

Abstract
Segmental duplications, which comprise ∼5%–10% of the human genome, are known to mediate medically relevant deletions, duplications, and inversions through nonallelic homologous recombination (NAHR) and have been suggested to be hot spots in chromosome evolution and human genomic instability. We report seven individuals with microdeletions at 17q23.1q23.2, identified by microarray-based comparative genomic hybridization (aCGH). Six of the seven deletions are ∼2.2 Mb in size and flanked by large segmental duplications of >98% sequence identity and in the same orientation. One of the deletions is ∼2.8 Mb in size and is flanked on the distal side by a segmental duplication, whereas the proximal breakpoint falls between segmental duplications. These characteristics suggest that NAHR mediated six out of seven of these rearrangements. These individuals have common features, including mild to moderate developmental delay (particularly speech delay), microcephaly, postnatal growth retardation, heart defects, and hand, foot, and limb abnormalities. Although all individuals had at least mild dysmorphic facial features, there was no characteristic constellation of features that would elicit clinical suspicion of a specific disorder. The identification of common clinical features suggests that microdeletions at 17q23.1q23.2 constitute a novel syndrome. Furthermore, the inclusion in the minimal deletion region of TBX2 and TBX4, transcription factors belonging to a family of genes implicated in a variety of developmental pathways including those of heart and limb, suggests that these genes may play an important role in the phenotype of this emerging syndrome.
Main Text
Segmental duplications comprise ∼5%–10% of the human genome.1 Misalignment of segmental duplications during meiosis can mediate genomic instability by nonallelic homologous recombination (NAHR). Depending on the orientation of the segmental duplications, NAHR can generate microdeletions, microduplications, and inversions of the intervening genomic sequence.2–7 Chromosomal rearrangements associated with segmental duplications include microdeletions at 3q29 (MIM 609425) and their reciprocal microduplications (MIM 611936);8 Williams syndrome (MIM 194050) and deletions at 7q11.23; Angelman (MIM 105830) and Prader-Willi syndromes (MIM 176270) and maternally and paternally derived deletions, respectively, of 15q11-q13; microdeletions at 16p11.2p12.2 and their reciprocal microduplications;9 Smith-Magenis syndrome (MIM 182290) and deletions at 17p11.2; duplication at 17p12 in Charcot-Marie-Tooth disease type 1A (MIM 604563); and the reciprocal deletion at 17p12 resulting in hereditary neuropathy with liability to pressure palsies (HNPP [MIM 162500]) (reviewed in 4). Recently, a recurrent microdeletion at 17q21.3 mediated by segmental duplications was identified by screening individuals with idiopathic mental retardation and congenital anomalies with a microarray targeting potential segmental duplication-rich rearrangement “hot spots” in the genome,5–7 suggesting that other such rearrangements may yet be identified.
Recent evidence suggests another segmental duplication “hub” on chromosome 17 at 17q23. Breakpoint analysis of a paracentric inversion that occurred in the human/chimpanzee/gorilla ancestor revealed that the distal breakpoint maps to the region syntenic to human 17q23, which suggests that the presence of this duplicon mediated the inversion in the Homo sapiens/Pan troglodytes/Gorilla gorilla ancestor.10 Furthermore, at least two insertions and deletions and one translocation have occurred within the past six million years since the divergence of chimpanzees and humans, indicating that these regions have continued to be subject to more-recent local rearrangements. Further data have demonstrated the structural complexity of the region, which has been shown to be polymorphic in the human population and associated with a multiple sclerosis susceptibility locus on 17q22-q24.11,12
Here we report the clinical and molecular characterization of seven individuals with microdeletions at 17q23.1q23.2. Patients 1–3 in this study were ascertained by Signature Genomic Laboratories. Patients 4 and 5 were ascertained by Nationwide Children's Hospital. Patient 6 was ascertained by Emory University School of Medicine. Patient 7 was ascertained by Baylor College of Medicine. Informed consent was obtained from patients 1, 4, 6, and 7 to publish photographs with consent forms approved by the Institutional Review Board of respective institutions.
All seven individuals with microdeletions at 17q23 were initially identified by microarray-based comparative genomic hybridization (aCGH) with various microarray platforms, one of which, patient 6, was previously reported (Figure 1).13 Whole-genome bacterial artificial chromosome (BAC)-based microarray analysis was originally performed on DNA from patients 1 and 2 with a >4600 clone custom microarray as previously described.14 Oligonucleotide-based microarray analysis was originally performed on DNA from patients 3–5 with a custom 105K-feature whole-genome microarray (Agilent Technologies), as previously described.14 The deletion in patient 6 was identified with a custom 44K oligonucleotide-based microarray (Agilent Technologies), as previously described.13,15 The deletion in patient 7 was originally identified with a custom-designed 105K-feature whole-genome oligonucleotide microarray (Agilent Technologies), as previously described.16 In addition, all patients for whom DNA was available (1–4, 6, and 7) were reanalyzed at higher resolution with an off-the-shelf 244K-feature whole-genome microarray (Agilent Technologies), as previously described.14 One individual, patient 2, had a 2.8 Mb deletion (chromosome 17: 54.8–57.6 Mb); the remaining six individuals had 2.2 Mb deletions (chromosome 17: 55.4–57.6 Mb).
Figure 1.
