

Abstract
Herpesviruses have been previously correlated to vascular disease and shown to cause thrombogenic and atherogenic changes to host cells. Herein we show that even in the absence of cells, purified cytomegalovirus (CMV) and herpes simplex virus type 1 (HSV-1) and type 2 (HSV-2) can initiate thrombin production. Functional assays demonstrated that purified HSV-1 and HSV-2 provide the necessary phospholipid (proPL) for assembling the coagulation factors Xa and Va into prothrombinase, which is responsible for generating thrombin. These observations are consistent with our earlier studies involving CMV. The presence of proPL on all three herpesviruses was confirmed directly by flow cytometry and electron microscopy by using annexin V and factor Va, respectively, as proPL-specific probes. Of equal importance, we found that CMV, HSV-1, and HSV-2 were also able to facilitate factor Xa generation from the inactive precursor factor X, but only when factor VII/VIIa and Ca2+ were present. Monoclonal antibodies specific for tissue factor (TF), the coagulation initiator, inhibited this factor X activation and, furthermore, enabled identification of TF antigen on each virus type by flow cytometry and electron microscopy. Collectively, these data show that CMV, HSV-1, and HSV-2 can initiate the generation of thrombin by having essential proPL and TF activities on their surface. Unlike the normal cellular source, the viral activity is constitutive and, therefore, not restricted to sites of vascular injury. Thus cell-independent thrombin production may be the earliest event in vascular pathology mediated by herpesviruses.
Keywords: factor X, tissue factor, cytomegalovirus, herpes simplex virus
Thrombin is the final enzyme produced in the blood coagulation pathway. It functions directly to generate fibrin clot and is also a potent cell agonist that can induce important intracellular and extracellular regulatory processes (for review, see ref. 1). The normal initiation of thrombin production is triggered by vascular damage and is strictly limited to neighboring cells where the transmembrane protein, tissue factor (TF; for reviews, see refs. 2 and 3), and procoagulant phospholipid [proPL; e.g., phosphatidylserine (PS)] (4, 5) become accessible to plasma clotting factors. The role of TF is to bind and accelerate the protease factor VIIa (FVIIa). This “tenase” complex starts the pathways involved in thrombin generation by activating the zymogen factor X (FX) to factor Xa (FXa). FXa may then combine with its cofactor Va (FVa) in the presence of Ca2+ to form “prothrombinase” (for reviews, see refs. 6 and 7). The significance of prothrombinase is that it is the only physiological enzyme that can convert prothrombin to thrombin. ProPL is an essential component of both tenase and prothrombinase and is needed to orient and colocalize the respective enzymes, cofactors, and substrates.
Cytomegalovirus (CMV) and herpes simplex virus (HSV), members of the herpesvirus family, can evade the usual cellular control programs by stimulating the expression of thrombogenic activity on cells independent of vascular damage (8–13). Therefore, a herpesvirus contribution to vascular disease has been suggested. A number of biochemical changes that render the host cell procoagulant in response to virus are known and include the expression of TF (12, 14) and a functional analogue encoded by HSV type 1 (HSV-1), termed glycoprotein C (10, 15). Recently, smooth muscle cell proliferation (16) and accumulation of oxidized low density lipoprotein (17), the morphological hallmarks of atherosclerotic plaques, were linked to the expression of CMV gene products. This biochemical evidence provides an explanation for clinical studies that correlate CMV to vascular thickening (18) and restenosis after coronary atherectomy (19) and HSV to fibrin deposition in microvasculature (20–22). CMV and HSV genetic materials have also been identified in atherosclerotic plaque (23–25). Although the clinical data are arguably circumstantial, a distinct cause-and-effect relationship was established in avian and rodent models, where viral infection induced atherosclerosis and thrombosis (26–28).
In a previous study from our laboratory, accessible proPL was identified on CMV (29). This suggests that herpesviruses could contribute to vascular disease through the production of thrombin, even before affecting the host cell. We now report that HSV-1 and HSV type 2 (HSV-2) also possess proPL and that all three herpesviruses have endogenous mechanisms to activate FX. Thus, CMV, HSV-1, and HSV-2 can generate thrombin independent of cells, which may represent the earliest influence these viruses have on host vasculature.
