

Abstract
Glucocorticoids exert multiple anti-inflammatory activities, one of which is the inhibition of transcription dependent on the nuclear factor (NF)-κB. It has been suggested that the effect of dexamethasone (DEX), a glucocorticoid analog, is attributed to an increased production of the inhibitory IκB molecule, which in turn would bind and remove activated, DNA-bound NF-κB complexes in the cell nucleus. Upon investigating DEX-mediated repression of interleukin-6 expression induced by tumor necrosis factor, DEX treatment was found to act directly on NF-κB-dependent transcription, without changing the expression level of IκB. Neither the mRNA of IκB nor the protein was significantly elevated by a combined treatment with tumor necrosis factor and DEX of murine endothelial or fibroblast cells. The DNA-binding activity of induced NF-κB also remained unchanged after stimulation of cells with DEX. Evidence for a direct nuclear mechanism of action was obtained by analysis of cell lines stably expressing a fusion protein between the DNA-binding domain of the yeast Gal4 protein and the transactivating p65 subunit of NF-κB. Expression of a Gal4-dependent luciferase reporter gene activated by this nuclear fusion protein was also strongly repressed after addition of DEX. Because the DNA-binding activity of the Gal4 fusion protein was not affected by DEX, it can be concluded that the reduction of gene activation was caused by interference of the activated glucocorticoid receptor with the transactivation potential of the NF-κB p65 subunit.
Glucocorticoids are potent immunosuppressive agents that are indispensable in development, cell proliferation, and differentiation processes and are also believed to be implicated in the inhibition of lymphocyte migration (1). Their effects are exerted by binding to the intracellular glucocorticoid receptor (GR), which belongs to the family of steroid hormone receptors containing several functional domains. The modular structure consists of a variable N-terminal domain in which a transactivation function is localized, a DNA-binding domain comprising transactivation and dimerization functions, and a C-terminal ligand-binding domain that is also involved in transactivation, and dimerization as well as in nuclear translocation and binding to hsp90 (2). In the resting state, GR is associated with two hsp90 molecules and one p59 immunophilin molecule. After binding of glucocorticoid hormone, this complex dissociates and the hormone-loaded receptor refolds into a homodimer. This new complex translocates to the nucleus and affects gene transcription either positively or negatively (3).
Glucocorticoids can down-regulate the expression of several genes, including cytokines such as interleukin (IL)-6 induced by various inflammatory stimuli such as lipopolysaccharide, IL-1, or tumor necrosis factor (TNF) (4–6). Mutational analysis identified various GR regions responsible for this repression (7–11). Transcriptional repression of some genes, such as the pro-opiomelanocortin (12), osteocalcin (13), prolactin (14), and proliferin (15) genes, has been reported to be mediated by a negative glucocorticoid response element (14, 16). In the case of other genes, such as IL-6 (17), IL-8 (5), glycoprotein α-subunit (18), and collagenase (19), these elements could not be identified in the promoter. Recent studies, however, emphasize the importance of interactions between domains of GR and other transcription factors, such as AP-1, nuclear factor (NF)-κB, CREB, OTF-1, and STAT5, as a possible way to explain the suppression of proinflammatory genes by glucocorticoids (4, 20–24).
The promoter of the IL-6 gene contains a variety of binding sites for known transcription factors (25, 26), which may cooperate or synergize to give a full response of gene expression in different cell types and conditions of induction (27, 28). The main transcription factor for response to inflammatory cytokines is, however, NF-κB (29–31). This factor is a heterodimer that typically consists of a p65 (Rel A) and a p50 subunit. In a latent form, NF-κB is sequestered in the cytoplasm through the ankyrin repeats of inhibitory κB IκB proteins (32, 33). Activation of NF-κB may occur through a variety of extracellular signals, which induce phosphorylation and ubiquitinylation events on IκB-α, resulting in proteolytic degradation (34–36). Subsequently, NF-κB is released from the inhibitor molecule and enters the nucleus, where it activates gene transcription. One such gene is the gene coding for IκB-α itself, which is resynthesized and will resequester the transcriptionally active NF-κB complexes, thus functioning in an autoregulatory fashion (33, 37).
Here we investigated the molecular mechanism of down-regulation of IL-6 synthesis by glucocorticoids in fibroblasts and endothelial cells, which are major producers of IL-6. It appeared that IL-6 repression did not result from IκB-α up-regulation, as previously proposed (38, 39), but is directly caused by down-modulation, through the activated GR, of the transactivation potential of p65 NF-κB.
MATERIALS AND METHODS
Cell Culture.
Murine L929sA fibrosarcoma cells (40) were maintained in DMEM supplemented with 10% newborn calf serum, 100 units/ml penicillin, and 0.1 mg/ml streptomycin. Transgenic endothelial murine heart cells, TC10, which express immortalizing polyomavirus large T-antigen, were generously provided by L. E. Chalifour (Department of Microbiology and Immunology, McGill University, Montreal). They were cultured in DMEM containing 10% fetal calf serum (FCS), 100 units/ml penicillin, and 0.1 mg/ml streptomycin. The cells were irreversibly differentiated to a mature endothelial character by addition of 10−6 M retinoic acid to the culture medium for 3 days (41).
Cytokines and Reagents.
Recombinant murine TNF, produced in our laboratory, had a specific biological activity of 1.3 × 108 units/mg protein and contained <1.8 ng endotoxin/mg protein. The specific activity was determined by a standardized cytotoxic assay on 164 WEHI cl 13 cells compared with an international standard TNF preparation (National Institute for Biological Standards and Control, Potters Bar, U.K.).
