

Abstract
Proliferative vasculopathy and hydranencephaly-hydrocephaly syndrome (PVHH), also known as Fowler syndrome, is an autosomal-recessively inherited prenatal lethal disorder characterized by hydranencephaly; brain stem, basal ganglia, and spinal cord diffuse clastic ischemic lesions with calcifications; glomeruloid vasculopathy of the central nervous system and retinal vessels; and a fetal akinesia deformation sequence (FADS) with muscular neurogenic atrophy. To identify the molecular basis for Fowler syndrome, we performed autozygosity mapping studies in three consanguineous families. The results of SNP microarrays and microsatellite marker genotyping demonstrated linkage to chromosome 14q24.3. Direct sequencing of candidate genes within the target interval revealed five different germline mutations in FLVCR2 in five families with Fowler syndrome. FLVCR2 encodes a transmembrane transporter of the major facilitator superfamily (MFS) hypothesized to be involved in regulation of growth, calcium exchange, and homeostasis. This is the first gene to be associated with Fowler syndrome, and this finding provides a basis for further studies to elucidate the pathogenetic mechanisms and phenotypic spectrum of associated disorders.
Main Text
In 1972, Fowler et al.1 described a family in which five sisters were affected by a prenatally lethal disorder diagnosed between 26 and 33 wks of gestation. Examination showed “bubble-like” cerebral hemispheres with no discernible gyral pattern, as well as a distinctive glomeruloid vascular proliferation in the central nervous system and retina. Polyhydramnios, fetal akinesia deformation sequence (FADS [MIM 208150]), and muscular neurogenic atrophy were also apparent. The disorder has subsequently been reported as Fowler syndrome, proliferative vasculopathy and hydranencephaly-hydrocephaly (PVHH), or encephaloclastic proliferative vasculopathy (EPV [MIM 225790]) and is inherited as an autosomal-recessive trait. Fowler syndrome is rare (fewer than 40 cases have been described1–13), but it is likely to be underdiagnosed because neuropathology is not available for many cases of FADS. Hence, the availability of accurate genetic testing for Fowler syndrome would facilitate diagnosis and improve family management. In order to map and identify a gene for Fowler syndrome, we initially investigated three consanguineous families of Pakistani origin (families 1–3) by using an autozygosity mapping strategy. A total of five kindreds (containing seven fetuses with Fowler syndrome) were analyzed by direct sequencing of candidate genes within the linked autozygous region. The major clinical findings in the seven fetuses with Fowler syndrome are summarized in Table 1.
Table 1.
Summary of Major Clinical Findings in the Seven Fetuses with Fowler Syndrome
| Case | Gestation at Diagnosis | Ultrasound Findings | Pathological Findings | Karyotype |
|---|---|---|---|---|
| Family 2 | ||||
| 3 | 19 wks | Ventriculomegaly, joint contractures upper and lower limbs, talipes | Hydrocephalus, cortical thinning, pterygia, polymicrogyria, muscle hypoplasia, cleft palate. Glomeruloid vascular proliferation (cerebral cortex, cerebellum, brain stem). | 46XX |
| 4 | 21 wks | Intrauterine death | Cortical thinning, hydranencephaly. Glomeruloid vascular proliferation (cerebral cortex, cerebellum, brain stem). Joint contractures upper and lower limbs. | Not undertaken. Phenotypically male |
| Family 1 | ||||
| 1 | 21 wks | Ventriculomegaly, joint contractures upper and lower limbs, talipes, non-visualized cerebellum | Hydrocephalus, cortical thinning. Glomeruloid vascular proliferation (cerebral cortex, cerebellum, brain stem, spinal cord). | 46XY |
| 2 | 16 wks | Cystic hygroma, ventriculomegaly, joint contractures upper and lower limbs, talipes, hydranencephaly | Hydrocephalus, cortical thinning, hydranencephaly. Glomeruloid vascular proliferation (cerebral cortex, cerebellum, brain stem, spinal cord). | 46XY |
| Family 3 | ||||
| 5 | 20 wks | Ventriculomegaly and a posterior fossa cyst, cerebellum not seen | Dandy-Walker malformation (1.5cm posterior fossa cyst, small cerebellum); marked bilateral ventriculomegaly, cortical thinning, glomeruloid vascular proliferation (cerebral cortex, cerebellum, brain stem). | 46XY |
| Family 4 | ||||
| 6 | 13 wks | Ventriculomegaly, joint contractures upper and lower limbs, pterygia, talipes | Hydrocephalus, cortical thinning. Severely hypoplastic brain stem, cerebellum and spinal cord. Glomeruloid vascular proliferation (cerebral cortex, cerebellum, brain stem, spinal cord). | 46XY |
| Family 5 | ||||
| 7 | 24 wks | Ventriculomegaly, joint contractures upper and lower limbs, talipes, hydranencephaly | Hydrocephalus, cortical thinning, hydranencephaly. Severely hypoplastic brain stem, cerebellum and spinal cord. Glomeruloid vascular proliferation (cerebral cortex, cerebellum, brain stem, spinal cord). | 46XX |
All parents gave written informed consent. The study was approved by South Birmingham Local Research Ethics Committee and was performed in accordance with the Declaration of Helsinki. Genomic DNA from the affected fetuses was extracted from fetal tissues, placenta material, and amniotic fluid, respectively. Furthermore, genomic DNA from parents and some unaffected siblings of two of the five families (families 1 and 2) was extracted from peripheral lymphocytes and buccal swabs by standard techniques, respectively.