MATERIALS AND METHODS
Reagents.
H-d-Phe-Pip-Arg-p-nitroaniline dichloride (S-2238, Chromogenix, Molndal, Sweden; where Pip is piperidinyl), EDTA, and Hepes were from Sigma. All experiments were conducted in Hepes-buffered saline (HBS; 20 mM Hepes/150 mM NaCl, pH 7.2).
Virus Preparation.
CMV (strain AD169) was propagated in human foreskin fibroblasts (HFF) and purified as described (29, 30). Commercial sources of purified HSV-1 (MacIntyre strain) and HSV-2 (strain G) propagated in Vero cells were used (Advanced Biotechnologies, Columbia, MD). Virions were quantified and evaluated for purity by electron microscopy (29, 30). Less than 10% of particles in the virus preparations were attributed to cellular debris.
Protein Preparation.
Human coagulation proteins FVIIa, factor VII (FVII), and FVa were from Haematologic Technologies (Burlington, VT) and prothrombin was from Enzyme Research Laboratories (South Bend, IN). Human FX and FXa were prepared as described (31). FX was radioiodinated on ice by using Iodogen (Pierce) according to manufacturers instructions. The specific activity was typically 1.2 × 105 cpm/μg of FX. Annexin V (AnV; supplied by T. Yokoyama, Kowa, Tokyo) was labeled with fluorescein isothiocyanate (FITC), as described (32). By using the extinction coefficients for AnV and FITC (33), the conjugate (AnV-F) was purified to a 1:2 molar ratio of AnV to FITC by FPLC on a Mono Q column developed with a salt gradient in HBS containing EDTA (1 mM).
Procoagulant Phospholipid-Dependent Clotting Assay.
In manual-tilt clotting assays, CMV [3.8 × 109 virus particles (vp) per ml], HSV-1 (1.7 × 108 vp per ml), and HSV-2 (6.6 × 107 vp per ml) were incubated with FX-deficient plasma (Sigma) for 1 min at 37°C. At this point FXa (20 nM) was added and clot production was initiated with Ca2+ (2 mM). The functional amount of proPL was determined by using a PS standard curve based on synthetic small unilamellar vesicles composed of 75% phosphatidylcholine and 25% PS (PCPS) that were prepared and quantified as described (7). An amount of virus was chosen to produce activity within the linear region of the standard curve. Values were corrected for background activity generated in the absence of virus.
Procoagulant Phospholipid-Dependent Chromogenic Assay.
CMV (7.2 × 107 vp per ml), HSV-1 (6.9 × 107 vp per ml), and HSV-2 (6.6 × 107 vp per ml) were incubated for 30 min with FXa (5 nM), FVa (5 nM), and prothrombin (1.4 μM). The formation of thrombin was initiated by the addition of Ca2+ (2 mM). Thrombin generation was monitored by cleavage of the chromogenic substrate S-2238 in a kinetic microplate reader (Vmax, Molecular Devices). The amount of proPL was determined by using a PS standard curve. Values were corrected for residual activity in the absence of phospholipid.
FXa-Dependent Clotting Activity.
In manual tilt assays, CMV (3.8 × 109 vp per ml), HSV-1 (1.7 × 108 vp per ml), and HSV-2 (6.6 × 107 vp per ml) were incubated with excess PCPS (300 μM) in either FX- or FX/VII-deficient plasma (Sigma) for 1 min at 37°C. FX (200 nM) was added and clot formation was initiated with Ca2+ (2 mM). The amount of FXa generated was determined from a standard curve obtained with purified FXa. As before, the amount of virus was adjusted to produce activity within the linear region of the standard curve. Values were corrected for residual FXa activity produced in the absence of virus.
FXa-Dependent Chromogenic Assay.
CMV (7.2 × 107 vp per ml), HSV-1 (6.9 × 107 vp per ml), and HSV-2 (6.6 × 107 vp per ml) were incubated with or without each of FX (100 nM), FVIIa (1 nM), and Ca2+ (2 mM) for 5 min at room temperature. FVa (3 nM) and prothrombin (1.4 μM) were then added, and the reaction mixture was incubated for a further 25 min. Thrombin generation was measured by using the chromogenic substrate S-2238. The complete reaction mixture was taken as 100% activity. To determine whether TF was involved in the generation of FXa activity by viruses, CMV, HSV-1, and HSV-2 were preincubated with the inhibitory anti-TF mAb (40 μg/ml; American Diagnostica, Greenwich, CT, product 4508) or nonimmune mouse IgG (Sigma) at 4°C for 2 hr.
