

Abstract
Targeted genome capture combined with next-generation sequencing was used to analyze 2.9 Mb of the DFNB79 interval on chromosome 9q34.3, which includes 108 candidate genes. Genomic DNA from an affected member of a consanguineous family segregating recessive, nonsyndromic hearing loss was used to make a library of fragments covering the DFNB79 linkage interval defined by genetic analyses of four pedigrees. Homozygosity for eight previously unreported variants in transcribed sequences was detected by evaluating a library of 402,554 sequencing reads and was later confirmed by Sanger sequencing. Of these variants, six were determined to be polymorphisms in the Pakistani population, and one was in a noncoding gene that was subsequently excluded genetically from the DFNB79 linkage interval. The remaining variant was a nonsense mutation in a predicted gene, C9orf75, renamed TPRN. Evaluation of the other three DFNB79-linked families identified three additional frameshift mutations, for a total of four truncating alleles of this gene. Although TPRN is expressed in many tissues, immunolocalization of the protein product in the mouse cochlea shows prominent expression in the taper region of hair cell stereocilia. Consequently, we named the protein taperin.
Introduction
The initial identification of pathogenic mutations at a genetic locus can be challenging when the linkage interval includes a large number of genes. The conventional strategy has been to sequence the coding regions of each gene in a critical interval one by one, after rank ordering them on the basis of hypotheses about gene function or expression pattern. We previously applied this approach to the nonsyndromic recessive deafness locus DFNB79, which we mapped to a 3.84 Mb linkage interval on chromosome 9q34.3.1 This interval contains 113 predicted and reported genes. We excluded eight candidate genes, including NOTCH1 (MIM 190198) and CACNA1B (MIM 601012), by using dideoxy-terminator sequencing.1
Continuing this hierarchical approach of sequencing exons of candidate genes seemed impractical because of the large number of genes at the DFNB79 locus. Moreover, our experience with other nonsyndromic deafness genes is that they often are the ones that initially seem unlikely because of a nearly ubiquitous pattern of expression, such as ACTG1 (MIM 102560),2,3 TRIC (MIM 610572),4 TMPRSS3 (MIM 605511),5 LRTOMT (MIM 612414),6 and HGF (MIM 142409),7 and pathogenic alleles are sometimes found in a noncoding region, such as those that were found to occur in HGF.7
Recent advances in technology have made it possible to quickly and cost-effectively sequence all of the genes, including the noncoding regions, at a specified locus.8–11 We applied targeted genome capture by using a NimbleGen array and high-throughput Roche 454 sequencing to the genomic DNA of an affected individual from a DFNB79-linked family. We identified a nonsense mutation in a predicted gene, C9orf75, designated TPRN, and subsequently identified three additional frameshift mutations in the same gene in the other three DFNB79-linked families. RT-PCR analysis revealed that transcripts from this gene are expressed in many tissues, including the cochlea. Our immunolocalization results demonstrate that, in the mouse cochlea, the protein encoded by Tprn is localized prominently at the taper regions of hair cell stereocilia. This is the region near the base of stereocilia where the diameter of stereocilia gradually narrows before insertion into the cuticular plate. On the basis of the immunolocalization in inner ear hair cells, we have named the protein taperin.
Subjects and Methods
Family Enrollment and Diagnosis
Ascertainment and enrollment methodology for family PKDF1129 was the same as that which has been described for families PKDF741, PKDF517, and PKDF280.1 Written informed consent was obtained for all study participants after approval from the Combined Neuroscience Institutional Review Board (IRB) at the National Institutes of Health (NIH), Bethesda, MD, USA, and the IRB at the National Centre of Excellence in Molecular Biology (NCEMB), University of the Punjab, Lahore, Pakistan. Hearing in affected individuals was evaluated by pure-tone audiometry. Funduscopic, tandem gait, and Romberg tests were performed to assess vision loss and vestibular dysfunction. Affected individuals from DFNB79-linked pedigrees exhibited prelingual, bilateral, severe to profound sensorineural hearing loss, and apparently normal vestibular function on the basis of Romberg and tandem gait testing. There were no other obvious clinical findings.1 Blood samples were obtained from study participants and DNA was extracted as previously described.12
NimbleGen Target-Region Capture
A custom Sequence Capture 385K Human Array was designed and manufactured by Roche NimbleGen. A total of 385,000 unique, overlapping probes 60–90 nucleotides in length were designed across the DFNB79 target region (chromosome 9: 137,372,128–140,273, 252; NCBI build 36.1, hg18). Probe uniqueness was determined by the Sequence Search and Alignment by Hashing Algorithm (SSAHA).13 Probe tiling of the target region excludes repetitive DNA, and adjacent coding regions are sometimes sacrificed. Approximately 20 μg of genomic DNA from affected individual IV-4 of family PKDF741 (Figure 1A) was fragmented to a size range of 300–500 base pairs (bp) with the use of a GS Nebulizer Kit (Roche Applied Science). The fragmented DNA was purified (DNA Clean & Concentrator-25, Zymo Research) and analyzed on an Agilent Bioanalyzer 2100 DNA Chip 7500 according to the manufacturer's instructions. Fragment ends were polished with the use of T4 DNA Polymerase and T4 Polynucleotide Kinase, and adapters were ligated onto the polished ends with T4 DNA Ligase. Small fragments (< 100 bp) were removed with the use of AMPure Beads (Agencourt). The resulting library was hybridized to a custom 385K array with the use of the NimbleGen Sequence Capture Hybridization System 4. The hybridized DNA from the DFNB79 target region was washed and eluted with the use of a NimbleGen Wash and Elution Kit according to the manufacturer's instructions. There is a tradeoff in adjusting the stringency of hybridization and wash conditions in order to retain sufficient on-target sequence while reducing the capture of off-target sequence.14 The eluted sample was amplified by ligation-mediated PCR with the use of primers complementary to the sequence of the adaptors.
Figure 1.