We first performed a genome-wide linkage scan by using Affymetrix 250k SNP microarrays (Affymetrix), in cases 1 and 2 of family 1 and case 3 of family 2, to identify regions of homozygosity (> 2 Mb) that were common to all cases. DNA was amplified and hybridized to the Affymetrix SNP chip according to the manufacturer's instructions. Two extended overlapping regions of homozygosity on chromosome 14 (12.56 Mb [from rs10134110 to rs1205233; 57,642,936–70,202,512] and 24.65 Mb [from rs7160623 to rs1952299; 72,140,408–96,793,024 according to NCBI build 36.3]) shared by the three fetuses from families 1 and 2 were identified. Further genotyping with microsatellite markers was then undertaken in all available members of families 1 and 2, as well as in the more recently ascertained case 4 of family 2 and case 5 of family 3 (information on primer sequences and the physical order of the markers was obtained from the NCBI database and the UCSC browser, respectively). Linkage to the 12.56 Mb region on chromosome 14 was excluded by the finding of heterozygous alleles in case 4 of family 2 (data not shown). However, genotyping of microsatellite markers within the second region confirmed that affected fetuses were homozygous and that one unaffected sibling was heterozygous (Figure 1). Upon comparison of the microsatellite marker data of cases 1–5, a common haplotype was identified in the three consanguineous families of Pakistani ancestry (from 74.87 Mb to 78.97 Mb). This common haplotype was confirmed, and the candidate region was narrowed by the SNP microarray results showing for cases 1–3 an identical homozygous genotype between SNPs rs4148077 and rs11622335 (from 73.83 Mb to 75.87 Mb). Multipoint linkage analysis for families 1 and 2 gave a combined maximum LOD score of 3.69 at D14S61 (family 1, 1.81; family 2, 1.88).
Figure 1.
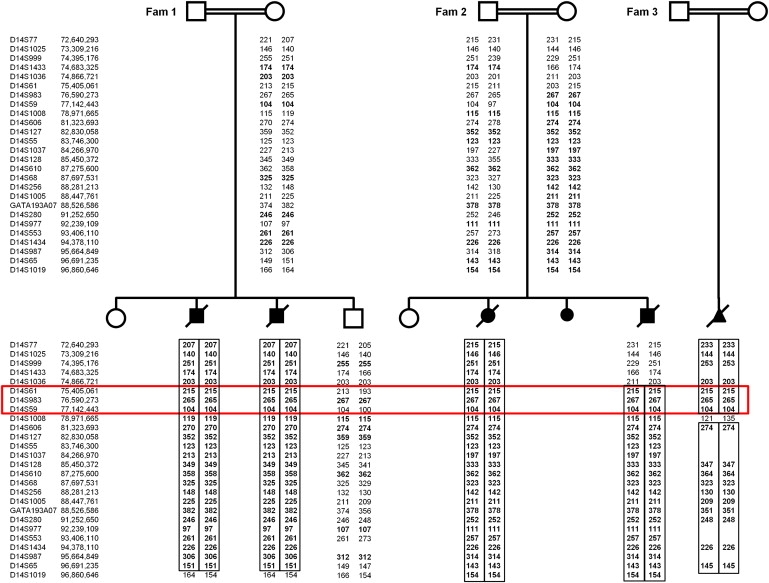
Pedigrees and Linkage Analysis Results for Three Consanguineous Families with Fowler Syndrome
The results of the microsatellite marker analysis for chromosome 14 of the three Pakistani families 1, 2 and 3 are shown (localization of markers according to NCBI build 36.3). The markers of the assumed common homozygous haplotype are highlighted (red box).