Cycloheximide (CHX) and dexamethasone (DEX) were purchased from Sigma. Stock solutions of the reagents were routinely prepared in culture medium or dimethyl sulfoxide (DMSO) (Sigma–Aldrich). Control experiments showed that the final concentration of organic solvent did not interfere with any of the assays.
Luciferase (luc) assay reagent comprised 270 μM CoA (Sigma), 470 μM luciferin (Sigma), and 530 μM ATP (Boehringer Mannheim) in 10 mM tricine, 0.54 mM (MgCO3)4Mg(OH)2, 1.34 mM MgSO4, 0.05 mM EDTA, 16.7 mM DTT (all from Sigma).
Plasmids.
pGal4, pGal4-p65, pGal4-p651–285, pGal4-p65286–551, and pGal4-VP16 were described previously (42, 43). The full IL-6 promoter of p1168hu.IL6P-luc+ corresponds to a 1,168 bp HindIII–XhoI fragment of the human IL-6 promoter (44), whereas p50hu.IL6P-luc only contains the corresponding TATA box region (45). p(Gal)2-50hu.IL6P-luc+ and p(IL6-κB)3-50hu.IL6P-luc+ are synthetic promoter constructs containing two palindromic GAL4 linkers or three NF-κB response elements, respectively, preceding a 50-bp TATA box-containing IL-6 promoter fragment coupled to luc cDNA (figure 8 in ref. 45). pPGKβgeobpA, which contains a β-galactosidase (β-gal)/neomycin fusion gene, was a gift from P. Soriano (Fred Hutchinson Cancer Research Center, Seattle). pBR325 (46) was used as carrier DNA in transfections of eukaryotic cells.
The murine IL-6 cDNA fragment was made by a BamHI and EcoRI double-restriction digestion of pUC8 mIL-6 (47). The IκB-α cDNA fragment was obtained by HindIII digestion of pRC/CMV-MAD-3. The cDNA fragment of the glyceraldehyde-3-phosphate dehydrogenase gene was isolated by digestion of pBluMGAPDHf (obtained from W. Kruijer, Dutch Institute for Developmental Biology, Utrecht, The Netherlands) with EcoRI. All cDNA fragments were used as probes in Northern blot analysis.
Site-Directed Mutagenesis.
Site-directed mutagenesis of the IL-6 promoter was carried out following the gapped heteroduplex method (48), by using a transformer site-directed mutagenesis kit (CLONTECH). The mutator oligonucleotide 5′-CTCCAACAAAGATTCTAGAAATGTGG-3′, which contains a specific BglII restriction site (altered nucleotides are underlined), was used to construct p1168hu.IL6P-luc+ NFκBmut. Mutant clones were screened for the presence of a newly created BglII restriction site and confirmed by sequence analysis. This mutation was already described to abolish NF-κB binding with the purified recombinant protein (27) and was also tested for the biological effect on gene induction in corresponding, stably transfected clones (unpublished results).
Biological IL-6 Assay.
Secreted IL-6 was quantitated according to its growth-stimulatory effect on 7TD1 cells. Cell proliferation was assessed by colorimetric determination of hexosaminidase levels (49, 50).
Electrophoretic Mobility-Shift Assay (EMSA).
L929sA or TC10 cells were seeded in medium-sized Petri dishes at 106 cells per dish at day −1. After appropriate induction, the cells were washed with PBS, scraped off, and transferred to precooled Eppendorf tubes. EMSA were performed essentially as described (51). DNA-binding activity was tested with an oligonucleotide containing the NF-κB or Gal4-binding site. Equal amounts of protein were incubated with 2 μg of poly(dI-dC) (Pharmacia), 20 μg BSA, 0.1% (vol/vol) Nonidet P-40, 2 mM Hepes/KOH (pH 7.9), 2% (vol/vol) glycerol, 10 mM KCl, and 0.05 mM EDTA in combination with 4 μl of 5× binding buffer [20% (wt/vol) Ficoll 400/100 mM Hepes/KOH, pH 7.9/1 mM DTT/300 mM KCl] in a total volume of 20 μl. After 10-min incubation, 10,000 cpm of [32P]-labeled oligonucleotide was added and incubated on ice for 10 min. The free and protein-bound oligonucleotides were separated by electrophoresis on a native 4% polyacrylamide gel. Gel and running buffers contained 25 mM Tris, 25 mM boric acid, and 0.5 mM EDTA. After electrophoresis, the gel was dried and exposed to a PhosphorImager screen (Molecular Dynamics).
Labeling of the oligonucleotides was performed with [γ-32P]-ATP by using T4 polynucleotide kinase or with [α-32P]-dCTP by using Klenow enzyme (Boehringer Mannheim). The NF-κB oligonucleotide comprises the sequences 5′-AGCTATGTGGGATTTTCCCATGAGC-3′ (underlined: single κB motif derived from the IL-6 promoter) and 3′-TACACCCTAAAAGGGTACTCGTCGA-5′. The GAL4 oligonucleotide consists of the sequences 5′-GGCGGGTCGGAGTACTGTCCTCCGACTGC-3′ (underlined: single Gal4-binding site) and 3′-GCCCAGCCTCATGACAGGAGGCTGACGGAC-5′.
Western Blot Analysis.