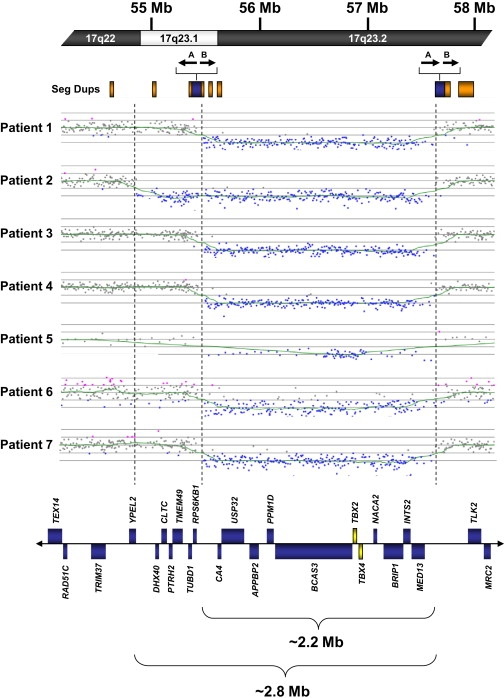
Summary of Microarray Analysis of Individuals with Copy Number Imbalances at 17q23.1q23.2
At the top of the figure is a partial idiogram showing chromosome bands 17q22-17q23.2 with genomic coordinates corresponding to the hg18 build of the human genome. Orange boxes represent segmental duplications (seg dups), with paired segmental duplications indicated by blue boxes. The location and orientation of the 18 kb inverted duplication and the 15 kb direct duplication contained within the paired segmental duplications are indicated by arrows marked A and B, respectively. The complex segmental duplication structure of this region has been simplified for illustrative purposes. Diagrams show the deletions in seven individuals with microdeletions at 17q23.1q23.2. Six of the seven deletions were refined with a high-density, 244K oligonucleotide microarray (patients 1–4, 6, and 7). A 105K oligonucleotide microarray was used for patient 5. Vertical dashed lines indicate the 2.2 and 2.8 Mb deletion sizes in the individuals. Blue boxes represent all known genes annotated in OMIM in the deletion region. OMIM genes TBX2 and TBX4 are shown as yellow boxes.
All seven deletions were confirmed and visualized by fluorescence in situ hybridization (FISH) with BAC clones, as previously described (Figure 2).17 Parental FISH testing in five of the seven cases confirmed an apparently de novo origin (patients 2 and 4–7). The deletions in patients 1–3 and 5 were confirmed by FISH with BAC probe RP11-289K16 from 17q23.1. For case 4, BAC RP11-119J7 was used to confirm the deletion. The deletion in patient 6 was confirmed by FISH with BAC RP11-436E15, as previously described,13,15 and the deletion in patient 7 was confirmed by FISH with BAC clone RP11-615P24. Microsatellite marker analysis with the Identifiler kit (Applied Biosystems) showed correct paternity in patient 6, confirming the de novo origin of the deletion (Rudd et al.13). All other parental samples were unavailable.
Figure 2.
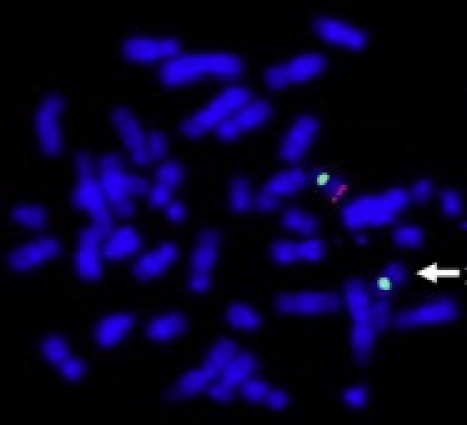
Representative FISH Image Showing Deletion at 17q23 in Patient 2
BAC clone RP11-289K16 from 17q23.1 is labeled in red, and chromosome 17 centromere probe D17Z1 is labeled in green as a control. The absence of a red signal on one homolog (arrow) indicates deletion at 17q23.1.
Computational analysis of the 17q23.1q23.2 region with the hg18 build of the UCSC genome browser18 and the hg17 build of the Human Genome Segmental Duplication Database identified a complex arrangement of segmental duplications, some of which directly flank the 2.2 Mb deletion breakpoints and the distal 2.8 Mb breakpoint (Figure 1). These flanking segmental duplications are ∼100 kb in size and have a complex evolutionary structure10 that includes an ∼15 kb segment of >98% sequence identity present in the same orientation. In addition, there is an ∼18 kb segment of >94% sequence identity that is inverted in orientation with respect to its duplication partner. The 2.2 Mb deletions are flanked by the homologous segmental duplications present in the same orientation. Highly identical (>98% sequence identity) segmental duplications in direct orientation flanking the deletion breakpoints are consistent with NAHR-mediated rearrangements (Figure 1). Although six of seven deletions were flanked by segmental duplications, the proximal breakpoint of the 2.8 Mb deletion falls between two segmental duplications (Figure 1). Atypical breakpoints have been reported for other recurrent rearrangements mediated by segmental duplications: for example, some of the rarer rearrangements of 17p11.2 associated with Smith-Magenis syndrome do not have breakpoints flanked by the typical paired segmental duplications and are not associated with known genomic architectural features,19 and some of the breakpoints in the recently identified 16p11.2p12.2 microdeletion syndrome are not flanked by segmental duplications.9
Clinical characterization of the seven individuals with microdeletions at 17q23.1q23.2 revealed multiple common clinical features (Table 1; see also Supplemental Discussion available online). All individuals had mild to moderate developmental delay. A majority had low birth weight (5 of 7); microcephaly or relative microcephaly (5 of 7), including one with overall small growth parameters; postnatal growth retardation (5 of 7); heart defect(s) (6 of 7), in most cases either patent ductus arteriosus or atrial septal defect (ASD); hand and foot anomalies including long, thin fingers and toes (7 of 7); and musculoskeletal abnormalities of varying severity (4 of 7). Features identified in more than one individual include aggressive behavior (2 of 7) and hearing loss (2 of 7). Although five of seven individuals had eye anomalies such as chalazion, stellate pattern of the irises, retinopathy of prematurity, and esotropia, the anomalies affect different components of the eye and are unlikely to be related; therefore, these anomalies are likely incidental to the 17q23.1q23.2 microdeletion phenotype. Although all individuals had at least mild dysmorphic facial features, there was no characteristic constellation of features that would elicit clinical suspicion of a specific disorder (Figure 3). Nonetheless, the common clinical features in these individuals together with the common size and gene content of the deleted regions suggest that deletion at 17q23.1q23.2 is causative of the individuals' phenotypes and may constitute a novel microdeletion syndrome.