We hypothesized that the three consanguineous families of Pakistani origin (families 1–3) might share a common mutation, and we focused on the region of the shared haplotype at 14q24.3 (from 74.87 Mb to 75.87 Mb) that contained ten known genes, pseudogenes, and hypothetical proteins. Mutation analysis of genes in this region was undertaken by direct sequencing with the use of the Big Dye Terminator Cycle Sequencing System and with use of an ABI PRISM 3730 DNA Analyzer (Applied Biosystems). The genomic DNA sequence of the genes was taken from Ensembl, and primer pairs for the translated exons were designed with primer3 software. Sequencing of Jun dimerization protein 2 (JDP2 [MIM 608657]), basic leucine zipper transcription factor, ATF-like (BATF [MIM 612476]), chromosome 14 open reading frame 1 (C14orf1 [MIM 604576]), transforming growth factor, beta 3 (TGFB3 [MIM 190230]), chromosome 14 open reading frame 179 (C14orf179), and chromosome 14 open reading frame 118 (C14orf188) did not reveal any pathogenic variants. However, a homozygous missense mutation, c.1289C>G (p.Thr430Arg), was detected in exon 7 of feline leukemia virus subgroup C cellular receptor family, member 2 (FLVCR2 or C14orf58 or FLJ20371 [MIM 610865]) in all five affected fetuses in families 1–3 (Figure 2A). This mutation cosegregated with the disease in families 1 and 2 and was not detected in 646 control chromosomes (354 alleles from Asian individuals and 292 alleles from individuals of European descent). Sequence alignment showed Thr430 to be conserved down to C. elegans (Figure 3A), and the missense mutation at this position was predicted to be damaging by bioinformatic analysis with PolyPhen (PSIC score 2.256) and SIFT. After the identification of the putative missense mutation in FLVCR2, mutation analysis was performed in two additional recently ascertained Fowler syndrome cases (cases 6 and 7) from two nonconsanguineous kindreds of Northern European origin. Case 6 (family 4) was compound heterozygous for a 6 bp deletion, c.329_334del (p.Asn110_Phe112delinsIle), in exon 1 and a missense mutation, c.1192C>G (p.Leu398Val), in exon 6 (Figure 2B). Case 7 (family 5) was compound heterozygous for a nonsense mutation, c.473C>A (p.Ser158X), in exon 1 and a missense mutation, c.839C>G (p.Pro280Arg), in exon 3 (Figure 2C). Both the Leu398 and the Pro280 residues are highly conserved (Leu398 is conserved in Danio rerio and C. elegans but not in Drosophila melanogaster [Figure 3A], and Pro280 is conserved in all FLVCR2 orthologs [Figure 3B]). The 6 bp deletion removed three conserved amino acids (three conserved to Danio rerio and two to C. elegans [Figure 3C]) and produced a missense substitution. None of the identified mutations was detected in at least 936 control chromosomes (346 alleles from Asian individuals and 590 alleles from individuals of European descent).
Figure 2.
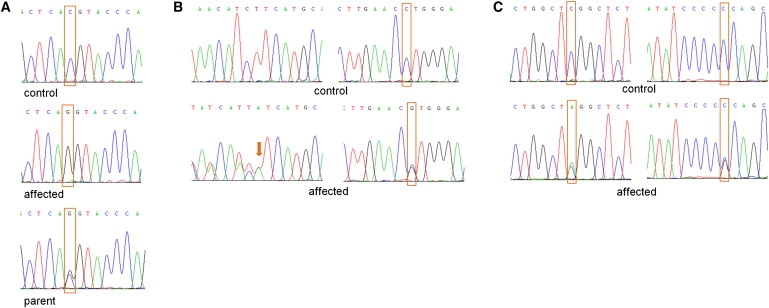
Sequences of the FLVCR2 Mutations
(A) Chromatograms of mutation c.1289C>G in Pakistani families 1, 2 and 3. Top row: The sequence chromatogram of a control sample with wild-type allele. Middle row: The sequence chromatogram of an affected fetus with the homozygous variant (c.1289G/G). Bottom row: The sequence chromatogram of a parent with the heterozygous mutation (c.1289C/G).
