. Insertion of beta-satellite repeats identifies a transmembrane protease causing both congenital and childhood onset autosomal recessive deafness. Nat. Genet. 2001;27:59–63. doi: 10.1038/83768. [DOI] [PubMed] [Google Scholar]
- 19.Schwander M., Sczaniecka A., Grillet N., Bailey J.S., Avenarius M., Najmabadi H., Steffy B.M., Federe G.C., Lagler E.A., Banan R. A forward genetics screen in mice identifies recessive deafness traits and reveals that pejvakin is essential for outer hair cell function. J. Neurosci. 2007;27:2163–2175. doi: 10.1523/JNEUROSCI.4975-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grillet N., Schwander M., Hildebrand M.S., Sczaniecka A., Kolatkar A., Velasco J., Webster J.A., Kahrizi K., Najmabadi H., Kimberling W.J. Mutations in LOXHD1, an evolutionarily conserved stereociliary protein, disrupt hair cell function in mice and cause progressive hearing loss in humans. Am. J. Hum. Genet. 2009;85:328–337. doi: 10.1016/j.ajhg.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lai N.S., Wang T.F., Wang S.L., Chen C.Y., Yen J.Y., Huang H.L., Li C., Huang K.Y., Liu S.Q., Lin T.H., Huang H.B. Phostensin caps to the pointed end of actin filaments and modulates actin dynamics. Biochem. Biophys. Res. Commun. 2009;387:676–681. doi: 10.1016/j.bbrc.2009.07.086. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.