Table 1.
Clinical Features in Subjects with Microdeletions at 17q23.2
| Patient Number | 1 | 2a | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| Age | 3 yrs | 5 yrs, 1 mo. | 4 yrs | 16 yrs, 6 mo. | 8 mo. | 4 yrs | 1 yr, 10 mo. |
| Sex | Female | Female | Male | Female | Female | Female | Female |
| Low birth weight (≤25th percentile) | + | + | − | + | + | + | − |
| Postnatal growth retardation | + | − | − | + | + | + | + |
| Microcephaly (<5th percentile) | + | + | + (relative) | − | +b | − | + |
| Mild to moderate developmental delays | + | + | + | + | + | + | + |
| Heart defects | PDA | ASD secundum | Pulmonary hypertension | PDA with pulmonary hypertension | PDA; bicuspid aortic valve | None; no echocardiography | ASD secundum with pulmonary hypertension |
| Musculoskeletal | Tibial torsion | Abnormally laterally positioned ossification centers of the patella, delayed for age | Underossified femoral heads; hips deeply seated in acetabulae; mottled femoral epiphyses and metaphyses; small and abnormal lower-limb epiphyses | Short stature with leg length discrepancy; hypoplasia of patellae and tibial epiphyses; shallow acetabulae; right side short femoral neck with coxa vara and magna; scoliosis | NR | None | Scoliosis; limited extension of knees and elbows |
| Hearing loss | + | − | − | + | − | − | − |
| Eyes | Chalazion | Left sided esotropia | Normal | Bilateral esotropia | Retinopathy of prematurity | Stellate pattern of irises | Normal |
| Facial features | High eyebrows; right epicanthal fold | Hypertelorism; flattened nasal bridge and midface; simple right ear | Plagiocephaly; bilateral inner epicanthal folds; bulbous, bifid nose; posteriorly rotated, prominent ears; long eyelashes | Prominence of forehead; moderate dental crowding requiring orthodontic appliances | Box-shaped cranium; open anterior fontanelle; prominent forehead; wide-set eyes, small nose with bulbous nasal tip; microstomia; small ears; recessed jaw | Mild frontal bossing; rounded nasal tip; wide space between front two teeth | Mild micrognathia; long eyelashes; protuberant ears; small mouth; long neck |
| Hands and feet | Thin fingers; deep, grooved space between first and second toes; familial 2–3 syndactyly | Long, thin fingers and toes | Long, thin fingers and toes; second toes longer than great toes | Long, thin fingers and toes; digital clubbing; bilateral pes planus; slightly shortened foot rays | Long fingers and toes | Long fingers and toes; overlapping toes | Congenital contractures; long fingers and toes; overlapping toes 4 and 5; pes planus; fifth finger clinodactyly |
| Other | Behavioral abnormalities; intention tremors; asthma | NR | Tethered cord; sacral dimples; shawl scrotum | Mild increased DTR at knees and ankles; migraines | Cutis aplasia at birth; central hypotonia; brisk DTRs | Behavioral abnormalities | NR |
The following abbreviations are used: +, feature present; −, feature absent; PDA, patent ductus arteriosus; ASD, atrial septal defect; DTR, deep tendon reflexes; NR, none reported.
Patient 2 has an atypical ∼2.8 Mb deletion. All other patients described in this table have ∼2.2 Mb deletions.
All growth parameters were at the fifth percentile.
Figure 3.
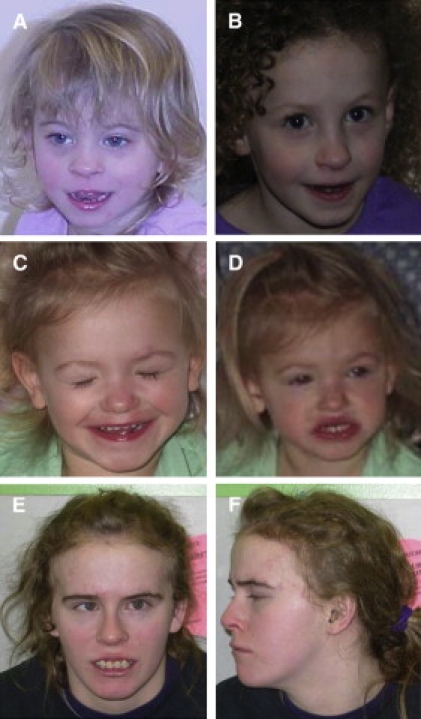
Facial Features of Individuals with Microdeletion at 17q23.1q23.2
(A) Patient 1 at 3 years. Note high eyebrows and right epicanthal fold.