(B) Chromatograms of mutations c.329_334 del (panel left) and c.1192C>G (panel right) in families 4. Top row: The sequence chromatogram of a control sample with wild-type alleles. Bottom row: The sequence chromatogram of an affected fetus with the heterozygous deletion and heterozygous C to G substitution (c.1192C/G).
(C) Chromatograms of variants c.473C>A (panel left) and c.839C>G (panel right) in families 5. Top row: The sequence chromatogram of a control sample with wild-type alleles. Bottom row: The sequence chromatogram of an affected fetus with the heterozygous changes (c.473C/A and c.839C/G).
Figure 3.

Conservation of Mutated FLVCR2 Amino Acid Residues
Seven representative sequences of Bilateria for FLVCR2 are aligned (∗predicted amino acid sequences). The affected amino acid residues are shown in red and bold. Residues matching the human FLVCR2 sequence are in gray and those not matching the sequence are in white. Residues which are conserved in all seven sequences are in dark gray.
(A) Leu398 in exon 6 and Thr430 in exon 7.
(B) Pro280 in exon 3.
(C) Asn110_Phe112 in exon 1.
In total, five different mutations, including three missense variants, one nonsense mutation, and one deletion/insertion change, have been found in FLVCR2 (Table 2). FLVCR2 is a protein with 12 predicted transmembrane domains that belongs to the major facilitator superfamily (MFS) of secondary carriers that transport small solutes in response to chemiosmotic ion gradients.14 The complex amino acid deletion/insertion mutation occurs in the predicted first extracellular loop, whereas the missense substitutions at amino acid residues 280 and 430 arise within intracellular loops, and the amino acid residue 398 is located in or in close proximity to transmembrane domain 9. The nonsense mutation occurs in the predicted transmembrane domain 3 and results (in the absence of nonsense-mediated decay) in a 158 aa protein that lacks the remaining nine transmembrane domains (Figure 4). The precise consequence of these mutations is difficult to establish, but it is conceivable that they may cause local conformational changes altering the function of the protein, such as selectivity or transport performance.
Table 2.
FLVCR2 Mutations Identified in the Five Families with Fowler Syndrome
| Patient | Nucleotide Level | Protein Level | Type of Mutation | Exon |
|---|---|---|---|---|
| Cases 1 and 2 (family 1) | c.1289C>G | p.Thr430Arg | missense | 7 |
| Cases 3 and 4 (family 2) | c.1289C>G | p.Thr430Arg | missense | 7 |
| Case 5 (family 3) | c.1289C>G | p.Thr430Arg | missense | 7 |
| Case 6 (family 4) | c.329_334del | p.Asn110_Phe112delinsIle | deletion/insertion | 1 |
| c.1192C>G | p.Leu398Val | missense | 6 | |
| Case 7 (family 5) | c.473C>A | p.Ser158X | nonsense | 1 |
| c.839C>G | p.Pro280Arg | missense | 3 |
Figure 4.
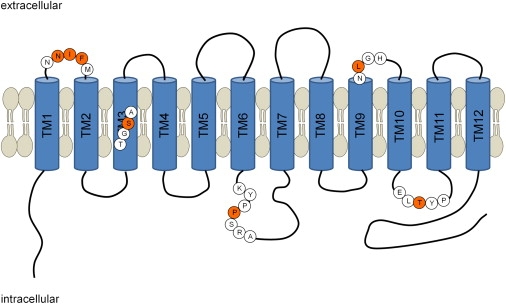
Localization of Mutations in a Schematic Diagram of the FLVCR2 Gene Product that Comprises Twelve Transmembrane Domains
Asn110_Phe112delinsIle and Leu398Val are predicted to be in an extracellular loop whereas Pro280Arg and Thr430Arg are probably in an intracellular loop. The nonsense mutation Ser158X is probably located in transmembrane domain 3. The mutated amino acid residues are shown in red circles and the surrounding amino acids are in white circles.