Cells were pretreated with 10−6 M DEX, followed by incubation with 2,000 units/ml TNF. Cells were harvested and lysed in TOTEX buffer [20 mM Hepes/KOH, pH 7.9/0.35 M NaCl/20% (vol/vol) glycerol/1% (vol/vol) Nonidet P-40/1 mM MgCl2/0.5 mM EDTA/0.1 mM EGTA/2 mM Pefabloc/5 mM DTT]. After centrifugation and addition of Laemmli buffer, the samples were loaded onto a reducing SDS/12% polyacrylamide gel and subjected to electrophoresis. Equal amounts of protein, determined according to Bradford (52), were transferred to a polyvinylidene difluoride membrane (Bio-Rad). The membrane was washed twice in TBS/0.1% Tween, followed by incubation for at least 1 hr in the same compound supplemented with 5% nonfat, dry milk powder. The membrane was incubated in a small volume of TBS/0.1% Tween containing a 1:500 dilution of anti-IκB-α antibody (Santa Cruz Biotechnology). After 4-hr incubation at room temperature, the membrane was washed 8 times in TBS/0.1% Tween and incubated for 1 hr in TBS/0.1% Tween, containing a 1:3,000 dilution of secondary anti-rabbit antibody coupled to horseradish peroxidase (Amersham). After extensive washing, the bound antibodies were detected by using enhanced chemiluminescence (Amersham).
Northern Blot Analysis.
After appropriate inductions, RNA isolations were obtained by using TRIzol (Life Technologies, Paisley, U.K.). Briefly, 5 × 106 TC10 cells were grown to subconfluency in 143-cm2 Petri dishes and cells were induced with different combinations of DEX (10−6 M), TNF (2,000 units/ml), and/or CHX (25 ng/ml). DEX and CHX were administered to the cells 1 hr before TNF induction, which lasted another 6 hr. Total RNA was isolated with TRIzol; denaturation was achieved with a combination of DMSO and deionized glyoxal (Sigma-Aldrich). A total of 20 μg RNA was separated on a 1% agarose gel in 20 mM phosphate buffer (pH 7). RNA was transferred to Hybond-N+ membranes (Amersham) by standard capillary blotting and cross-linked by UV irradiation. Hybridization was done by sequentially probing with murine IL-6, human IκB-α, and murine glyceraldehyde-3-phosphate dehydrogenase cDNA fragments. The membrane was stripped (0.5% SDS for 5 min at 100°C) before each new hybridization step. All fragments were labeled with [α-32P]-dCTP by using a Random Primed labeling kit (Boehringer Mannheim).
Transfections.
TC10 endothelial cells were seeded at 105 cells per 24-well on day −1. The cells were transiently transfected with lipofectamine (Life Technologies), the total amount being 600 ng of DNA per well. Lipofectamine was dissolved at a concentration of 12% in DMEM without serum. The amount of reporter plasmid p(Gal)2-50hu.IL6P-luc+ was kept constant at 275 ng. pPGKβgeobpA allows to correct for transfection efficiencies and was also used at 275 ng. Fifty nanograms of pGal4, or 10, 25, or 50 ng of pGal4-p65 was used in various transfection assays. The total amount of DNA was adjusted to 600 ng with pBR325. One day later, the medium was replaced with fresh medium with or without DEX (10−6 M) for 24 hr. The cells were then washed with PBS and lysed with 120 μl of lysis buffer (Tropix, Bedford, MA). luc assays were performed according to the manufacturer’s instructions (Promega). The light emission was quantified in a TiterTek Luminoscan UV 91–6 (Labsystems, Helsinki, Finland). Detection of β-gal was achieved with a Chemiluminescent Reporter Assay Galacto-Light kit (Tropix).
L929sA cells were transiently transfected with DEAE-dextran (Pharmacia) essentially as described (53). For stable transfection by calcium phosphate coprecipitation (54), 2 × 106 actively growing L929sA cells were seeded per 75-cm2 flask 24 hr before transfection. pGal4, pGal4-p65, or pGal4-VP16 were transfected together with pPGKβgeobpA. Cells were split on the first day and selection started with 500 μg/ml G418 (Life Technologies) for at least 2 weeks.
RESULTS
Induction of IL-6 Transcription by TNF Is Repressed by DEX.
The inhibition by DEX of TNF-induced IL-6 gene expression in TC10 cells is shown in Fig. 1A. To ascertain whether this down-modulatory effect of DEX was also evident at the IL-6 mRNA level and whether it was still present in the absence of novel protein synthesis, Northern blot analysis was performed. In the endothelial cell system, the up-regulation of IL-6 mRNA levels was indeed suppressed by DEX (Fig. 1B). The enhanced levels of IL-6 mRNA by treatment with CHX, an inhibitor of protein synthesis, were further up-regulated by TNF; the high induction level, obtained by a combination of CHX + TNF, could still be suppressed by DEX (Fig. 1B). This indicates that the repressive activity of DEX does not require new protein synthesis.
Figure 1.