(B) Patient 6 at 4 years. Note mild frontal bossing and rounded nasal tip.
(C and D) Patient 7 at 22 months. Note long eyelashes, protuberant ears, small mouth, and mild micrognathia.
(E and F) Patient 4 at 16 years. With the exception of some prominence of the forehead, this patient was not noted to be dysmorphic.
From November 2007 to October 2009, Signature Genomic Laboratories tested 19,912 patients with whole-genome microarray platforms that had coverage over 17q23.1q23.2 and identified three microdeletions of 17q23.1q23.2, for a frequency of 0.015% among its patient population. By comparison, during this same period, the laboratory identified 23 Smith-Magenis syndrome (SMS) deletions (0.12% of patient population), which has a frequency in the general population of ∼1 in 15,000,20 suggesting that the population frequency of 17q23.1q23.2 microdeletions may be ∼1 in 115,000. This may be an overestimate of frequency because some cases of SMS are diagnosed by other methods, and therefore not all individuals with these syndromes will have aCGH testing, whereas 17q23.1q23.2 microdeletions would not be expected to be diagnosed by other methods that require clinical suspicion of a specific disorder.
Because the phenotype of the individual with the larger 2.8 Mb deletion does not appear to differ from that of the individuals with the smaller 2.2 Mb deletion, the critical region for this syndrome likely lies inside this 2.2 Mb region. Of the 11 OMIM and RefSeq genes within the 2.2 Mb smallest region of overlap (Figure 1), at least two, TBX2 (MIM 600747) and TBX4 (MIM 601719), are strong candidates that might play a role in some of the features of individuals with this microdeletion.
TBX2 and TBX4 belong to an ancient family of genes present in divergent multicellular organisms, such as sponges and humans, that encode transcription factors characterized by a strongly conserved, sequence-specific DNA-binding domain (or T-box domain). More specifically, TBX2 and TBX4 are members of two closely related T-box subfamilies, TBX2/3 and TBX4/5. Interestingly, at least 17 evolutionary rearrangements, including gene cluster insertions and deletions, one inversion, and one local translocation, have occurred within the region since the divergence of humans and chickens ∼300 million years ago. Despite the instability of the region in which the gene pair resides, TBX2 and TBX4 (as well as TBX3 and TBX5) have remained flanked by several loci present in all amniotes, BCAS33 and BRIP1 (MIM 605822) (THRAP2 [MIM 608711] and RBM19 flank TBX3 [MIM 601621] and TBX5 [MIM 601620]).21 The high conservation of these gene clusters indicates a functional significance. Alternatively, the presence of large segmental duplications in the region of 17q23 may predispose the region to rearrangements that conserve the linkage of these genomic segments.
Targeted gene deletions in mouse have demonstrated that T-box genes play numerous roles in development; consequently, disruptions of many of these genes have been associated with human disease. Mutations in the coding sequence of TBX1 (MIM 602054), which maps within the DiGeorge syndrome (DGS) region (MIM 188400) on chromosome 22q11.2,22 have been identified in individuals with sporadic DGS/conotruncal anomaly face syndrome/velocardiofacial syndrome (VCFS),23–25 which has a characteristic clinical feature of outflow tract heart defects. In addition, heterozygous mutations in TBX3 cause ulnar-mammary syndrome (UMS [MIM 181450]),26,27 which has a characteristic feature of posterior upper-limb abnormalities; heterozygous loss-of-function mutations of TBX4 in 15 individuals from five families and one sporadic patient were found to cause the autosomal-dominant Scott-Taor or small patella syndrome (SPS [MIM 147891]);28 heterozygous mutations in TBX5 cause anterior limb abnormalities and ASD in Holt-Oram syndrome (MIM 142900);27 and mutations of TBX20 (MIM 606061) have been associated with congenital heart disease and cardiac developmental anomalies, including defects in septation, chamber growth, and valvulogenesis and cardiomyopathy.29
The TBX2/3 and TBX4/5 subfamilies of genes are especially interesting as haploinsufficiency candidates in microdeletions at 17q23 because of their role in the evolution of novel vertebrate structures, including mouse limbs.30
Tbx2 is expressed in the anterior and posterior margins of forelimbs and hindlimbs,31–33 and loss-of function and misexpression studies suggest a role for Tbx2 in the anteroposterior patterning of digits.34 All individuals reported here had long fingers and toes, and some had additional digital anomalies (Table 1); patient 4, for example, had slightly shortened fourth and fifth rays of both feet (Figure 4). The presence of subtle digital anomalies in individuals with microdeletions at 17q23.1q23.2 suggests that haploinsufficiency of TBX2 plays a role in the phenotype.
Figure 4.
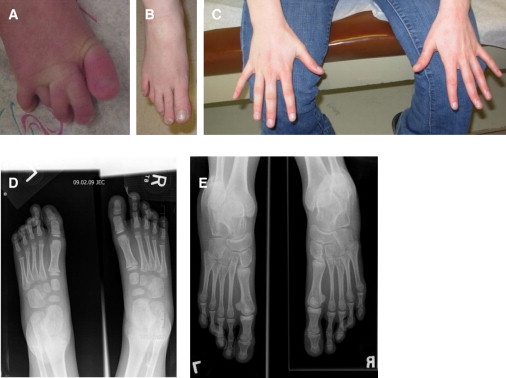
Hand and Foot Abnormalities in Individuals with Microdeletion at 17q23.1q23.2
(A) Feet of patient 1. In this individual, 2–3 syndactyly is familial and likely independent of this syndrome.