The exact function of the putative transporter FLVCR2 and its specific substrate is still unclear. FLVCR2 and its paralog feline leukemia virus subgroup C cellular receptor 1 (FLVCR1 [MIM 609144]) share 60% amino acid sequence identity across their 12 transmembrane domains and were apparently involved in chromosomes 1q/14q block duplication.15 FLVCR1 is a human exporter of heme that is essential for erythropoiesis and serves as a receptor for feline leukemia virus subgroup C.16 Feline leukemia viruses (FeLVs) are pathogenic retroviruses of domestic cats that induce proliferative, degenerative, and immunosuppressive disorders. FeLV-A is the primary strain, and FeLV-C was formed by mutations in the FeLV-A Env gene. FeLV-C is tightly associated with red blood cell aplasia caused by specific disruption in erythroid progenitor cell development. Whereas FeLV-A viruses infect cells through the solute carrier family 19 (thiamine transporter), member 2 (SLC19A2 or THTR1 [MIM 603941]), FeLV-C strains utilize the heme exporter FLVCR1.15–17 Disruption of FLVCR1 specifically disrupts early erythropoiesis and mimics FeLV-C-induced anemia in cats. Recently, a FeLV-C strain was identified that caused pure red cell aplasia and, as a result of a Env mutation, can use FLVCR1, THTR1, and FLVCR2 for infection.18 An interesting inference of this finding is that, because retroviruses often use structurally and functionally related proteins as receptors, FLVCR1 and FLVCR2 may have related functions. FLVCR1 null mice die in utero at one of two embryonic times (at or before embryonic day [E]7.5 and between E14.5 and E16.5) and demonstrate developmental defects most prominently at E14.5, including abnormal limb, hand, and digit maturation, flattened faces, and hypertelorism (in addition to defective erythropoiesis).19
It has been previously suggested that the FLVCR2 transporter is specific for a calcium-chelate complex and is involved in the regulation of growth and calcium metabolism.20 We note that the proliferation and motility of vascular endothelial cells (ECs), which are major processes in angiogenesis and the induction of CNS vascularization, are calcium-dependent events. These processes require interaction of endothelial cells with the extracellular matrix and associated support cells (pericytes),21 and it is interesting that a recent study suggested that the pathogenesis of the cerebral proliferative glomeruloid vasculopathy might be related to a possible deficit of pericytes.12
The glomeruloid vasculopathy seen in Fowler syndrome is pathognomonic, can occur throughout the brain and spinal cord, and is associated with varying degrees of calcification and necrosis in the white matter, basal ganglia, brainstem, cerebellum, and spinal cord.4 The proliferative glomeruloid vasculopathy can also occur in the optic nerve but does not appear outside of the CNS. Nevertheless, although there is a close association between the vascular and CNS lesions, it is unclear which process is the primary event. Thus, the proliferative vasculopathy might be a response to loss of brain tissue. Vascular proliferation (neovascularisation) occurs as part of the reparative process following hypoxic-ischemic and traumatic brain injury. However, the proliferation occurs only in areas of damage to the brain tissue. An alternative hypothesis is that the CNS proliferative vasculopathy is the primary event and produces focal ischemic necrosis of adjacent white and gray matter, with neuronal loss, calcification, and gliosis. In Fowler syndrome, the proliferative vasculopathy seems to be invariable and the key diagnostic feature, being found in areas of otherwise preserved brain tissue, suggesting that the vascular proliferation is the primary abnormality.2,4,13 Although expression of the mouse Flvcr2 ortholog (Mfsd7c; NM_145447; BC011209) has not yet been annotated in publicly available databases, closer examination of in situ hybridization images in Genepaint22 and the Allen Brain Atlas23 revealed that the mouse Flvcr2 ortholog has strong and widespread expression throughout the adult brain and spinal cord (Figure 5). Of particular note, with regard to human glomeruloid vasculopathy, is the similar localization of Endoglin and Flvcr2 in endothelial-like cells (Figure 5).
Figure 5.

Embryonal Expression of Mfsd7c the Mouse Ortholog of FLVCR2
(A) Endoglin expression at E12.5 by in situ hybridization showing expression in endothelial cells (blue) throughout the embryo.
(B) Magnification of the embryo in panel A showing the form of endothelial cells at the hindbrain level.
(C) Flvcr2 expression in an E14.5 embryo taken from Genepaint22 (ID: EH275 Gene: BC011209). Note the similarity in form to endoglin expression.
(D) Sagittal brain image taken from the Allen Brain Atlas23 (Allen Brain Atlas [Internet]. Seattle (WA): Allen Institute for Brain Science. © 2004– [cited 1/2/10], (Section ID BC011209_80)).
(E) Transverse image of the spinal cord taken from the Allen Brain Atlas23 (Allen Brain Atlas [Internet]. Seattle (WA): Allen Institute for Brain Science. © 2004– [cited 1/2/10], (Section ID 100009650-BC011209-4.0-116)).