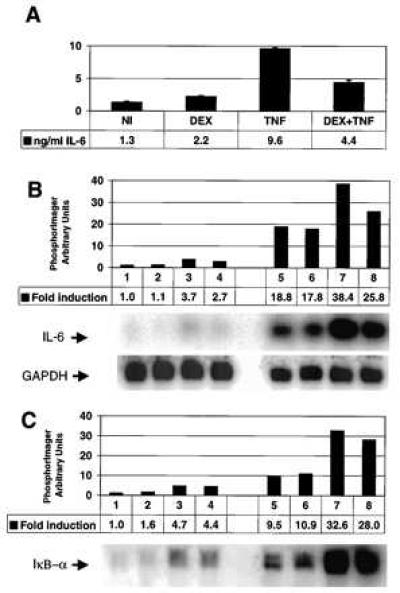
(A) Regulation of IL-6 expression in TC10 cells. After induction for 6 hr with 2,000 units/ml TNF and/or 10−6 M DEX (added at −1 hr), the medium was assayed for IL-6 biological activity. NI, noninduced cell culture. (B) Repression by DEX is independent of new protein synthesis. TC10 cells were noninduced (lane 1), treated with 10−6 M DEX (lanes 2 and 6), or induced for 6 hr with 2,000 units/ml TNF in the absence (lanes 3 and 7) or presence (lanes 4 and 8) of 10−6 M DEX. The same series of treatments was performed in the presence of 25 ng/ml CHX (lanes 5–8). DEX and/or CHX were added, where appropriate, 1 hr before TNF. The respective induction levels were quantified and compared with the mRNA level in the noninduced state, which was arbitrarily set to 1. Rehybridization with a glyceraldehyde-3-phosphate dehydrogenase probe served as a control for equal loading. (C) DEX treatment does not influence the amount of TNF-activated IκB-α mRNA. The filter shown in B was stripped and reprobed with human IκB-α.
DEX Does Not Affect TNF-Activated IκB-α mRNA.
The Northern blots used for testing the effect of DEX on TNF-induced IL-6 mRNA synthesis were reprobed with IκB-α cDNA (Fig. 1C). The level of IκB-α mRNA was increased by TNF or CHX. However, the level of induced IκB-α mRNA was not enhanced further by the use of glucocorticoids.
DEX Treatment Does Not Interfere with the Formation of Active NF-κB and Has No Effect on TNF-Activated IκB-α Protein Levels.
The potential influence of DEX on TNF-activated NF-κB in TC10 cells was tested by band-shift analysis. As evident from Fig. 2A, DEX had no effect on the DNA-binding activity of NF-κB at any time point. Also, the levels of recombination binding protein-Jκ (45) were not altered. Similar results were obtained in HeLa cells (data not shown) and L929sA cells (55). IκB-α protein levels tested at the same time points revealed that a combined treatment of TNF with DEX gave the same results as TNF alone (Fig. 2B). Twenty minutes after TNF administration, IκB-α was almost completely degraded, whereas it fully reappeared after 2 hr. At later time points, when repression was clearly visible at the level of IL-6 mRNA expression, the amount of IκB-α protein was certainly not up-regulated by DEX treatment compared with induction with TNF alone.
Figure 2.
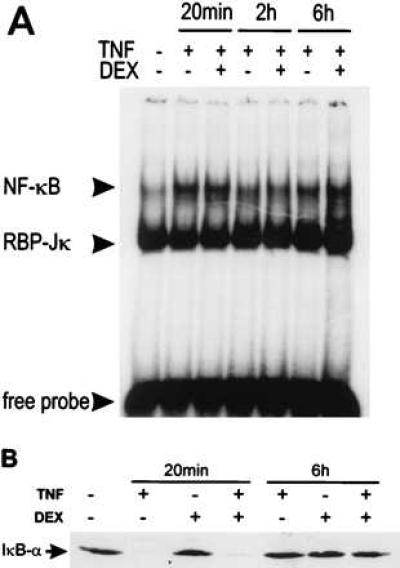
(A) The NF-κB/DNA complex is still formed in the presence of DEX. TC10 cells were left untreated, or were treated with TNF alone (2,000 units/ml) or with TNF + DEX (10−6 M) for various periods. The total protein extract was incubated with a 32P-labeled IL-6-κB response element; protein/DNA complexes were analyzed in an EMSA. Arrowheads indicate the activated κB complex, the constitutively expressed recombination binding protein (RBP)-Jκ and the free probe. (B) DEX treatment does not up-regulate the amount of IκB-α protein. TC10 cells were left untreated or were treated with DEX (10−6 M) alone, with 2,000 units/ml TNF, or with TNF + 10−6 M DEX, added at −1 hr. After cell lysis, equal amounts of protein were loaded onto an SDS/polyacrylamide gel for Western blot analysis.
DEX Repression Acts on κB-Driven Promoters.
To study further the specificity of DEX repression, p1168hu.IL6P-luc+ was stably transfected in L929sA cells. In the clones selected, luc gene expression by TNF treatment could reproducibly be induced and again repressed by treatment with DEX (Fig. 3). For p(IL6-κB)3-50hu.IL6P-luc+, TNF inducibility and repression by DEX showed a pattern similar to that obtained with p1168hu.IL6P-luc+. However, for the promoter constructs lacking a functional κB site (i.e., p1168hu.IL6P-luc+ NFκBmut and p50hu.IL6P-luc+), no induction by TNF or repression by DEX could be observed. Although the levels of activated NF-κB were not changed after addition of DEX (Fig. 2A), these experiments indicate that the repressive mechanism exerted by DEX was clearly directed against κB-driven promoter constructs.
Figure 3.