(B and C) Right foot and hands of patient 4. Note the long, thin fingers and toes, with the second toe longer than the great toe.
(D) X-rays of feet of patient 3. The second digit of both feet is elongated compared to the great toe, and multiple bipartite growth plates are seen throughout the metatarsal heads.
(E) X-rays of feet of patient 4. There is slight shortening of the fourth and fifth rays bilaterally.
Expression studies in chicken and mice show limb-specific expression of Tbx4 in forelimbs and hindlimbs, which suggests that Tbx4 plays a role in lower-limb development.32,35–37 Although recent studies in mice have suggested that Tbx4 expression does not confer limb-specific morphology to the limb that subsequently develops,38,39 it is clear that the gene plays a vital role in the maintenance of limb outgrowth.
The association between mutations of TBX4 and SPS further suggests that the gene plays a role in the musculoskeletal phenotypes of the patients reported here. SPS is better characterized as ischiopatellar dysplasia because clinical diagnosis includes essential features in addition to patellar aplasia or hypoplasia.40 These essential additional features are absent, delayed, or irregular ossification of the ischiopubic junctions and/or the infra-acetabular axe-cut notches.28 Individuals with SPS and known TBX4 mutations may also have femur and foot anomalies.28 In contrast, some sporadic cases reported that predate TBX4 mutation analysis had, in addition to dysplasia of the lower limbs and pelvis, dysmorphic facial features and digital anomalies.40–43 Thus, these sporadic cases may have TBX4 mutations or, based on the features in addition to essential SPS features, may actually represent deletions at 17q23.1q23.2. Likewise, the musculoskeletal abnormalities identified in patients 3 and 4 of this study are reminiscent of, but not specific to, SPS (Figure 5). For example, patient 4 has lower limb abnormalities similar to patients with TBX4 mutations28 including small patellae, infra-acetabular axe-cut notches, abnormal femoral heads, lesser trochanter hypoplasia, and pes planus. In contrast, patient 4 has shortened rather than elongated femoral neck and no ischiopubic ossification abnormality. It is unknown whether the absence of SPS-like musculoskeletal abnormalities in the other individuals in this study is because detailed skeletal surveys were not performed, although patient 6 was specifically noted to lack any patellar, pelvic, or foot anomalies characteristic of SPS.13 Therefore, deletions of TBX4 may show incomplete penetrance for SPS. Concerning the dysmorphic features, there does not seem to be a common constellation of features among the SPS patients in the literature and our patients, although this does not rule out the possibility of the previously reported sporadic cases being deletions at 17q23.1q23.2.
Figure 5.
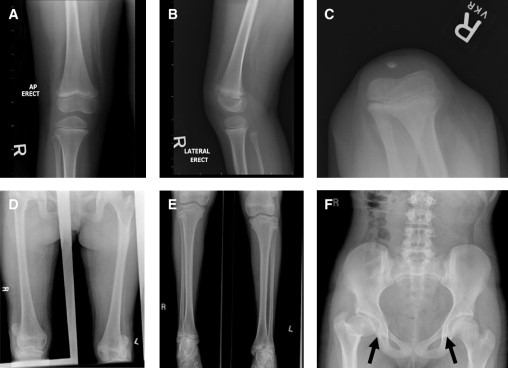
X-rays Showing Musculoskeletal Findings Reminiscent of Mutations of TBX4 and Small Patella Syndrome
(A–C) Patellar X-rays for patient 2. Note abnormally laterally positioned ossification centers of the patella, which is delayed for age. The patella is normally located on the sunrise view, suggesting intermittent subluxation.
(D–F) X-rays for patient 4.
(D) There is lateral subluxation of the patella bilaterally. The patellae are small. The medial tibial plateau is mildly hypoplastic, with mild relative overgrowth of the femoral condyle medially.
(E) The lateral portion of the distal tibial epiphysis is hypoplastic bilaterally. There is a tilting of the talar dome.
(F) The anteroposterior pelvis shows mildly shallow right acetabulum with slight widening of the supra-acetabular iliac bone. There is coxa magna and a short femoral neck on the right with varus deformity, with the greater trochanter slightly higher than the superior right femoral head. The right femoral head is not completely covered by the acetabulum, with the lateral third uncovered. There is minimal superior subluxation. The left femoral head is approximately 15% uncovered by the bony acetabulum. The femoral head is normal in contour. There are axe-cut notches of the infra-acetabulae (arrows).
Interestingly, it has been suggested that an ancestral function of Tbx4/Tbx5 in chordates involved heart cell specification and that, during the evolution of vertebrates, the genes were co-opted to play a role in the initiation of limb outgrowth.21 Furthermore, studies of Tbx2 in the developing mouse heart show that Tbx2 is expressed in areas complementary to that of Anf, expression of which is specific to formation of the ventricular and atrial chambers; Tbx2 also cooperatively functions with Nkx2.5 on the Anf promoter to repress Anf activity.44 The presence of “heart-hand” syndromes in human, including Holt-Oram syndrome, the characteristic features of which are thumb anomalies and ASD, provide further evidence that T-box genes play dual roles in limb outgrowth and heart specification. Such a mechanism may also explain the presence of heart and limb anomalies in individuals with microdeletions at 17q23.1q23.2.