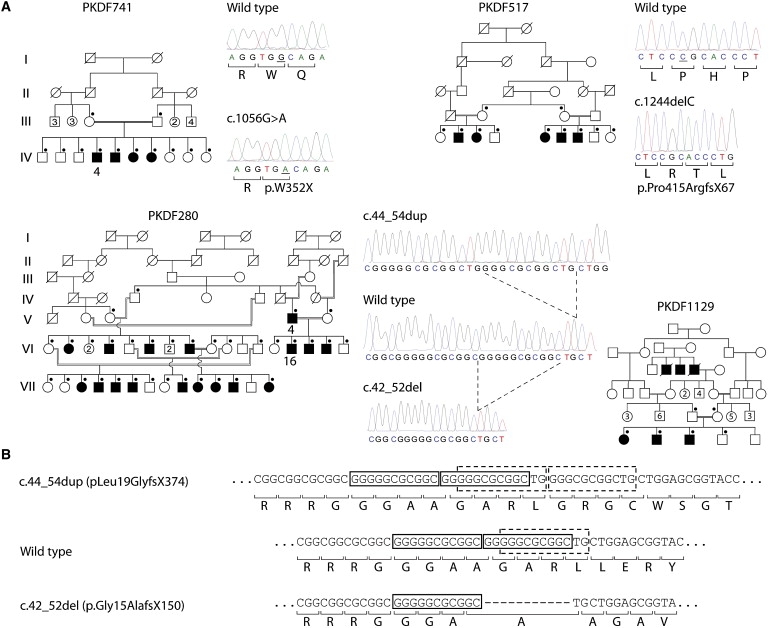
DFNB79-Linked Pedigrees and Sequence Traces Showing Pathogenic Variants
(A) Affected individual IV-4 from family PKDF741 was selected for NimbleGen capture and enrichment of genomic DNA from the DFNB79 locus and next-generation sequencing. Filled symbols in each pedigree represent affected individuals. A dot in the upper right corner of symbols for females (circles) and males (squares) indicates an individual whose DNA sequence of TPRN was determined via dideoxy-terminator chemistry. Bars below the DNA sequence indicate altered nucleotide(s). In family PKDF280, the sequence trace from an affected individual shows a homozygous 11 bp duplication. In an affected individual from family PKDF1129, dotted lines indicate the location of the 11 bp deletion. NCBI Reference Sequences (NM_001128228.1 for nucleotide change and NP_001121700 for amino acid change) were used to indicate positions of mutations in cDNA and protein, respectively.
(B) The 11 bp deletion and 11 bp duplication found in affected members of families PKDF1129 and PKDF280, respectively, occur in sequence that is already duplicated in the wild-type.
454 Sequencing
Sequencing was performed on a Roche 454 FLX Titanium instrument at SeqWright. Three micrograms of the eluted library, now enriched for fragments from the DFNB79 interval, were size selected (> 400 bp) with AMpure beads (Agencourt). Library immobilization and single-strand isolation was performed with the use of a GS FLX Titanium General Library Preparation Kit (Roche). The library was purified with the use of MinElute PCR Purification Kit (QIAGEN) and then amplified on the sequencing beads by emulsion PCR. After bead recovery, bead enrichment, and sequencing-primer annealing, the beads were deposited on a 454 Picotiter Plate and sequenced with the use of a GS FLX Titanium Kit XLR70 (Roche). The reads were assembled with Newbler Assembler (454 Life Sciences) and then mapped to the human hg18 assembly reference sequence (NCBI build 36.1). Fold enrichment of the target region was calculated as described previously,15 with use of the formula ∑REMTrm/STrm: ∑RMG/SG, in which REMTrm is the number of reads mapped to the repeat masked target region, STrm is the size of the repeat masked target region, RMG is the number of reads mapped to the human genome, and SG is the size of the human genome. Putative variants were detected by GS Amplicon Variant Analyzer software (454 Life Sciences) with a filtering parameter such that a variant should be supported by at least three nonduplicate forward and reverse 454 reads, or at least five unidirectional reads, with quality scores > 20 (or > 30 if the difference involved a homopolymer of five or more nucleotides or larger).
Dideoxy-Terminator Sequencing
Primers (Integrated DNA Technologies) were designed with Primer3 software. GC-rich genomic templates were amplified with the use of Advantage GC Genomic LA Polymerase Mix (Clontech) and the FailSafe PCR System (Epicenter Biotechnologies). All previously unreported variants discovered in the transcribed sequences of the DFNB79 target region and all exons of SNHG7 and TPRN were sequenced with the use of conventional dideoxy-terminator as described previously.16 Sequencing products were resolved on an ABI3730 capillary sequencing instrument (Applied Biosystems), and sequence traces were analyzed with Lasergene 8 software (DNASTAR).
Genotyping
Linkage to DFNB79 was established for family PKDF1129 during the screening for linkage to all currently known nonsyndromic deafness loci (DFNB), including DFNB79,1 with the use of STR markers as previously described.17 Amplicons were resolved on ABI Prism 3100 or ABI3730 instruments. Genotypes were analyzed using GeneMapper software (Applied Biosystems).
RT-PCR Analysis of Tprn Expression
C430004E15Rik (RefSeq NM_175286.4), the mouse ortholog of human TPRN, was amplified from two mouse multitissue cDNA panels (Clontech MTC Panels I and III) and from mouse cochlear cDNA that was prepared with a SMART kit and oligo-dT primers (Clontech). The PCR primers (forward, 5′-GTTCATGCGACTTGAGTCGGAGCGG-3′; reverse, 5′-GGGCAGGAGCTTGGATTTCGGCAAA-3′) are located in exon 1 and in the 3′ untranslated region (UTR), respectively.
Expression Plasmids
Mouse full-length Tprn cDNA was PCR amplified from postnatal day 5 inner ear cDNA with LA Taq DNA polymerase (Takara Mirus), cloned into XL-TOPO vector (Invitrogen), and transformed into DH5α bacteria (Invitrogen). A 2250 bp cDNA product encoding the Tprn open reading frame was amplified with Pfu Ultra DNA polymerase (Stratagene) and ligated into the EcoRI and SalI sites of pAcGFP1-C2. A 1785 bp cDNA product encoding the open reading frame of mouse phostensin was also amplified with Pfu Ultra DNA polymerase from a full-length mouse cDNA clone (Open Biosystems). The PCR product was ligated into the EcoRI and SalI sites of the AcGFP1-C2 plasmid, and this plasmid was transformed into XL10-Gold cells (Stratagene). For expression plasmids, both strands of the cDNA inserts were verified with Sanger sequencing.
Antibody Validation
To validate anti-taperin antibody (HPA020899; C9orf75, Sigma), we performed a colocalization assay by using green fluorescent protein (GFP)-taperin- and GFP-phostensin-transfected COS7cells. The cells were grown on coverslips in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen) until ∼80% confluency was achieved, then the cells were transfected with Lipofectamine 2000 (Invitrogen). After incubation at 37°C with 5% CO2 for 24–36 hr, transfected COS7 cells were fixed with 4% paraformaldehyde (EMS) in PBS for 20 min and permeabilized for 15 min in 0.2% Triton X-100 in 1× PBS. Nonspecific binding sites were blocked by incubation in 2% BSA and 5% normal goat serum in PBS for 30 min. Primary taperin antibody was diluted in blocking solution to a final concentration of ∼5 μg per ml. COS7 cells were incubated with taperin antibody for 2 hr at room temperature, followed by incubation with Alexa Fluor 568 goat anti-rabbit immunoglobulin G (Molecular Probes) for 20 min at room temperature. Samples were washed in PBS, mounted with ProLong Gold Reagent (Molecular Probes), and imaged with the use of a LSM710 confocal microscope equipped with a Zeiss 100X objective that has a numerical aperature of 1.4.