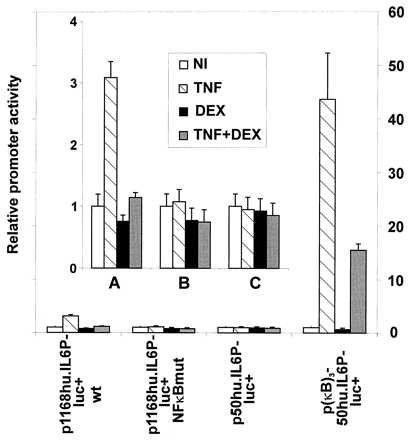
Repression by DEX acts on κB-driven promoter constructs. L929sA cells with stably integrated p1168hu.IL6P-luc+ (A), p1168hu.IL6P-luc+ NFκBmut (B), p50hu.IL6P-luc+ (C), or p(IL6-κB)3-50hu.IL6P-luc+ were left untreated (NI), or were treated with DEX alone (10−6 M) or with TNF (2,000 units/ml) for 6 hr, either alone or with DEX (10−6 M), added at −1 hr. The relative promoter activity indicates the ratio between the expression levels of the treated and the untreated state, the latter being arbitrarily set to 1. (Inset) Enlargement of the respective induction profiles.
pGal4-p65-Mediated Expression of a Reporter Gene Can Be Repressed by DEX.
A Gal4-based system was used to test whether the activated GR interfered with the transactivation potential of the p65 κB subunit in the nucleus. L929sA cells were stably transfected with pGal4, pGal4-p65, pGal4-p651–285, pGal4-p65286–551, or pGal4-VP16. These proteins are exclusively localized in the cell nucleus, as revealed by immunofluorescence studies (data not shown), and allow the monitoring of regulatory events, independently of IκB effects. The corresponding transactivation capacity of the pGal4 plasmids and the down-modulation by DEX were measured after transient transfection of the respective cell lines with p(Gal)2-50hu.IL6P-luc+. As shown in Fig. 4A, the basal activation level mediated by pGal4-p65 was strongly suppressed by the addition of DEX, whereas no effect of DEX treatment was observed by using the control plasmid expressing Gal4 only. Moreover, activation by the strong, acidic VP16 in the cell line expressing the pGal4-VP16 fusion was also not repressed by DEX, showing that the repressive effect of glucocorticoids on p65-mediated transactivation is very specific. Furthermore, it is also clear from the experiments with the Gal4 deletion variants that repression by DEX acts as effectively with the C-terminal half only of the p65 molecule, i.e., where the domains for transcriptional activation are localized.
Figure 4.
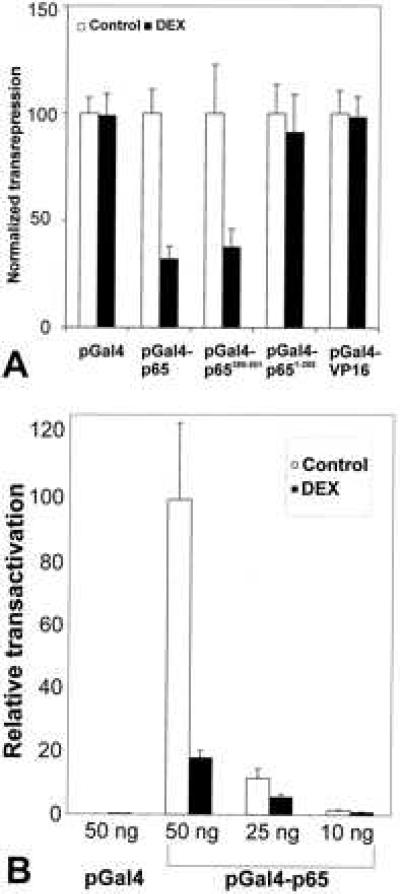
(A) DEX specifically represses p65-mediated transactivation in L929sA cells. Cells stably transfected with pGal4, pGal4-p65, pGal4-p651–285, pGal4-p65286–551, or pGal4-VP16 were further transiently transfected with 700 ng p(Gal)2-50hu.IL6P-luc+, 700 ng of a plasmid containing β-gal, and carrier DNA, the total being 1.6 μg DNA. After 72 hr, lysates were made and the concentration of luc was determined by using β-gal values as a basis for normalization. In appropriate assays, 10−6 M DEX was added 24 hr before analysis. The normalized luc activity of the untreated samples was arbitrarily set to 100. Error bars show standard deviation of four independent experiments. (B) DEX represses p65 transactivation in TC10 cells. Cells were transiently transfected with pGal4 or pGal4-p65, together with a constant amount of p(Gal)2-50hu.IL6P-luc+, and treated with 10−6 M DEX or untreated. After 24 hr, luc activity was determined by using β-gal values as a basis for normalization. Error bars show standard deviation of four independent transfections.
A similar pattern of gene repression by DEX in the Gal4 system was observed in TC10 cells transiently transfected with pGal4-p65 in combination with p(Gal)2-50hu.IL6P-luc+ (Fig. 4B). Also in this case, expression of the DNA-binding domain of Gal4 alone transactivated only marginally the Gal4-driven luc reporter gene.
The potential influence of DEX on the DNA-binding activity of the Gal4 fusion proteins was tested in a control experiment. After treatment with DEX for 12 or 24 hr, lysates were prepared from L929sA cells that stably expressed Gal4-p65. The DNA-binding activities of the fusion protein were tested in an EMSA assay by using a 32P-labeled oligonucleotide containing a Gal4-binding site (Fig. 5). The DNA-binding activity of the Gal4 fusion protein was unchanged after treatment with DEX for up to 24 hr, indicating that the repressive effect of DEX was because of down-modulation of p65 transactivation, and not to a decrease in the expression levels of Gal4-p65 protein or in DNA-binding capacity.
Figure 5.