We have identified a previously unknown microdeletion syndrome at 17q23.1q23.2 by analyzing individuals with mental retardation, developmental delay, or dysmorphic features by array CGH. The common clinical features of these individuals and the presence of segmental duplications flanking the deletion breakpoints suggest that deletions at 17q23.1q23.2 represent a novel recurrent microdeletion. The identification of additional cases with microdeletions at 17q23.1q23.2 should further elucidate the clinical features of this emerging syndrome.
Acknowledgments
The authors wish to recognize the contributions of many clinicians, genetic counselors, and families who have participated in this study. B.C.B., A.T., J.A.R., and R.N.T. are employees of Signature Genomic Laboratories. L.G.S. and B.A.B. own shares in, are employees of, and sit on the Board of Directors of Signature Genomic Laboratories. P.S. was supported in part by grant R13-0005-04/2008 from the Polish Ministry of Science and Higher Education.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man, http://www.ncbi.nlm.nih.gov/Omim/
UCSC Genome Browser, http://genome.ucsc.edu/
Human Genome Segmental Duplication Database, http://projects.tcag.ca/humandup/
Accession Numbers
The GEO accession numbers for the probands in this study are GSM498356 (patient 1), GSM498357 (patient 2), GSM498358 (patient 3), GSM498359 (patient 4), GSM506250 (patient 5), GSM498360 (patient 6), and GSM498361 (patient 7).
References
- 1.Bailey J.A., Gu Z., Clark R.A., Reinert K., Samonte R.V., Schwartz S., Adams M.D., Myers E.W., Li P.W., Eichler E.E. Recent segmental duplications in the human genome. Science. 2002;297:1003–1007. doi: 10.1126/science.1072047. [DOI] [PubMed] [Google Scholar]
- 2.Mefford H.C., Eichler E.E. Duplication hotspots, rare genomic disorders, and common disease. Curr. Opin. Genet. Dev. 2009;19:196–204. doi: 10.1016/j.gde.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shaffer L.G., Lupski J.R. Molecular mechanisms for constitutional chromosomal rearrangements in humans. Annu. Rev. Genet. 2000;34:297–329. doi: 10.1146/annurev.genet.34.1.297. [DOI] [PubMed] [Google Scholar]
- 4.Stankiewicz P., Lupski J.R. Genome architecture, rearrangements and genomic disorders. Trends Genet. 2002;18:74–82. doi: 10.1016/s0168-9525(02)02592-1. [DOI] [PubMed] [Google Scholar]
- 5.Koolen D.A., Vissers L.E., Pfundt R., de Leeuw N., Knight S.J., Regan R., Kooy R.F., Reyniers E., Romano C., Fichera M. A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nat. Genet. 2006;38:999–1001. doi: 10.1038/ng1853. [DOI] [PubMed] [Google Scholar]
- 6.Sharp A.J., Hansen S., Selzer R.R., Cheng Z., Regan R., Hurst J.A., Stewart H., Price S.M., Blair E., Hennekam R.C. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat. Genet. 2006;38:1038–1042. doi: 10.1038/ng1862. [DOI] [PubMed] [Google Scholar]
- 7.Shaw-Smith C., Pittman A.M., Willatt L., Martin H., Rickman L., Gribble S., Curley R., Cumming S., Dunn C., Kalaitzopoulos D. Microdeletion encompassing MAPT at chromosome 17q21.3 is associated with developmental delay and learning disability. Nat. Genet. 2006;38:1032–1037. doi: 10.1038/ng1858. [DOI] [PubMed] [Google Scholar]
- 8.Ballif B.C., Theisen A., Coppinger J., Gowans G.C., Hersh J.H., Madan-Khetarpal S., Schmidt K.R., Tervo R., Escobar L.F., Friedrich C.A. Expanding the clinical phenotype of the 3q29 microdeletion syndrome and characterization of the reciprocal microduplication. Mol Cytogenet. 2008;1:8. doi: 10.1186/1755-8166-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ballif B.C., Hornor S.A., Jenkins E., Madan-Khetarpal S., Surti U., Jackson K.E., Asamoah A., Brock P.L., Gowans G.C., Conway R.L. Discovery of a previously unrecognized microdeletion syndrome of 16p11.2-p12.2. Nat. Genet. 2007;39:1071–1073. doi: 10.1038/ng2107. [DOI] [PubMed] [Google Scholar]
- 10.Cardone M.F., Jiang Z., D'Addabbo P., Archidiacono N., Rocchi M., Eichler E.E., Ventura M. Hominoid chromosomal rearrangements on 17q map to complex regions of segmental duplication. Genome Biol. 2008;9:R28. doi: 10.1186/gb-2008-9-2-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen D.C., Saarela J., Clark R.A., Miettinen T., Chi A., Eichler E.E., Peltonen L., Palotie A. Segmental duplications flank the multiple sclerosis locus on chromosome 17q. Genome Res. 2004;14:1483–1492. doi: 10.1101/gr.2340804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saarela J., Schoenberg Fejzo M., Chen D., Finnilä S., Parkkonen M., Kuokkanen S., Sobel E., Tienari P.J., Sumelahti M.L., Wikström J. Fine mapping of a multiple sclerosis locus to 2.5 Mb on chromosome 17q22-q24. Hum. Mol. Genet. 2002;11:2257–2267. doi: 10.1093/hmg/11.19.2257. [DOI] [PubMed] [Google Scholar]