Fluorescence Immunostaining and Confocal Microscopy
Whole-mounted organ of Corti and vestibular epithelium samples were immunostained as described previously,18 with some modifications. In brief, cochleae were fixed in 4% paraformaldehyde for 1.5 hr at room temperature and permeabilized in 0.5% Triton X-100 for 30 min. Nonspecific binding sites were blocked in 1× PBS-based solution containing 2% BSA and 5% normal goat serum for 1 hr at room temperature. Antiserum against a fragment of human taperin 70 amino acids in length was obtained from Sigma-Aldrich (HPA020899; C9orf75). The specimens were incubated overnight at 4°C with this primary antibody diluted in blocking solution. This was followed by several washes in 1× PBS and 20 min of incubation at room temperature in AlexaFluor fluorescein isothiocyanate (FITC)-conjugated secondary antibody (Molecular Probes) at a 1:500 dilution and in rhodamine-phalloidin at 1:100 dilution in blocking solution. The specimens were then processed as described above.
Results
SNP Genotyping Refines the DFNB79 Interval
We refined the DFNB79 linkage region1 from 3.84 Mb to 2.9 Mb through analysis of additional SNP genotypes. This reduced the number of candidate genes from 113 to 108 (Table S1, available online). These 108 genes have 870 protein-coding exons comprising a total of 150 kb. The UTRs, introns, and noncoding genes in this interval span an additional 1731 kb, and intergenic regions encompass 989 kb of genomic DNA (Table 1).
Table 1.
NimbleGen Probe and 454 Sequencing Coverage Summary of 2.9 Mb Targeted DFNB79 Interval
| Category | Total | Protein Coding | UTRs, Introns, and Noncoding Genes | Intergenic Region |
|---|---|---|---|---|
| Number of bases | 2,871,073a | 150,439 | 1,731,069 | 989,565 |
| Probe coverage in bases | 2,132,062 (74.26%) | 141,019 (93.74%) | 1,297,527 (74.95%) | 693,516 (70.08%) |
| Sequence coverage in bases | 2,220,820 (77.35%) | 142,108 (94.46%) | 1,338,676 (77.33%) | 740,036 (74.78%) |
| Gaps in probe coverage | 2498 | 99 | 1,508 | 958 |
| Gaps in sequencing coverage | 1264 | 50 | 812 | 493 |
| Probe gap length range (mean) | 1–25,940 (295.84) | 1–1058 (95.15) | 1–19,898 (287.49) | 1–9,989 (309.03) |
| Sequencing gap range (mean) | 2–49,014 (514.44) | 8–886 (166.62) | 1–35,906 (483.24) | 4–9,989 (506.14) |
See Table S1 as well.
A gap of 31,051 bp in the DFNB79 interval is present in the NCBI 36 reference human genome sequence (hg18).
Targeted Genome Capture and Next-Generation Sequencing
Using a custom NimbleGen array with probes covering 93.74% of the protein-coding sequence, 74.95% of UTRs, introns, and noncoding genes, and 70.08% of intergenic regions (Table 1), we enriched DNA for the refined DFNB79 locus and sequenced the captured library. A total of 443,047 reads with an average length of 278 nucleotides were produced, of which 405,799 (91.59%) were assembled after adaptor and quality trimming (Table 2). We found that 99.20% of the assembled reads mapped to the human genome, of which 35.50% mapped to multiple off-target locations and were not included in our downstream analyses. Of the assembled reads, 38.75% uniquely mapped to the DFNB79 target region, whereas 25.75% uniquely mapped across the human genome outside the target region (Table 2 and Table S2). This represents a 561-fold enrichment of the repeat-masked target region. Incidentally, the off-target reads included 94% coverage of the mitochondrial genome, which allowed us to inspect for reported mitochondrial mutations associated with hearing loss,19 but we found none.
Table 2.
Summary Statistics of 454 Reads
| Category | Reads | Base Calls |
|---|---|---|
| Total reads generated by 454 sequencing | 443,047 | 123,249,163 |
| Read length range (mean) | 14–761 (278) | - |
| Reads used for assembly after quality trimming | 405,799 (91.59%)a | 113,353,237 (91.97%) |
| Reads mapped to human genome | 402,554 (99.20%)b | 111,498,382 (98.36%) |
| Reads not mapped to human genome | 3,245 (0.80%)b | - |
| Mapped to multiple places in human genome | 142,881 (35.50%)c | - |
| Mapped to DFNB79 target region | 156,019 (38.75%)c | 42,752,954 (38.34%) |
| Off-target | 103,654 (25.75%)c | - |
See Table S2 as well.
Percentage of 443,047 reads generated by 454 sequencing.
Percentage of quality filtered reads.
Percentage of quality filtered reads mapped to the human genome.
Sequence coverage was, on average, 20-fold for 77.35% of the DFNB79 target region. This included 94.46% of the protein-coding region, 77.33% of the region covered by introns, UTRs, and noncoding genes, and 74.78% of the intergenic regions (Table 1). Of the remaining DFNB79 target region with limited coverage, one third was from a subtelomeric region, which is difficult to sequence.20 Moreover, there were protein-coding regions that were represented by probes on the NimbleGen array but for which no sequence was obtained. All of these regions have high (average = 75%) GC content (Table S3). Of the 108 genes in the DFNB79 interval, 52 were completely sequenced and 53 were partially sequenced. Only three genes were less than 10% sequenced: one was located very close to the telomere and consequently could not be represented on the NimbleGen array, and two were small single-exon genes adjacent to repetitive sequences (Table S1).
After filtering for quality, there appeared to be a total of 2231 variants in the DFNB79 interval. Of these, 1928 were reported in dbSNP, leaving 303 previously unreported variants (Figure 2). Seventeen of these were located in transcribed regions of annotated or predicted genes. We chose these variants for further analysis. Given the consanguinity in pedigree PKDF741 and our extensive genotyping data, we expected that affected individual IV-4 would be homozygous for all variants at the DFNB79 locus. In fact, the reads in contigs spanning the variants were not always congruent. Nine of the 17 variants appeared to be heterozygous because less than 85% of the contig reads showed a difference from the hg18 reference sequence. This apparent heterozygosity was most likely due to poor-quality sequencing. We designed PCR primers to amplify these 17 variants from the genomic DNA of individual IV-4, and we sequenced the PCR products via conventional Sanger sequencing. Only eight variants were confirmed as real, and all eight were homozygous in individual IV-4. At the other nine sites of putative sequence variation, individual IV-4 was homozygous for the reference sequence. We assume that these nine false-positive variants were 454-derived sequencing artifacts.
Figure 2.