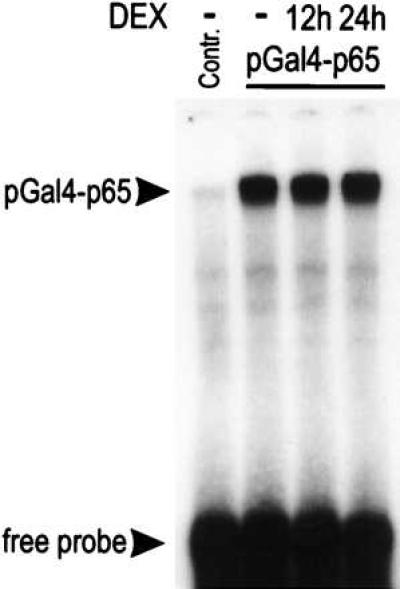
DEX does not interfere with Gal4 binding to its DNA motif. L929sA cells containing a stably integrated Gal4-p65 cDNA were left untreated (solid line) or were treated with DEX (10−6 M) for 12 or 24 hr. Total protein extracts were incubated with 32P-labeled GAL4 response element for analysis in an EMSA. No complex was observed when a protein extract of parental L929sA cells was incubated with the probe (contr.).
DISCUSSION
Glucocorticoids are widely used in medicine for their anti-inflammatory and immunosuppressive activities. It is generally accepted that their mechanism of action is based on repression of particular inflammatory (mostly cytokine) genes by the activated complex formed by the binding of the ligands to their corresponding intracellular receptors. However, the molecular mechanism of this gene repression is not yet fully understood. Recently, it has been reported that glucocorticoids enhance the expression of IκB-α and a model has been proposed in which activation of NF-κB, driving the inflammatory genes, could be counteracted by the reappearance of the inhibitory partner, thus slowing down and finally terminating the ongoing gene expression (38, 39).
This study shows that in endothelial cells the amount of TNF-activated NF-κB was not affected by the addition of the synthetic glucocorticoid DEX, although there was clearly repression of IL-6 gene expression under these conditions. Also, in a previous study with murine fibroblast cells, it was shown that the levels of IL-6 mRNA, following induction with TNF, could be increased or decreased, irrespective of the amount of activated NF-κB (55). Furthermore, we found that by treatment with DEX, the level of IκB-α was unchanged at the mRNA and protein levels as compared with untreated or TNF-induced cells. This again demonstrates that removal of activated NF-κB by an enhanced expression of IκB-α cannot be the underlying mechanism for gene repression, at least not in endothelial cells. Such a mechanism is also not compatible with the finding that gene repression still occurred in the absence of protein synthesis.
Several reports suggest that down-modulation of κB-driven genes results from a physical association between activated GR and the p65 κB subunit, following overexpression of one or both of these proteins in transient transfection assays (4, 22, 56). Our results indicate that gene activation by NF-κB is repressed by glucocorticoids at the “transcriptional” level, both in murine fibroblasts and endothelial cells, and that this repression mechanism constitutes an exclusively nuclear mechanism. The activated, endogenous GR was able to specifically repress p65 κB-dependent transactivation in a completely artificial Gal4 system, independently of the level of NF-κB. Moreover, this effect was obtained by a normal physiological activation of endogenous GR. It may therefore well be that direct physical interaction between GR and p65 accounts for the observed down-modulation of gene expression. In this scenario, direct interaction between p65 and activated GR would mask the involved activation domain(s) of p65. Alternatively, direct interaction may also lead to modifications or conformational changes of p65, which would result in down-modulation of transcription. Furthermore, there is growing evidence that p65 exerts its transactivating function by interacting with components of the transcriptional machinery (57), as well as with a number of coactivating factors (43, 58–60). Thus, the model of induced gene activation has become more complex, and multiple protein/protein interactions might contribute to the repressive effects of activated GR on NF-κB. Further studies will help to clarify whether these proteins are also involved in the repressive action of glucocorticoids on gene transcription.
Acknowledgments
We thank W. Burm and I. Van Rompaey for technical assistance. K.D.B. holds a fellowship from the Vlaams Instituut voor de Bevordering van het Wetenschappelijk-Technologisch Onderzoek in de Industrie. M.L.S. was supported by a European Union Biomed-2 grant. G.H. is a Research Director with the Fonds voor Wetenschappelijk Onderzoek–Vlaanderen. Research was supported by the Vlaams Actiecomité voor Biotechnologie, the Interuniversitaire Attractiepolen, and the Vlaams Interuniversitair Instituut voor Biotechnologie.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: β-gal, β-galactosidase; CHX, cycloheximide; DEX, dexamethasone; GR, glucocorticoid receptor; IκB, inhibitory κB; IL, interleukin; luc, luciferase; NF-κB, nuclear factor κB; TNF, tumor necrosis factor; EMSA, electrophoretic mobility-shift assay.