Electrophoretic Detection of FX Activation.
125I-labeled FX (100 nM) was incubated with CMV (1.4 × 108 vp per ml), HSV-1 (6.9 × 107 vp per ml), or HSV-2 (6.6 × 107 vp per ml) or without virus in the presence of combinations of FVIIa (1 nM) and Ca2+ (2 mM) at 37°C for 30 min. The amount of virus was chosen to produce comparable amounts of FX activation based on the chromogenic assay. The reaction mixtures were then subjected to SDS/PAGE using a 12% polyacrylamide gel under reducing conditions (34). The electrophoretic pattern of FX under each condition was visualized by autoradiography on Kodak X-Omat film.
Flow Cytometry.
Before analysis by flow cytometry, purified CMV, HSV-1, and HSV-2 were inactivated for 1 hr on ice at a distance of 5 cm from a UV germicidal lamp (GE 030T8, 30 W). UV inactivation was confirmed by plaque assay (30). Three methods were used to ensure the identity of virus particles detected by flow cytometry. (i) Microspheres (0.1 μm; Fluospheres, Molecular Probes) were used to define the boundaries of forward scattering expected for the similarly sized virus particles. (ii) Various concentrations of purified virions were tested to indicate the location of the virions on the side vs. forward scatter dot plot relative to background. (iii) CMV was directly detected by binding of fluorescein-conjugated glycoprotein B-specific (CMVB-1) mAb (B. Brodeur, Laboratory Centre for Disease Control, Ottawa). These procedures showed that the side-to-side scatter pattern was relatively homogenous, which allowed us to collect data ungated for all flow cytometry.
To detect proPL, UV-inactivated CMV (2.3 × 108 vp), HSV-1 (1.3 × 107 vp), and HSV-2 (5.3 × 106 vp) were incubated with AnV-F (0.35 μM) in the presence of Ca2+ (5 mM) or as a negative control in EDTA (5 mM). After incubation for 1 hr at room temperature, samples were diluted to 500 μl in the appropriate Ca2+- or EDTA-containing Hepes (20 mM)/NaCl (150 mM), pH 7.4 (HBS). Data were immediately acquired by using a Becton Dickinson flow cytometer and analyzed by using lysys ii software.
For TF detection, UV-inactivated CMV (4.1 × 107 vp), HSV-1 (8.0 × 107 vp), and HSV-2 (7.7 × 107 vp) were incubated for 1 hr at room temperature with either anti-TF mAb (American Diagnostica, product 4503) at a dilution of 1:28, 1:12, or 1:24, respectively, or the same amount of nonimmune mouse IgG (Cappell) as a negative control. Bound primary antibody was detected by incubation with goat anti-mouse IgG R-phycoerythrin-conjugated secondary antibody (Molecular Probes) at a dilution of 1:110, 1:55, or 1:180, respectively, for 1 hr at room temperature. The samples were then diluted in 1 ml of filtered HBS, and the data were acquired immediately after dilution by using an Epics XL flow cytometer (Coulter) and analyzed by using facsolv software.
Electron Microscopy.
For detection of proPL and direct FVa binding, virions were incubated with FVa at a final concentration of 50 ng/ml for 30 min at 4°C. The virus was then adsorbed onto carbon/Formvar-coated grids in HBS/2 mM Ca2+ (HBS/Ca). After washing five times with HBS/Ca/0.1% fish gelatin, the grids were treated with anti-FVa-heavy-subunit mAb (Haematologic Technologies, AHV-5146) or, as a negative control, with anti-actin mAb (Sigma, A-2547), both at 50 ng/ml for 45 min at 22°C. Washing was repeated, followed by incubation with 10-nm gold-conjugated secondary antibody (goat anti-mouse IgG; Sigma) at 100 ng/ml for 45 min at 22°C and another round of washing. Negative staining was then conducted as described (29, 30).