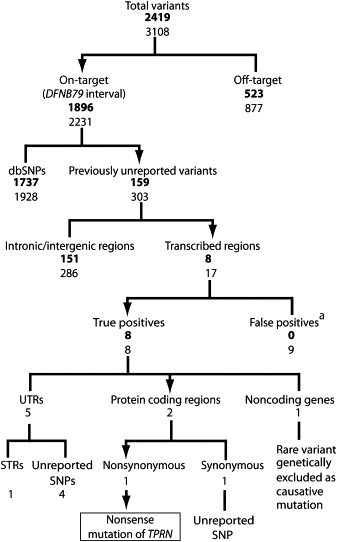
Summary of Variants Found by Next-Generation Sequencing
Values in regular font are variants supported by at least three nonduplicate forward and reverse 454 reads or at least five unidirectional reads with a quality score greater then 20 (or 30 if the difference involved a pentamer or larger). Values in bold are restricted to those variants supported by at least 85% of the reads mapped at that location. Abbreviations are as follows: UTR, untranslated region; STR, short tandem repeat; SNP, single-nucleotide polymorphism.
a False positives demonstrated by Sanger sequencing.
All of the eight confirmed sequence variants cosegregate with DFNB79 in family PKDF741, as determined by Sanger sequencing of PCR products. We also evaluated these eight variants in PCR products from at least 176 healthy Pakistani control chromosomes, and we found that six are polymorphisms with allele frequencies ranging from 1.72% to 69.4% (Table 3).
Table 3.
Variants Found in Transcribed Regions of DFNB79 Interval and their Allele Frequencies
| Family | Gene | Mutation Location | Pakistani Control Chromosomes | Coriell HD200CAU Panel | Comment |
|---|---|---|---|---|---|
| PKDF280 | TPRN | c.44_54dup (NM_001128228.1) | 0/488 | 0/400 | Pathogenic variant |
| PKDF517 | TPRN | c.1244 delC (NM_001128228.1) | 0/500 | 0/400 | Pathogenic variant |
| PKDF741 | TPRN | c.1056G>A (NM_001128228.1) | 0/500 | 0/400 | Pathogenic variant |
| PKDF1129 | TPRN | c.42_52 del (NM_001128228.1) | 0/488 | 0/400 | Pathogenic variant |
| PKDF741 | QSOX2a | c.∗587_∗588delGT (NM_181701.3) | 4/174 | - | SNPc |
| PKDF741 | C9orf167 | c.-73G>A (NM_017723.2) | 4/174 | - | SNPc |
| PKDF741 | C9orf167 | c.∗1641G>A (NM_017723.2) | 3/174 | - | SNPc |
| PKDF741 | LOC100133077 | c.∗483C>T (AK128414.1) | 2/174 | - | SNPc |
| PKDF741 | GPSM1a | c.1959C>T (NM_001145638.1) | 6/160 | - | SNPc |
| PKDF741 | GPSM1a | c.∗207GGGCT[56] (NM_015597.4) | 111/160 | - | STR |
| PKDF741 | SNHG7b | c.729C>T (NR_024542.1) | 0/176 | - | Rare variant |
| PKDF280 | SNHG7b | c.190_209dup (NR_024542.1) | 13/172 | - | SNPd |
The following abbreviations are used: SNP, single-nucleotide polymorphism; STR, short tandem repeats.
Nucleotides for SNHG7, a non-protein-coding gene, are numbered relative to the first base in the cDNA sequence of NR_024542.1.
Previously unreported SNP.
This previously unreported SNP found in SNHG7 does not cosegregate with deafness in family PKDF280. A meiotic break point in two affected subjects in this family refined the proximal end of the DFNB79 linkage interval and excludes SNHG7 as a DFNB79 candidate gene.
Two of the variants, c.729C>T (NR_024542.1) and c.1056G>A (NM_001128228.1), were not found in the Pakistani control chromosomes. c.729C>T (NR_024542.1) is located in exon 4 of SNHG7, Small Nucleolar RNA Host Gene 7, a four-exon, non-protein-coding gene. DNA sequence of the four SNHG7 exons and approximately 50 bp of intronic region flanking each exon was determined by Sanger sequencing of PCR products amplified from genomic DNA of one affected individual from two other DFNB79-linked pedigrees, PKDF280 and PKDF517. We did not find any variant in PKDF517. However, in PKDF280, we found a 20 bp duplication, c.190_209dup (NR_024542.1), in the first exon of SNHG7. We determined that c.190_209dup (NR_024542.1) is a polymorphism in the Pakistani control group (Table 3), and it does not cosegregate with deafness in family PKDF280. Two of the 18 affected individuals (V-4 and VI-16, Figure 1A) of family PKDF280 are heterozygous for c.190_209dup (NR_024542.1). These data provide evidence of a meiotic breakpoint, which further refines the DFNB79 linkage interval and excludes SNHG7 as a candidate gene. Prior to ascertainment of family PKDF1129, SNHG7 was excluded as a candidate DFNB79 gene and therefore was not sequenced in family PKDF1129.
Identifying the DFNB79 Causative Gene
The remaining variant is c.1056G>A (NM_001128228.1), located in a GC-rich first exon (74% GC content) of TPRN (C9orf75). The predicted open reading frame of TPRN spans four exons and encodes 711 amino acids (ENST00000409012). Exon 1 spans 1725 nucleotides and encodes 81% (575 of the 711 residues) of the predicted protein. The c.1056G>A (p.W352X) mutation is predicted to introduce a premature stop codon and results in a truncated protein of 412 amino acids, renders the transcript susceptible to nonsense-mediated RNA decay,21 or both. PCR products encompassing all coding exons, including approximately 50 bp of flanking intronic sequence, of TPRN were amplified from genomic DNA of individuals in the other three DFNB79-linked pedigrees (PKDF517, PKDF280, and PKDF1129) and were sequenced via dideoxy-terminator chemistry. We found a 1 bp deletion (c.1244delC), an 11 bp duplication (c.44_54dup), and an 11 bp deletion (c.42_52) cosegregating with deafness in families PKDF517, PKDF280, and PKDF1129, respectively (Figure 1A). Each of the three mutations alters the predicted open reading frame and introduces 66, 374, and 149 missense amino acids followed by a stop codon, respectively. The long missense open reading frames are due to high GC content, because stop codons are AT-rich. This phenomenon of long, open, altered reading frames following a frameshift mutation in GC-rich exons has been noted by others.22 The 11 bp duplication and 11 bp deletion found in families PKDF280 and PKDF1129, respectively, are overlapping. The 11 bp insertion produces a nearly perfect tandem triplication of this sequence (Figure 1B). The 11 bp deletion removes one of two wild-type tandem 11 bp duplications (Figures 1A and 1B) and appears to be identical to a mutation of TPRN segregating in an unrelated Moroccan family.23 All four mutations of TPRN are located in exon 1 (Figure 3) and were not observed in 488–500 control chromosomes from the Pakistani population and 400 chromosomes from the Coriell HD200CAU human variation panel (Table 3).
Figure 3.