References
- 1.Cronstein B N, Kimmel S C, Levin R I, Martiniuk F, Weissmann G. Trans Assoc Am Physicians. 1992;105:25–35. [PubMed] [Google Scholar]
- 2.Beato M. Cell. 1989;56:335–344. doi: 10.1016/0092-8674(89)90237-7. [DOI] [PubMed] [Google Scholar]
- 3.Muller M, Renkawitz R. Biochim Biophys Acta. 1991;1088:171–182. doi: 10.1016/0167-4781(91)90052-n. [DOI] [PubMed] [Google Scholar]
- 4.Caldenhoven E, Liden J, Wissink S, van de Stolpe A, Raaijmakers J, Koenderman L, Okret S, Gustafsson J A, van der Saag P T. Mol Endocrinol. 1995;9:401–412. doi: 10.1210/mend.9.4.7659084. [DOI] [PubMed] [Google Scholar]
- 5.Mukaida N, Morita M, Ishikawa Y, Rice N, Okamoto S, Kasahara T, Matsushima K. J Biol Chem. 1994;269:13289–13295. [PubMed] [Google Scholar]
- 6.van de Stolpe A, Caldenhoven E, Raaijmakers J A, van der Saag P T, Koenderman L. Am J Respir Cell Mol Biol. 1993;8:340–347. doi: 10.1165/ajrcmb/8.3.340. [DOI] [PubMed] [Google Scholar]
- 7.Beato M, Herrlich P, Schütz G. Cell. 1995;83:851–857. doi: 10.1016/0092-8674(95)90201-5. [DOI] [PubMed] [Google Scholar]
- 8.Helmberg A, Auphan N, Caelles C, Karin M. EMBO J. 1995;14:452–460. doi: 10.1002/j.1460-2075.1995.tb07021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ray A, LaForge K S, Sehgal P B. Proc Natl Acad Sci USA. 1991;88:7086–7090. doi: 10.1073/pnas.88.16.7086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oro A E, Hollenberg S M, Evans R M. Cell. 1988;55:1109–1114. doi: 10.1016/0092-8674(88)90255-3. [DOI] [PubMed] [Google Scholar]
- 11.Liden J, Delaunay F, Rafter I, Gustafsson J-Å, Okret S. J Biol Chem. 1997;272:21467–21472. doi: 10.1074/jbc.272.34.21467. [DOI] [PubMed] [Google Scholar]
- 12.Drouin J, Sun Y L, Chamberland M, Gauthier Y, De Lean A, Nemer M, Schmidt T J. EMBO J. 1993;12:145–156. doi: 10.1002/j.1460-2075.1993.tb05640.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stromstedt P E, Poellinger L, Gustafsson J A, Carlstedt- Duke J. Mol Cell Biol. 1991;11:3379–3383. doi: 10.1128/mcb.11.6.3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sakai D D, Helms S, Carlstedt-Duke J, Gustafsson J A, Rottman F M, Yamamoto K R. Genes Dev. 1988;2:1144–1154. doi: 10.1101/gad.2.9.1144. [DOI] [PubMed] [Google Scholar]
- 15.Mordacq J C, Linzer D I. Genes Dev. 1989;3:760–769. doi: 10.1101/gad.3.6.760. [DOI] [PubMed] [Google Scholar]
- 16.Akerblom I E, Slater E P, Beato M, Baxter J D, Mellon P L. Science. 1988;241:350–353. doi: 10.1126/science.2838908. [DOI] [PubMed] [Google Scholar]
- 17.Waage A, Slupphaug G, Shalaby R. Eur J Immunol. 1990;20:2439–2443. doi: 10.1002/eji.1830201112. [DOI] [PubMed] [Google Scholar]
- 18.Chatterjee V K, Madison L D, Mayo S, Jameson J L. Mol Endocrinol. 1991;5:100–110. doi: 10.1210/mend-5-1-100. [DOI] [PubMed] [Google Scholar]
- 19.Yang Yen H F, Chambard J C, Sun Y L, Smeal T, Schmidt T J, Drouin J, Karin M. Cell. 1990;62:1205–1215. doi: 10.1016/0092-8674(90)90396-v. [DOI] [PubMed] [Google Scholar]
- 20.Stöcklin E, Wissler M, Gouilleux F, Groner B. Nature (London) 1996;383:726–728. doi: 10.1038/383726a0. [DOI] [PubMed] [Google Scholar]
- 21.Liu W, Hillmann A G, Harmon J M. Mol Cell Biol. 1995;15:1005–1013. doi: 10.1128/mcb.15.2.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ray A, Prefontaine K E. Proc Natl Acad Sci USA. 1994;91:752–756. doi: 10.1073/pnas.91.2.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kutoh E, Stromstedt P E, Poellinger L. Mol Cell Biol. 1992;12:4960–4969. doi: 10.1128/mcb.12.11.4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stauber C, Altschmied J, Akerblom I E, Marron J L, Mellon P L. New Biol. 1992;4:527–540. [PubMed] [Google Scholar]
- 25.Ray A, Tatter S B, May L T, Sehgal P B. Proc Natl Acad Sci USA. 1988;85:6701–6705. doi: 10.1073/pnas.85.18.6701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tanabe O, Akira S, Kamiya T, Wong G G, Hirano T, Kishimoto T. J Immunol. 1988;141:3875–3881. [PubMed] [Google Scholar]
- 27.Dendorfer U, Oettgen P, Libermann T A. Mol Cell Biol. 1994;14:4443–4454. doi: 10.1128/mcb.14.7.4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haegeman G, Fiers W. In: Signaling Mechanisms: From Transcription Factors to Oxidative Stress. Packer L, Wirtz K, editors. Berlin: Springer; 1995. pp. 375–382. [Google Scholar]