- 13.Rudd M.K., Keene J., Bunke B., Kaminsky E.B., Adam M.P., Mulle J.G., Ledbetter D.H., Martin C.L. Segmental duplications mediate novel, clinically relevant chromosome rearrangements. Hum. Mol. Genet. 2009;18:2957–2962. doi: 10.1093/hmg/ddp233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ballif B.C., Theisen A., McDonald-McGinn D.M., Zackai E.H., Hersh J.H., Bejjani B.A., Shaffer L.G. Identification of a previously unrecognized microdeletion syndrome of 16q11.2q12.2. Clin. Genet. 2008;74:469–475. doi: 10.1111/j.1399-0004.2008.01094.x. [DOI] [PubMed] [Google Scholar]
- 15.Baldwin E.L., Lee J.Y., Blake D.M., Bunke B.P., Alexander C.R., Kogan A.L., Ledbetter D.H., Martin C.L. Enhanced detection of clinically relevant genomic imbalances using a targeted plus whole genome oligonucleotide microarray. Genet. Med. 2008;10:415–429. doi: 10.1097/GIM.0b013e318177015c. [DOI] [PubMed] [Google Scholar]
- 16.El-Hattab A.W., Smolarek T.A., Walker M.E., Schorry E.K., Immken L.L., Patel G., Abbott M.A., Lanpher B.C., Ou Z., Kang S.H. Redefined genomic architecture in 15q24 directed by patient deletion/duplication breakpoint mapping. Hum. Genet. 2009;126:589–602. doi: 10.1007/s00439-009-0706-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shaffer L.G., McCaskill C., Han J.Y., Choo K.H., Cutillo D.M., Donnenfeld A.E., Weiss L., Van Dyke D.L. Molecular characterization of de novo secondary trisomy 13. Am. J. Hum. Genet. 1994;55:968–974. [PMC free article] [PubMed] [Google Scholar]
- 18.Bailey J.A., Yavor A.M., Massa H.F., Trask B.J., Eichler E.E. Segmental duplications: Organization and impact within the current human genome project assembly. Genome Res. 2001;11:1005–1017. doi: 10.1101/gr.187101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stankiewicz P., Shaw C.J., Dapper J.D., Wakui K., Shaffer L.G., Withers M., Elizondo L., Park S.S., Lupski J.R. Genome architecture catalyzes nonrecurrent chromosomal rearrangements. Am. J. Hum. Genet. 2003;72:1101–1116. doi: 10.1086/374385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elsea S.H., Girirajan S. Smith-Magenis syndrome. Eur. J. Hum. Genet. 2008;16:412–421. doi: 10.1038/sj.ejhg.5202009. [DOI] [PubMed] [Google Scholar]
- 21.Horton A.C., Mahadevan N.R., Minguillon C., Osoegawa K., Rokhsar D.S., Ruvinsky I., de Jong P.J., Logan M.P., Gibson-Brown J.J. Conservation of linkage and evolution of developmental function within the Tbx2/3/4/5 subfamily of T-box genes: Implications for the origin of vertebrate limbs. Dev. Genes Evol. 2008;218:613–628. doi: 10.1007/s00427-008-0249-5. [DOI] [PubMed] [Google Scholar]
- 22.Chieffo C., Garvey N., Gong W., Roe B., Zhang G., Silver L., Emanuel B.S., Budarf M.L. Isolation and characterization of a gene from the DiGeorge chromosomal region homologous to the mouse Tbx1 gene. Genomics. 1997;43:267–277. doi: 10.1006/geno.1997.4829. [DOI] [PubMed] [Google Scholar]
- 23.Paylor R., Glaser B., Mupo A., Ataliotis P., Spencer C., Sobotka A., Sparks C., Choi C.H., Oghalai J., Curran S. Tbx1 haploinsufficiency is linked to behavioral disorders in mice and humans: Implications for 22q11 deletion syndrome. Proc. Natl. Acad. Sci. USA. 2006;103:7729–7734. doi: 10.1073/pnas.0600206103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zweier C., Sticht H., Aydin-Yaylagül I., Campbell C.E., Rauch A. Human TBX1 missense mutations cause gain of function resulting in the same phenotype as 22q11.2 deletions. Am. J. Hum. Genet. 2007;80:510–517. doi: 10.1086/511993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yagi H., Furutani Y., Hamada H., Sasaki T., Asakawa S., Minoshima S., Ichida F., Joo K., Kimura M., Imamura S. Role of TBX1 in human del22q11.2 syndrome. Lancet. 2003;362:1366–1373. doi: 10.1016/s0140-6736(03)14632-6. [DOI] [PubMed] [Google Scholar]
- 26.Li Q.Y., Newbury-Ecob R.A., Terrett J.A., Wilson D.I., Curtis A.R., Yi C.H., Gebuhr T., Bullen P.J., Robson S.C., Strachan T. Holt-Oram syndrome is caused by mutations in TBX5, a member of the Brachyury (T) gene family. Nat. Genet. 1997;15:21–29. doi: 10.1038/ng0197-21. [DOI] [PubMed] [Google Scholar]
- 27.Bamshad M., Lin R.C., Law D.J., Watkins W.C., Krakowiak P.A., Moore M.E., Franceschini P., Lala R., Holmes L.B., Gebuhr T.C. Mutations in human TBX3 alter limb, apocrine and genital development in ulnar-mammary syndrome. Nat. Genet. 1997;16:311–315. doi: 10.1038/ng0797-311. [DOI] [PubMed] [Google Scholar]