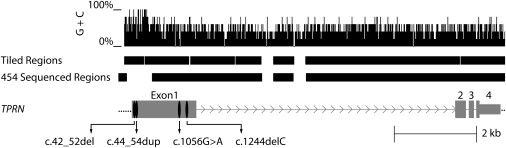
NimbleGen Probe Coverage and Sequencing Coverage across TPRN, GC Content and Locations of Four Pathogenic Variants Found in DFNB79-Linked Families
Approximately 10 kb spanning TPRN in the 2.9 Mb DFNB79 interval is shown. Vertical lines correspond to GC content in a five-base window. The upper black bars depict probe coverage (tiled regions) of TPRN. Regions for which 454 sequence was obtained are shown in the lower set of bars. Note that in exon 1, a region of 878 bp that has 81% GC content was not sequenced even though it was tiled on the NimbleGen array. We determined the sequence of this GC-rich region from PCR products amplified from genomic DNA of family members of the four DFNB79-linked pedigrees by using dideoxy-terminator chemistry. Grey-colored thick bars, lines joining them, and the thin gray bar represent exons, introns, and the 3′ UTR, respectively, of TPRN. See Table S3 as well.
The DFNB79 linkage region on chromosome 9q34.3 shows conserved synteny with mouse chromosome 2qA3 (Figure 4A). ClustalW alignment of human and mouse taperin (711 and 749 residues, respectively) shows that there is 68% identity and 75% similarity (Figure 5 and Figure S1). On the basis of conserved synteny and sequence similarity, mouse C430004E15Rik appears to be the ortholog of human TPRN. We designed primers for PCR amplification of cDNA of C430004E15Rik from two mouse multiple-tissue cDNA panels (Clontech MTC panels I and III) and from cochlear cDNA produced in our laboratory. A single band corresponding to a transcript of 2.4 kb was found in all cDNA samples tested (Figure 4B) and was sequence verified. Although Tprn is expressed in many organs and tissues, there were no other obvious clinical findings besides the hearing loss in the individuals who were homozygous for TPRN mutations.
Figure 4.
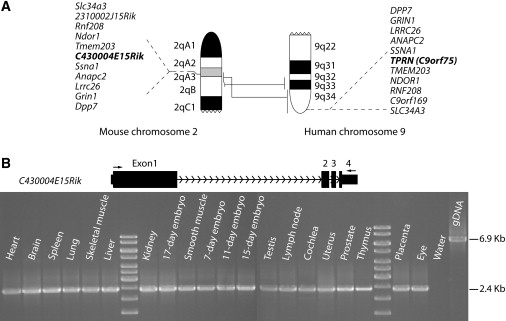
Conserved Synteny of a Part of Human Chromosome 9q with Mouse Chromosome 2 and Expression Profile of TPRN in Several Tissues
(A) Part of human chromosome 9q, 17.71 Mb at the telomeric end, is syntenic to 14.62 Mb of mouse chromosome 2, which also includes 2.9 Mb of the DFNB79 linkage interval. Vertical lines (not drawn to scale) between the two chromosomal segments show the syntenic region, with five closely linked orthologous genes on each side of the DFNB79 gene. On the basis of conserved synteny and sequence comparison (see Figure 5), C430004E15Rik is the mouse ortholog of human TPRN.
(B) Arrows indicate the position of the forward and reverse primer used to amplify the C430004E15Rik transcript from mouse multiple-tissue cDNA libraries. Expected cDNA and gDNA product sizes are 2.4 kb and 6.9 kb, respectively. Amplimers were sequence confirmed.
Figure 5.

ClustalW Alignment of Predicted Amino Acid Sequences Encoded by Human TPRN and Its Mouse Ortholog C430004E15Rik
Taperin (711 residues, Ensembl, ENSP00000387100) has 68% identity and 75% similarity with the orthologous mouse predicted protein of 749 amino acids deduced from genomic DNA and confirmed by sequencing cDNA. Residues located in the black-, gray-, and white-colored blocks are identical, similar and mismatched, respectively, and dots represent gaps. “M” on top of the black triangle is the translation start site predicted for human taperin (RefSeq, NP_001121700). Note that the additional N-terminal 61 residues are highly conserved in the mouse ortholog. Red triangles point to the first affected amino acid due to the one nonsense mutation and the three frameshift mutations segregating in the four DFNB79-linked pedigrees. The rectangle outlines the conservation of the human protein residues that were used as an immunogen for the commercially available antibody (Sigma-Aldrich) to taperin, which we used to immunostain mouse inner ear tissues. See also Figure S1 for a ClustalW alignment of taperin with multiple species.
Localization of Taperin in the Inner Ear
TPRN encodes an uncharacterized protein, which we named taperin. The human RefSeq transcript (NM_001128228.1) is predicted to encode 650 amino acids but lacks a 5′ UTR. In the Ensemble/Havana database of curated genes, there are four isoforms of TPRN. The longest isoform (ENST00000409012) extends the NM_001128228.1 RefSeq annotation at the 5′ end for an open reading frame encoding a total of 711 residues. The additional 61 residues show 74% identity and 82% similarity to the N terminus of mouse taperin (Figure 5). We consider the longer transcript to be a more accurate representation of the human and mouse ortholog on the basis of conservation of the open reading frame at the 5′ end.
Taperin appears to have no conserved domains or motifs in the amino acid sequence predicted by SMART or BLAST against the CDD database, although there are regions of low complexity and intrinsic disorder, and there is a low-probability predicted coiled-coil domain toward the C terminus. A BLAST search of the nonredundant (NR) protein database using the predicted 711-amino-acid-long sequence identified a small region of low similarity to only one other protein, phostensin (RefSeq NP_597728). Phostensin was reported to be an actin filament pointed-end-capping protein, which modulates actin dynamics by targeting protein phosphatase 1 to the F-actin cytoskeleton.24,25 A ClustalW alignment of taperin and phostensin shows an overall 22% identity and 34% similarity. In one region of 58 residues (543–600 of taperin, ENSP00000387100), there is 56% identity and 76% similarity between these two proteins (Figure S2).
A commercial antiserum from Sigma-Aldrich against human C9orf75 (taperin) was raised against a fragment 70 amino acids in length (Figure 5). These 70 residues are 75% identical to the residues of the mouse ortholog and have no obvious similarity to mouse phostensin. The antiserum to taperin was validated in heterologous cells, and no crossreactivity with phostensin was observed (Figures 6A–6D). The immunolocalization of taperin was examined in the mouse organ of Corti and vestibular sensory epithelium (Figures 6E–6H). We observed taperin immunoreactivity in the sensory epithelia of the organ of Corti and vestibular end organs and, to a lesser extent, in Reisner's membrane and the spiral ligament. In the organ of Corti, we found taperin within the supporting cells and in inner ear hair cell stereocilia, in which immunoreactivity was most prominent at the base of stereocilia where the diameter of stereocilia gradually narrows (Figures 6G and 6H). This region is referred to as the taper.26 Taperin was so named because it highlights the taper region of each stereocilium (Figures 6G and 6H).