(F) Magnification of the image of D at the brain stem level, showing expression in endothelial-like cells. The analysis of endoglin expression was performed as described previously.30
Fowler syndrome often presents with a FADS phenotype. FADS is a heterogeneous disorder. In some cases FADS may be caused by mutations in genes encoding components of the embryonal acetylcholine receptor,24–28 but in many cases the cause is unknown. Two cases of Fowler syndrome were detected in a series of 30 cases of FADS,29 but Fowler syndrome (with the characteristic histopathological findings) has also been reported in the absence of FADS. Hydrocephalus is usually diagnosed on prenatal ultrasonography, and although macroscopic examination at autopsy of Fowler syndrome cases generally suggests hydranencephaly (replacement of the forebrain by a fluid-filled sac bounded only by meninges), histopathological examination reveals severe hydrocephalus with (1) a remnant of the cortical mantle present around the cerebral ventricles and (2) in most cases, a normal underlying brain structure. The diagnosis of Fowler syndrome is complicated by variability of the clinical presentation. In addition to presentation with hydrocephalus or pterygia/FADS in the first trimester, other cases present in mid-second trimester. The differential diagnosis includes other causes of hydrocephalus and fetal akinesia (e.g., cerebro-ocular-dysplasia-muscular dystrophy, Walker-Warburg syndrome [MIM 236670] and Fukuyama congenital muscular dystrophy [MIM 253800]) and apparently isolated FADS or hydrocephalus/true hydranencephaly. Although the neuropathology is pathognomonic and can readily distinguish Fowler syndrome from isolated hydrocephalus, etc., an autopsy may be unavailable. Hence, the identification of germline mutations in FLVCR2 will enable genetic testing in suspected cases and will, in addition to allowing carrier testing and earlier prenatal diagnosis, provide a basis for determining the frequency and range of the phenotype of Fowler syndrome.
Acknowledgments
We thank the Wellcome Trust and WellChild for financial support. We are also grateful to the families for their help with this study.
Web Resources
The URLs for data presented herein are as follows:
Chromas software, http://www.technelysium.com.au/chromas.html
EmbryoExpress, http://www.embryoexpress.org/
Ensembl Genome Browser, http://www.ensembl.org/index.html
Genepaint, http://www.genepaint.org/
Mouse Brain Map, http://mouse.brain-map.org/
National Center for Biotechnology Information (NCBI) browser, http://www.ncbi.nlm.nih.gov
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/omim/
PolyPhen, http://genetics.bwh.harvard.edu/pph/
SIFT, http://sift.jcvi.org/
Superlink online, http://bioinfo.cs.technion.ac.il/superlink-online/
University of California, Santa Cruz (UCSC) Genome Browser, http://www.genome.ucsc.edu
References
- 1.Fowler M., Dow R., White T.A., Greer C.H. Congenital hydrocephalus-hydrencephaly in five siblings, with autopsy studies: a new disease. Dev. Med. Child Neurol. 1972;14:173–188. doi: 10.1111/j.1469-8749.1972.tb02575.x. [DOI] [PubMed] [Google Scholar]
- 2.Harper C., Hockey A. Proliferative vasculopathy and an hydranencephalic-hydrocephalic syndrome: a neuropathological study of two siblings. Dev. Med. Child Neurol. 1983;25:232–239. doi: 10.1111/j.1469-8749.1983.tb13747.x. [DOI] [PubMed] [Google Scholar]
- 3.Norman M.G., McGillivray B. Fetal neuropathology of proliferative vasculopathy and hydranencephaly-hydrocephaly with multiple limb pterygia. Pediatr. Neurosci. 1988;14:301–306. doi: 10.1159/000120409. [DOI] [PubMed] [Google Scholar]
- 4.Harding B.N., Ramani P., Thurley P. The familial syndrome of proliferative vasculopathy and hydranencephaly-hydrocephaly: immunocytochemical and ultrastructural evidence for endothelial proliferation. Neuropathol. Appl. Neurobiol. 1995;21:61–67. doi: 10.1111/j.1365-2990.1995.tb01029.x. [DOI] [PubMed] [Google Scholar]