- 29.Libermann T A, Baltimore D. Mol Cell Biol. 1990;10:2327–2334. doi: 10.1128/mcb.10.5.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shimizu H, Mitomo K, Watanabe T, Okamoto S, Yamamoto K. Mol Cell Biol. 1990;10:561–568. doi: 10.1128/mcb.10.2.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Y H, Lin J X, Vilček J. Mol Cell Biol. 1990;10:3818–3823. doi: 10.1128/mcb.10.7.3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Israel A. Trends Genet. 1995;11:203–205. doi: 10.1016/s0168-9525(00)89045-9. [DOI] [PubMed] [Google Scholar]
- 33.Baldwin A S. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 34.Verma I M, Stevenson J K, Schwarz E M, Van Antwerp D, Miyamoto S. Genes Dev. 1995;9:2723–2735. doi: 10.1101/gad.9.22.2723. [DOI] [PubMed] [Google Scholar]
- 35.Baeuerle P A, Baltimore D. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 36.Finco T S, Baldwin A S. Immunity. 1995;3:263–272. doi: 10.1016/1074-7613(95)90112-4. [DOI] [PubMed] [Google Scholar]
- 37.Chiao P J, Miyamoto S, Verma I M. Proc Natl Acad Sci USA. 1994;91:28–32. doi: 10.1073/pnas.91.1.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scheinman R I, Cogswell P C, Lofquist A K, Baldwin A S., Jr Science. 1995;270:283–286. doi: 10.1126/science.270.5234.283. [DOI] [PubMed] [Google Scholar]
- 39.Auphan N, DiDonato J A, Rosette C, Helmberg A, Karin M. Science. 1995;270:286–290. doi: 10.1126/science.270.5234.286. [DOI] [PubMed] [Google Scholar]
- 40.Vanhaesebroeck B, Decoster E, Van Ostade X, Van Bladel S, Lenaerts A, Van Roy F, Fiers W. J Immunol. 1992;148:2785–2794. [PubMed] [Google Scholar]
- 41.al Moustafa A E, Chalifour L E. Cell Growth Differ. 1993;4:841–847. [PubMed] [Google Scholar]
- 42.Schmitz M L, Baeuerle P A. EMBO J. 1991;10:3805–3817. doi: 10.1002/j.1460-2075.1991.tb04950.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schmitz M L, Stelzer G, Altmann H, Meisterernst M, Baeuerle P A. J Biol Chem. 1995;270:7219–7226. doi: 10.1074/jbc.270.13.7219. [DOI] [PubMed] [Google Scholar]
- 44.Haegeman G, Content J, Volckaert G, Derynck R, Tavernier J, Fiers W. Eur J Biochem. 1986;159:625–632. doi: 10.1111/j.1432-1033.1986.tb09931.x. [DOI] [PubMed] [Google Scholar]
- 45.Plaisance S, Vanden Berghe W, Boone E, Fiers W, Haegeman G. Mol Cell Biol. 1997;17:3733–3743. doi: 10.1128/mcb.17.7.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bolivar F. Gene. 1978;4:121–136. doi: 10.1016/0378-1119(78)90025-2. [DOI] [PubMed] [Google Scholar]
- 47.Van Snick J, Vink A, Uyttenhove C, Houssiau F, Coulie P. Curr Top Microbiol Immunol. 1988;141:181–184. doi: 10.1007/978-3-642-74006-0_24. [DOI] [PubMed] [Google Scholar]
- 48.Stanssens P, Opsomer C, McKeown Y M, Kramer W, Zabeau M, Fritz H-J. Nucleic Acids Res. 1989;17:4441–4453. doi: 10.1093/nar/17.12.4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Landegren U. J Immunol Methods. 1984;67:379–388. doi: 10.1016/0022-1759(84)90477-0. [DOI] [PubMed] [Google Scholar]
- 50.Van Snick J, Cayphas S, Vink A, Uyttenhove C, Coulie P G, Rubira M R, Simpson R J. Proc Natl Acad Sci USA. 1986;83:9679–9683. doi: 10.1073/pnas.83.24.9679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bork P, Schmitz M L, Weimann C, Kist M, Heinrich M. Phytomedicine. 1996;3:263–269. doi: 10.1016/S0944-7113(96)80064-X. [DOI] [PubMed] [Google Scholar]
- 52.Bradford M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 53.Lopata M A, Cleveland D W, Sollner-Webb B. Nucleic Acids Res. 1984;12:5707–5717. doi: 10.1093/nar/12.14.5707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Graham F L, van der Eb A J. Virology. 1973;52:456–467. doi: 10.1016/0042-6822(73)90341-3. [DOI] [PubMed] [Google Scholar]
- 55.Patestos N P, Haegeman G, Vandevoorde V, Fiers W. Biochimie. 1993;75:1007–1018. doi: 10.1016/0300-9084(93)90153-j. [DOI] [PubMed] [Google Scholar]
- 56.Scheinman R I, Gualberto A, Jewell C M, Cidlowski J A, Baldwin A S., Jr Mol Cell Biol. 1995;15:943–953. doi: 10.1128/mcb.15.2.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Paal K, Baeuerle P A, Schmitz M L. Nucleic Acids Res. 1997;25:1050–1055. doi: 10.1093/nar/25.5.1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gerritsen M E, Williams A E, Neish A S, Moore S, Shi Y. Proc Natl Acad Sci USA. 1997;94:2927–2932. doi: 10.1073/pnas.94.7.2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Perkins N D, Felzien L K, Betts J C, Leung K, Beach D H, Nabel G J. Science. 1997;275:523–526. doi: 10.1126/science.275.5299.523. [DOI] [PubMed] [Google Scholar]
- 60.Schmitz M L, Indorf A, Limbourg F P, Stadtler H, Traenckner E B B, Baeuerle P A. Mol Cell Biol. 1996;16:4052–4063. doi: 10.1128/mcb.16.8.4052. [DOI] [PMC free article] [PubMed] [Google Scholar]