Figure 6.
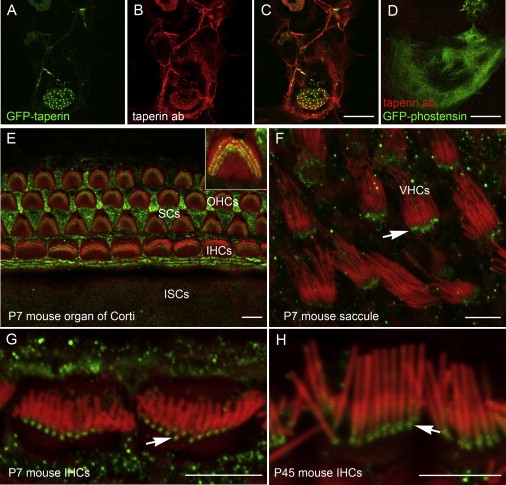
Antibody Validation and Immunolocalization of Taperin in the Inner Ear Sensory Epithelia
(A) COS7 cells transfected with an expression vector containing a cDNA construct for full-length mouse taperin epitope tagged with GFP.
(B and C) Staining of GFP-taperin-transfected COS7 cells with taperin antibody. The antibody immunofluorescence signal (red) colocalizes with GFP-taperin.
(D) COS7 cell transfected with full-length GFP-phostensin (green) and stained with taperin antibody (red). No colocalization signal is observed, revealing no crossreactivity of taperin antibody with phostensin.
(E–H) Rhodamine-phalloidin (red) in the mouse organ of Corti and vestibular sensory epithelium highlights filamentous actin of stereocilia core, hair cell cuticular plates, as well as cytoskeleton of nonsensory cells. (E) An optical section of P7 mouse organ of Corti at the surface of the reticular lamina. Taperin (green) is localized at the base of each stereocilium of outer hair cells (OHCs) and inner hair cells (IHCs) at the top of the actin-rich cuticular plate. Inset shows a higher-resolution image of an OHC apical surface with taperin (green) localized near the base of each stereocilium from all three rows. Taperin immunoreactivity is also present in the supporting cells (SCs). The inner sulcus cells (ISCs) and other cell types outside of the sensory epithelium have weaker or no obvious taperin immunoreactivity. (F) Taperin (green) is present in the taper region at the base of each stereocilium (arrow) of vestibular hair cells (VHCs) in the sensory epithelium of mouse saccule. (G and H) Higher-resolution images of IHC stereocilia reveal distinct localization of taperin along the taper region near stereocilia bases at the apical surface of P7 (G) and P45 (H) hair cells (arrows).
Scale bars in (C) and (D) represent 20 μm, and those in (E–H) represent 5 μm. See Figure S2 as well for a ClustalW alignment of human taperin and human phostensin.
Discussion
Targeted enrichment of a chromosomal interval in combination with next-generation sequencing has theoretically simplified human disease-gene identification, although there are caveats. For example, CpG islands are associated with the 5′ ends of many genes in vertebrates and have a high GC content.27 Highly GC-rich intervals covered by probes are difficult to capture and/or sequence. One such unsequenced GC-rich region represented on the NimbleGen array is shown in Figure 3. Underrepresentation of GC-rich regions from capture failure, sequencing dropout, or both was previously noted.14 It is also worth noting that two mutations of TPRN, c.42_52del and c.44_54dup, are in a region represented on the custom NimbleGen DFNB79 capture array, but no DNA sequence from this part of exon 1 was obtained (Figure 3). Thus, had we selected an affected individual from either family PKDF1129 or PKDF280 for NimbleGen capture and 454 sequencing, we probably would not have discovered a DFNB79 pathogenic mutation by using this particular disease-gene-discovery strategy. Therefore, to increase the probability of a successful disease-gene identification by using this technology, we suggest using genomic DNA from affected members of different families expected to have different allelic mutations. It will be of interest to determine which of the various alternative targeted capture and next-generation sequencing technologies work best for regions of high GC content.
There were 303 putative previously unreported variants found in the DFNB79 target region. We independently evaluated 17 of these variants by Sanger sequencing and found that only eight were true positives. Each of the eight authentic variants shared the characteristic of being supported by at least 85% of the 454 reads. We retrospectively applied this additional criterion to the original data set. The new criterion requires that variants be supported by at least 85% of the reads in a given contig, in addition to the original criterion that a contig comprise at least three nonduplicate forward and reverse 454 reads, or at least five unidirectional reads, with quality scores > 20 (or > 30 if the difference involves a homopolymer of five or more nucleotides or larger). These new criteria reduce the number of putative variants in the DFNB79 target region from 303 to 159 (values in bold in Figure 2). It is clear that it is helpful to have reference DNA sequences from ethnically matched individuals in order to distinguish polymorphisms from pathogenic mutations. Also, the likelihood of 159 novel variants in less than 2.9 Mb of genomic DNA of one subject highlights the importance of combining data from multiple families at a genetically well-defined locus. It also suggests that finding a causative mutation by whole-genome sequencing of a singleton is going to be problematic.
TPRN encodes taperin, which is concentrated at the taper region of stereocilia (Figure 6). Within the taper, the peripheral filaments of the actin core have pointed ends terminating at different levels, whereas actin filaments in the center of stereocilia core continue uninterrupted through the taper region.26 The most prominent immunolocalization signal for taperin was present at the taper of inner hair cells, but immunoreactivity was also detected in the less obvious taper regions of outer hair cell and vestibular hair cell stereocilia (Figures 6E and 6F). The specific concentration of taperin in the stereocilia taper region and the significant but limited sequence similarity to phostensin prompts speculation that taperin may also modulate actin dynamics through direct or indirect interaction with pointed ends of actin filaments specifically within the hair cell stereocilia. Also, we cannot exclude the possible involvement of taperin in the formation or maintenance of ankle links of stereocilia or interactions with proteins found near the base of stereocilia, such as PTPRQ,28,29 VLGR1,30 whirlin,31–33 usherin,34 and vezatin.33,35 Ankle links, filamentous connections between stereocilia, are transient structures that disappear after postnatal day 12 in mice.36 In contrast, taperin is present at the taper region in adult mouse stereocilia (Figure 6H), suggesting some other function in addition to, or unrelated to, ankle links.
We conclude that mutations of TPRN are the cause of hearing loss, because (1) a nonsense mutation c.1056G>A found in TPRN is the only potentially pathogenic variant identified by capture and sequencing of nearly the entire DFNB79 interval in one affected individual from family PKDF741, and (2) a total of four truncating mutations cosegregate with the deafness phenotype in four DFNB79-linked families and are not found in normal controls. Furthermore, despite a ubiquitous expression pattern, taperin appears to have only a crucial function in the inner ear. The distinct pattern of taperin expression in sensory hair cells and nonsensory cells of the organ of Corti suggests multiple possible pathogenic mechanisms causing DFNB79 deafness. Understanding the precise cause of deafness due to mutations of TPRN will require an appropriate mouse model and more detailed analyses of the expression pattern in different inner ear cell types, as well as a biochemical characterization of taperin function.