- 5.Castro-Gago M., Pintos-Martínez E., Forteza-Vila J., Iglesias-Diz M., Ucieda-Somoza R., Silva-Villar I., Codesido-López J., Viso-Lorenzo A., Campos Y., Arenas J., Eirís-Puñal J. Congenital hydranencephalic-hydrocephalic syndrome with proliferative vasculopathy: a possible relation with mitochondrial dysfunction. J. Child Neurol. 2001;16:858–862. doi: 10.1177/08830738010160111401. [DOI] [PubMed] [Google Scholar]
- 6.Laurichesse-Delmas H., Beaufrère A.M., Martin A., Kaemmerlen A.G., Déchelotte P., Lémery D. First-trimester features of Fowler syndrome (hydrocephaly-hydranencephaly proliferative vasculopathy) Ultrasound Obstet. Gynecol. 2002;20:612–615. doi: 10.1046/j.1469-0705.2002.00830.x. [DOI] [PubMed] [Google Scholar]
- 7.Witters I., Moerman P., Devriendt K., Braet P., Van Schoubroeck D., Van Assche F.A., Fryns J.P. Two siblings with early onset fetal akinesia deformation sequence and hydranencephaly: further evidence for autosomal recessive inheritance of hydranencephaly, fowler type. Am. J. Med. Genet. 2002;108:41–44. [PubMed] [Google Scholar]
- 8.Halder A., Panigrahi I., Pal L. Fowler-like syndrome with extreme oligohydramnios, growth restriction and without muscular hypoplasia. Indian Pediatr. 2003;40:418–423. [PubMed] [Google Scholar]
- 9.Usta I.M., AbuMusa A.A., Khoury N.G., Nassar A.H. Early ultrasonographic changes in Fowler syndrome features and review of the literature. Prenat. Diagn. 2005;25:1019–1023. doi: 10.1002/pd.1240. [DOI] [PubMed] [Google Scholar]
- 10.Ibrahim A., Murthy P., Arunkalaivanan A.S. A case of recurrent first-trimester Fowler syndrome. J. Obstet. Gynaecol. 2007;27:201–202. doi: 10.1080/01443610601138042. [DOI] [PubMed] [Google Scholar]
- 11.Al-Adnani M., Kiho L., Scheimberg I. Fowler syndrome presenting as a Dandy-Walker malformation: a second case report. Pediatr. Dev. Pathol. 2009;12:68–72. doi: 10.2350/07-09-0348.1. [DOI] [PubMed] [Google Scholar]
- 12.Bessières-Grattagliano B., Foliguet B., Devisme L., Loeuillet L., Marcorelles P., Bonnière M., Laquerrière A., Fallet-Bianco C., Martinovic J., Zrelli S. Refining the clinicopathological pattern of cerebral proliferative glomeruloid vasculopathy (Fowler syndrome): report of 16 fetal cases. Eur. J. Med. Genet. 2009;52:386–392. doi: 10.1016/j.ejmg.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 13.Williams D., Patel C., Fallet-Bianco C., Kalyanasundaram K., Yacoubi M., Déchelotte P., Scott R., Bazin A., Bessières B., Marton T., Cox P. Fowler syndrome—a clinical, radiological, and pathological study of 14 cases. Am. J. Med. Genet. A. 2010;152A:153–160. doi: 10.1002/ajmg.a.33094. [DOI] [PubMed] [Google Scholar]
- 14.Pao S.S., Paulsen I.T., Saier M.H., Jr. Major facilitator superfamily. Microbiol. Mol. Biol. Rev. 1998;62:1–34. doi: 10.1128/mmbr.62.1.1-34.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lipovich L., Hughes A.L., King M.C., Abkowitz J.L., Quigley J.G. Genomic structure and evolutionary context of the human feline leukemia virus subgroup C receptor (hFLVCR) gene: evidence for block duplications and de novo gene formation within duplicons of the hFLVCR locus. Gene. 2002;286:203–213. doi: 10.1016/s0378-1119(02)00457-2. [DOI] [PubMed] [Google Scholar]