- 28.Bongers E.M., Duijf P.H., van Beersum S.E., Schoots J., Van Kampen A., Burckhardt A., Hamel B.C., Losan F., Hoefsloot L.H., Yntema H.G. Mutations in the human TBX4 gene cause small patella syndrome. Am. J. Hum. Genet. 2004;74:1239–1248. doi: 10.1086/421331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kirk E.P., Sunde M., Costa M.W., Rankin S.A., Wolstein O., Castro M.L., Butler T.L., Hyun C., Guo G., Otway R. Mutations in cardiac T-box factor gene TBX20 are associated with diverse cardiac pathologies, including defects of septation and valvulogenesis and cardiomyopathy. Am. J. Hum. Genet. 2007;81:280–291. doi: 10.1086/519530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ruvinsky I., Gibson-Brown J.J. Genetic and developmental bases of serial homology in vertebrate limb evolution. Development. 2000;127:5233–5244. doi: 10.1242/dev.127.24.5233. [DOI] [PubMed] [Google Scholar]
- 31.Gibson-Brown J.J., Agulnik S.I., Silver L.M., Papaioannou V.E. Expression of T-box genes Tbx2-Tbx5 during chick organogenesis. Mech. Dev. 1998;74:165–169. doi: 10.1016/s0925-4773(98)00056-2. [DOI] [PubMed] [Google Scholar]
- 32.Gibson-Brown J.J., Agulnik S.I., Silver L.M., Niswander L., Papaioannou V.E. Involvement of T-box genes Tbx2-Tbx5 in vertebrate limb specification and development. Development. 1998;125:2499–2509. doi: 10.1242/dev.125.13.2499. [DOI] [PubMed] [Google Scholar]
- 33.Gibson-Brown J.J., Agulnik S.I., Chapman D.L., Alexiou M., Garvey N., Silver L.M., Papaioannou V.E. Evidence of a role for T-box genes in the evolution of limb morphogenesis and the specification of forelimb/hindlimb identity. Mech. Dev. 1996;56:93–101. doi: 10.1016/0925-4773(96)00514-x. [DOI] [PubMed] [Google Scholar]
- 34.Suzuki T., Takeuchi J., Koshiba-Takeuchi K., Ogura T. Tbx genes specify posterior digit identity through Shh and BMP signaling. Dev. Cell. 2004;6:43–53. doi: 10.1016/s1534-5807(03)00401-5. [DOI] [PubMed] [Google Scholar]
- 35.Rodriguez-Esteban C., Tsukui T., Yonei S., Magallon J., Tamura K., Izpisua Belmonte J.C. The T-box genes Tbx4 and Tbx5 regulate limb outgrowth and identity. Nature. 1999;398:814–818. doi: 10.1038/19769. [DOI] [PubMed] [Google Scholar]
- 36.Takeuchi J.K., Koshiba-Takeuchi K., Matsumoto K., Vogel-Höpker A., Naitoh-Matsuo M., Ogura K., Takahashi N., Yasuda K., Ogura T. Tbx5 and Tbx4 genes determine the wing/leg identity of limb buds. Nature. 1999;398:810–814. doi: 10.1038/19762. [DOI] [PubMed] [Google Scholar]
- 37.Isaac A., Rodriguez-Esteban C., Ryan A., Altabef M., Tsukui T., Patel K., Tickle C., Izpisúa-Belmonte J.C. Tbx genes and limb identity in chick embryo development. Development. 1998;125:1867–1875. doi: 10.1242/dev.125.10.1867. [DOI] [PubMed] [Google Scholar]
- 38.Minguillon C., Del Buono J., Logan M.P. Tbx5 and Tbx4 are not sufficient to determine limb-specific morphologies but have common roles in initiating limb outgrowth. Dev. Cell. 2005;8:75–84. doi: 10.1016/j.devcel.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 39.Naiche L.A., Papaioannou V.E. Tbx4 is not required for hindlimb identity or post-bud hindlimb outgrowth. Development. 2007;134:93–103. doi: 10.1242/dev.02712. [DOI] [PubMed] [Google Scholar]
- 40.Bongers E.M., Van Bokhoven H., Van Thienen M.N., Kooyman M.A., Van Beersum S.E., Boetes C., Knoers N.V., Hamel B.C. The small patella syndrome: Description of five cases from three families and examination of possible allelism with familial patella aplasia-hypoplasia and nail-patella syndrome. J. Med. Genet. 2001;38:209–214. doi: 10.1136/jmg.38.3.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kozlowski K., Nelson J. Small patella syndrome. Am. J. Med. Genet. 1995;57:558–561. doi: 10.1002/ajmg.1320570408. [DOI] [PubMed] [Google Scholar]
- 42.Azouz E.M., Kozlowski K. Small patella syndrome: A bone dysplasia to recognize and differentiate from the nail-patella syndrome. Pediatr. Radiol. 1997;27:432–435. doi: 10.1007/s002470050163. [DOI] [PubMed] [Google Scholar]
- 43.Habboub H.K., Thneibat W.A. Ischio-pubic-patellar hypoplasia: Is it a new syndrome? Pediatr. Radiol. 1997;27:430–431. doi: 10.1007/s002470050162. [DOI] [PubMed] [Google Scholar]
- 44.Habets P.E., Moorman A.F., Clout D.E., van Roon M.A., Lingbeek M., van Lohuizen M., Campione M., Christoffels V.M. Cooperative action of Tbx2 and Nkx2.5 inhibits ANF expression in the atrioventricular canal: Implications for cardiac chamber formation. Genes Dev. 2002;16:1234–1246. doi: 10.1101/gad.222902. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.