Acknowledgments
We are thankful to the family members who helped make this study possible. We thank Jonathan Bird, Byung Yoon Choi, Dennis Drayna, Meghan Drummond, Karen Friderici, Andrew Griffith, Changsoo Kang, Sue Lee, M. Hashim Raza, Mary Vendetti, and Barbara Zwiesler. This work was supported by National Institutes of Health intramural funds from the National Institute of Deafness and Other Communication Disorders (1 Z01 DC000039-12) to T.B.F.; by the Higher Education Commission, Islamabad; the Ministry of Science and Technology, Islamabad; and by the International Center for Genetic Engineering and Biotechnology, Trieste, project CRP/PAK08-01 (contract 08/009), to S.R.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Ensembl genome browser, http://www.ensembl.org/index.html
Galaxy, http://main.g2.bx.psu.edu/
Mutalyzer, a sequence variant nomenclature checker, version 1.0.4, http://eu.liacs.nl/mutalyzer/1.0.4/
National Center for Biotechnology Information (NCBI), http://www.ncbi.nlm.nih.gov/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim
Primer3, http://frodo.wi.mit.edu/primer3/
University of California-Santa Clara (UCSC) genome browser, http://genome.ucsc.edu/
References
- 1.Khan S.Y., Riazuddin S., Shahzad M., Ahmed N., Zafar A.U., Rehman A.U., Morell R.J., Griffith A.J., Ahmed Z.M., Riazuddin S., Friedman T.B. DFNB79: reincarnation of a nonsyndromic deafness locus on chromosome 9q34.3. Eur. J. Hum. Genet. 2010;18:125–129. doi: 10.1038/ejhg.2009.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Belyantseva I.A., Perrin B.J., Sonnemann K.J., Zhu M., Stepanyan R., McGee J., Frolenkov G.I., Walsh E.J., Friderici K.H., Friedman T.B., Ervasti J.M. Gamma-actin is required for cytoskeletal maintenance but not development. Proc. Natl. Acad. Sci. USA. 2009;106:9703–9708. doi: 10.1073/pnas.0900221106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu M., Yang T., Wei S., DeWan A.T., Morell R.J., Elfenbein J.L., Fisher R.A., Leal S.M., Smith R.J., Friderici K.H. Mutations in the gamma-actin gene (ACTG1) are associated with dominant progressive deafness (DFNA20/26) Am. J. Hum. Genet. 2003;73:1082–1091. doi: 10.1086/379286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Riazuddin S., Ahmed Z.M., Fanning A.S., Lagziel A., Kitajiri S., Ramzan K., Khan S.N., Chattaraj P., Friedman P.L., Anderson J.M. Tricellulin is a tight-junction protein necessary for hearing. Am. J. Hum. Genet. 2006;79:1040–1051. doi: 10.1086/510022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scott H.S., Kudoh J., Wattenhofer M., Shibuya K., Berry A., Chrast R., Guipponi M., Wang J., Kawasaki K., Asakawa S. Insertion of beta-satellite repeats identifies a transmembrane protease causing both congenital and childhood onset autosomal recessive deafness. Nat. Genet. 2001;27:59–63. doi: 10.1038/83768. [DOI] [PubMed] [Google Scholar]
- 6.Ahmed Z.M., Masmoudi S., Kalay E., Belyantseva I.A., Mosrati M.A., Collin R.W., Riazuddin S., Hmani-Aifa M., Venselaar H., Kawar M.N. Mutations of LRTOMT, a fusion gene with alternative reading frames, cause nonsyndromic deafness in humans. Nat. Genet. 2008;40:1335–1340. doi: 10.1038/ng.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schultz J.M., Khan S.N., Ahmed Z.M., Riazuddin S., Waryah A.M., Chhatre D., Starost M.F., Ploplis B., Buckley S., Velásquez D. Noncoding mutations of HGF are associated with nonsyndromic hearing loss, DFNB39. Am. J. Hum. Genet. 2009;85:25–39. doi: 10.1016/j.ajhg.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.D'Ascenzo M., Meacham C., Kitzman J., Middle C., Knight J., Winer R., Kukricar M., Richmond T., Albert T.J., Czechanski A. Mutation discovery in the mouse using genetically guided array capture and resequencing. Mamm. Genome. 2009;20:424–436. doi: 10.1007/s00335-009-9200-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Okou D.T., Steinberg K.M., Middle C., Cutler D.J., Albert T.J., Zwick M.E. Microarray-based genomic selection for high-throughput resequencing. Nat. Methods. 2007;4:907–909. doi: 10.1038/nmeth1109. [DOI] [PubMed] [Google Scholar]
- 10.Tucker T., Marra M., Friedman J.M. Massively parallel sequencing: the next big thing in genetic medicine. Am. J. Hum. Genet. 2009;85:142–154. doi: 10.1016/j.ajhg.2009.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wheeler D.A., Srinivasan M., Egholm M., Shen Y., Chen L., McGuire A., He W., Chen Y.J., Makhijani V., Roth G.T. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–876. doi: 10.1038/nature06884. [DOI] [PubMed] [Google Scholar]
- 12.Grimberg J., Nawoschik S., Belluscio L., McKee R., Turck A., Eisenberg A. A simple and efficient non-organic procedure for the isolation of genomic DNA from blood. Nucleic Acids Res. 1989;17:8390. doi: 10.1093/nar/17.20.8390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ning Z., Cox A.J., Mullikin J.C. SSAHA: a fast search method for large DNA databases. Genome Res. 2001;11:1725–1729. doi: 10.1101/gr.194201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choi M., Scholl U.I., Ji W., Liu T., Tikhonova I.R., Zumbo P., Nayir A., Bakkaloğlu A., Ozen S., Sanjad S. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc. Natl. Acad. Sci. USA. 2009;106:19096–19101. doi: 10.1073/pnas.0910672106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Volpi L., Roversi G., Colombo E.A., Leijsten N., Concolino D., Calabria A., Mencarelli M.A., Fimiani M., Macciardi F., Pfundt R. Targeted next-generation sequencing appoints c16orf57 as clericuzio-type poikiloderma with neutropenia gene. Am. J. Hum. Genet. 2010;86:72–76. doi: 10.1016/j.ajhg.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bork J.M., Peters L.M., Riazuddin S., Bernstein S.L., Ahmed Z.M., Ness S.L., Polomeno R., Ramesh A., Schloss M., Srisailpathy C.R. Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23. Am. J. Hum. Genet. 2001;68:26–37. doi: 10.1086/316954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khan S.Y., Ahmed Z.M., Shabbir M.I., Kitajiri S., Kalsoom S., Tasneem S., Shayiq S., Ramesh A., Srisailpathy S., Khan S.N. Mutations of the RDX gene cause nonsyndromic hearing loss at the DFNB24 locus. Hum. Mutat. 2007;28:417–423. doi: 10.1002/humu.20469. [DOI] [PubMed] [Google Scholar]