- 16.Quigley J.G., Yang Z., Worthington M.T., Phillips J.D., Sabo K.M., Sabath D.E., Berg C.L., Sassa S., Wood B.L., Abkowitz J.L. Identification of a human heme exporter that is essential for erythropoiesis. Cell. 2004;118:757–766. doi: 10.1016/j.cell.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 17.Brown J.K., Fung C., Tailor C.S. Comprehensive mapping of receptor-functioning domains in feline leukemia virus subgroup C receptor FLVCR1. J. Virol. 2006;80:1742–1751. doi: 10.1128/JVI.80.4.1742-1751.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shalev Z., Duffy S.P., Adema K.W., Prasad R., Hussain N., Willett B.J., Tailor C.S. Identification of a feline leukemia virus variant that can use THTR1, FLVCR1, and FLVCR2 for infection. J. Virol. 2009;83:6706–6716. doi: 10.1128/JVI.02317-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Keel S.B., Doty R.T., Yang Z., Quigley J.G., Chen J., Knoblaugh S., Kingsley P.D., De Domenico I., Vaughn M.B., Kaplan J. A heme export protein is required for red blood cell differentiation and iron homeostasis. Science. 2008;319:825–828. doi: 10.1126/science.1151133. [DOI] [PubMed] [Google Scholar]
- 20.Brasier G., Tikellis C., Xuereb L., Craigie J., Casley D., Kovacs C.S., Fudge N.J., Kalnins R., Cooper M.E., Wookey P.J. Novel hexad repeats conserved in a putative transporter with restricted expression in cell types associated with growth, calcium exchange and homeostasis. Exp. Cell Res. 2004;293:31–42. doi: 10.1016/j.yexcr.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 21.Plate K.H. Mechanisms of angiogenesis in the brain. J. Neuropathol. Exp. Neurol. 1999;58:313–320. doi: 10.1097/00005072-199904000-00001. [DOI] [PubMed] [Google Scholar]
- 22.Visel A., Thaller C., Eichele G. GenePaint.org: an atlas of gene expression patterns in the mouse embryo. Nucleic Acids Res. 2004;32(Database issue):D552–D556. doi: 10.1093/nar/gkh029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lein E.S., Hawrylycz M.J., Ao N., Ayres M., Bensinger A., Bernard A., Boe A.F., Boguski M.S., Brockway K.S., Byrnes E.J. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- 24.Morgan N.V., Brueton L.A., Cox P., Greally M.T., Tolmie J., Pasha S., Aligianis I.A., van Bokhoven H., Marton T., Al-Gazali L. Mutations in the embryonal subunit of the acetylcholine receptor (CHRNG) cause lethal and Escobar variants of multiple pterygium syndrome. Am. J. Hum. Genet. 2006;79:390–395. doi: 10.1086/506256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoffmann K., Muller J.S., Stricker S., Megarbane A., Rajab A., Lindner T.H., Cohen M., Chouery E., Adaimy L., Ghanem I. Escobar syndrome is a prenatal myasthenia caused by disruption of the acetylcholine receptor fetal gamma subunit. Am. J. Hum. Genet. 2006;79:303–312. doi: 10.1086/506257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vogt J., Harrison B.J., Spearman H., Cossins J., Vermeer S., ten Cate L.N., Morgan N.V., Beeson D., Maher E.R. Mutation analysis of CHRNA1, CHRNB1, CHRND, and RAPSN genes in multiple pterygium syndrome/fetal akinesia patients. Am. J. Hum. Genet. 2008;82:222–227. doi: 10.1016/j.ajhg.2007.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Michalk A., Stricker S., Becker J., Rupps R., Pantzar T., Miertus J., Botta G., Naretto V.G., Janetzki C., Yaqoob N. Acetylcholine receptor pathway mutations explain various fetal akinesia deformation sequence disorders. Am. J. Hum. Genet. 2008;82:464–476. doi: 10.1016/j.ajhg.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vogt J., Morgan N.V., Marton T., Maxwell S., Harrison B.J., Beeson D., Maher E.R. Germline mutation in DOK7 associated with fetal akinesia deformation sequence. J. Med. Genet. 2009;46:338–340. doi: 10.1136/jmg.2008.065425. [DOI] [PubMed] [Google Scholar]
- 29.Witters I., Moerman P., Fryns J.P. Fetal akinesia deformation sequence: a study of 30 consecutive in utero diagnoses. Am. J. Med. Genet. 2002;113:23–28. doi: 10.1002/ajmg.10698. [DOI] [PubMed] [Google Scholar]
- 30.Pimanda J.E., Chan W.Y., Wilson N.K., Smith A.M., Kinston S., Knezevic K., Janes M.E., Landry J.R., Kolb-Kokocinski A., Frampton J. Endoglin expression in blood and endothelium is differentially regulated by modular assembly of the Ets/Gata heS200mangioblast code. Blood. 2008;112:4512–4522. doi: 10.1182/blood-2008-05-157560. [DOI] [PMC free article] [PubMed] [Google Scholar]