- 18.Belyantseva I.A., Boger E.T., Friedman T.B. Myosin XVa localizes to the tips of inner ear sensory cell stereocilia and is essential for staircase formation of the hair bundle. Proc. Natl. Acad. Sci. USA. 2003;100:13958–13963. doi: 10.1073/pnas.2334417100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kokotas H., Petersen M.B., Willems P.J. Mitochondrial deafness. Clin. Genet. 2007;71:379–391. doi: 10.1111/j.1399-0004.2007.00800.x. [DOI] [PubMed] [Google Scholar]
- 20.Mewborn S.K., Lese Martin C., Ledbetter D.H. The dynamic nature and evolutionary history of subtelomeric and pericentromeric regions. Cytogenet. Genome Res. 2005;108:22–25. doi: 10.1159/000080798. [DOI] [PubMed] [Google Scholar]
- 21.Nicholson P., Yepiskoposyan H., Metze S., Zamudio Orozco R., Kleinschmidt N., Mühlemann O. Nonsense-mediated mRNA decay in human cells: mechanistic insights, functions beyond quality control and the double-life of NMD factors. Cell. Mol. Life Sci. 2009 doi: 10.1007/s00018-009-0177-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clamp M., Fry B., Kamal M., Xie X., Cuff J., Lin M.F., Kellis M., Lindblad-Toh K., Lander E.S. Distinguishing protein-coding and noncoding genes in the human genome. Proc. Natl. Acad. Sci. USA. 2007;104:19428–19433. doi: 10.1073/pnas.0709013104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Y., Pohl E., Redouane B., Schraders M., Nürnberg G., Charif M., Admiraal R.J.C., von Ameln S., Baessmann I., Kandil M. Mutations in TPRN cause a progressive form of autosomal-recessive nonsyndromic hearing loss. Am. J. Hum. Genet. 2010;86:479–484. doi: 10.1016/j.ajhg.2010.02.003. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kao S.C., Chen C.Y., Wang S.L., Yang J.J., Hung W.C., Chen Y.C., Lai N.S., Liu H.T., Huang H.L., Chen H.C. Identification of phostensin, a PP1 F-actin cytoskeleton targeting subunit. Biochem. Biophys. Res. Commun. 2007;356:594–598. doi: 10.1016/j.bbrc.2007.03.026. [DOI] [PubMed] [Google Scholar]
- 25.Lai N.S., Wang T.F., Wang S.L., Chen C.Y., Yen J.Y., Huang H.L., Li C., Huang K.Y., Liu S.Q., Lin T.H., Huang H.B. Phostensin caps to the pointed end of actin filaments and modulates actin dynamics. Biochem. Biophys. Res. Commun. 2009;387:676–681. doi: 10.1016/j.bbrc.2009.07.086. [DOI] [PubMed] [Google Scholar]
- 26.Tilney L.G., Derosier D.J., Mulroy M.J. The organization of actin filaments in the stereocilia of cochlear hair cells. J. Cell Biol. 1980;86:244–259. doi: 10.1083/jcb.86.1.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gardiner-Garden M., Frommer M. CpG islands in vertebrate genomes. J. Mol. Biol. 1987;196:261–282. doi: 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
- 28.Goodyear R.J., Legan P.K., Wright M.B., Marcotti W., Oganesian A., Coats S.A., Booth C.J., Kros C.J., Seifert R.A., Bowen-Pope D.F., Richardson G.P. A receptor-like inositol lipid phosphatase is required for the maturation of developing cochlear hair bundles. J. Neurosci. 2003;23:9208–9219. doi: 10.1523/JNEUROSCI.23-27-09208.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hirono M., Denis C.S., Richardson G.P., Gillespie P.G. Hair cells require phosphatidylinositol 4,5-bisphosphate for mechanical transduction and adaptation. Neuron. 2004;44:309–320. doi: 10.1016/j.neuron.2004.09.020. [DOI] [PubMed] [Google Scholar]
- 30.McGee J., Goodyear R.J., McMillan D.R., Stauffer E.A., Holt J.R., Locke K.G., Birch D.G., Legan P.K., White P.C., Walsh E.J., Richardson G.P. The very large G-protein-coupled receptor VLGR1: a component of the ankle link complex required for the normal development of auditory hair bundles. J. Neurosci. 2006;26:6543–6553. doi: 10.1523/JNEUROSCI.0693-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Belyantseva I.A., Boger E.T., Naz S., Frolenkov G.I., Sellers J.R., Ahmed Z.M., Griffith A.J., Friedman T.B. Myosin-XVa is required for tip localization of whirlin and differential elongation of hair-cell stereocilia. Nat. Cell Biol. 2005;7:148–156. doi: 10.1038/ncb1219. [DOI] [PubMed] [Google Scholar]
- 32.Delprat B., Michel V., Goodyear R., Yamasaki Y., Michalski N., El-Amraoui A., Perfettini I., Legrain P., Richardson G., Hardelin J.P., Petit C. Myosin XVa and whirlin, two deafness gene products required for hair bundle growth, are located at the stereocilia tips and interact directly. Hum. Mol. Genet. 2005;14:401–410. doi: 10.1093/hmg/ddi036. [DOI] [PubMed] [Google Scholar]
- 33.Michalski N., Michel V., Bahloul A., Lefèvre G., Barral J., Yagi H., Chardenoux S., Weil D., Martin P., Hardelin J.P. Molecular characterization of the ankle-link complex in cochlear hair cells and its role in the hair bundle functioning. J. Neurosci. 2007;27:6478–6488. doi: 10.1523/JNEUROSCI.0342-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Adato A., Lefèvre G., Delprat B., Michel V., Michalski N., Chardenoux S., Weil D., El-Amraoui A., Petit C. Usherin, the defective protein in Usher syndrome type IIA, is likely to be a component of interstereocilia ankle links in the inner ear sensory cells. Hum. Mol. Genet. 2005;14:3921–3932. doi: 10.1093/hmg/ddi416. [DOI] [PubMed] [Google Scholar]
- 35.Küssel-Andermann P., El-Amraoui A., Safieddine S., Nouaille S., Perfettini I., Lecuit M., Cossart P., Wolfrum U., Petit C. Vezatin, a novel transmembrane protein, bridges myosin VIIA to the cadherin-catenins complex. EMBO J. 2000;19:6020–6029. doi: 10.1093/emboj/19.22.6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goodyear R.J., Marcotti W., Kros C.J., Richardson G.P. Development and properties of stereociliary link types in hair cells of the mouse cochlea. J. Comp. Neurol. 2005;485:75–85. doi: 10.1002/cne.20513. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.