Virions were evaluated for endogenous TF antigen by immunogold staining with a TF-specific primary mAb (American Diagnostica, product 4503) and 10-nm gold-conjugated secondary antibody (goat anti-mouse IgG; Sigma), as described (29, 30).
RESULTS
Procoagulant Phospholipid on Herpesviruses.
Previous studies from our laboratory showed that proPL exists on the surface of CMV (29). To determine whether other herpesviruses also exhibit proPL activity on their surface, HSV-1 and HSV-2 were evaluated. By using proPL-dependent tilt clotting assays, CMV, HSV-1, and HSV-2 were included as the sole source of phospholipid. All three viruses were observed to reduce the clotting time of FX-deficient or combined FX- and FVII-deficient plasma by at least 30 sec. These experiments demonstrated that the HSVs had considerably more apparent proPL activity than CMV per virion (CMV, 0.23 ± 0.01 μM PS per 108 vp; HSV-1, 2.4 ± 0.1 μM PS per 108 vp; HSV-2, 9.1 ± 1.4 μM PS per 108 vp; n = 3).
A second functional method was used to further support the presence of proPL on the three enveloped viruses. In this system, virus was again added as the source of proPL but prothrombinase was assembled on the virions by using purified FXa and FVa with purified prothrombin as substrate. The addition of each virus resulted in chromogenic substrate cleavage, corresponding to thrombin generation. This method confirmed that all three viruses have accessible proPL activity (CMV, 0.48 ± 0.36 nM PS per 108 vp; HSV-1, 287.1 ± 70.8 nM PS per 108 vp; HSV-2, 194.4 ± 58.1 nM PS per 108 vp; n = 4). Although the presence of proPL for each virus type was clear and was at least comparable to that expected for activated platelets (29), the two assay methods reproducibly gave different quantitative results. These differences may have arisen from an as yet unidentified component(s) in plasma that influences the apparent proPL activity.
After obtaining functional evidence, a physical demonstration of proPL on the virus was obtained by flow cytometry using AnV-F, which is known to bind anionic phospholipid in a Ca2+-dependent manner (35–37). Fig. 1A shows that all particles present in the CMV, HSV-1, and HSV-2 preparations could specifically bind AnV-F in the presence of Ca2+, but not when a chelator (EDTA) was included as a negative control. Chelation had no significant effect on the side-to-side scatter pattern of any virus preparation. Therefore, the demonstrated proPL activity was associated with the virus particles and not due to the potentially copurifying cellular debris (found to represent <10% of all particles observed by electron microscopy). It should be noted that a number of particles were accumulated at a value of 104 on the x-axis channel axis for the CMV and HSV-1 samples containing Ca2+ (data not shown).
Figure 1.
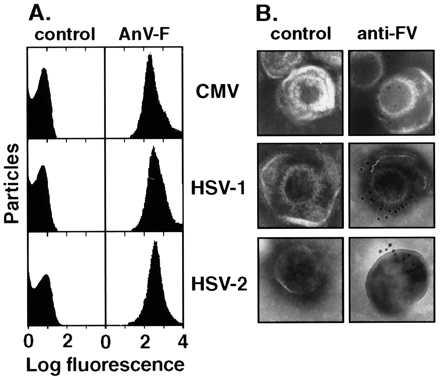
Demonstration of proPL on herpesviruses. (A) Flow cytometry to detect fluorescein-conjugated AnV binding to viral particles in the presence (AnV-F) or absence (control) of Ca2+. (B) Immunogold electron microscopy to detect FVa binding to viral particles with a specific mAb (anti-FV) or nonimmune IgG (control) as primary antibodies and goat anti-mouse IgG-conjugated to gold as the secondary antibody.
The flow cytometry data were corroborated by immunogold electron microscopy (Fig. 1B) using exogenous FVa and a FVa-heavy-subunit-specific mAb to probe for proPL. The electron-dense spots indicate the recognition of FVa on the virus. Both lysed (“fried-egg” shaped) and intact (spherical) virus particles were found to bind FVa, whereas no positive particles were found in control experiments using a nonimmune primary antibody. Endogenous FV/Va was not detectable (data not shown). Combined with our earlier electron microscopy demonstrating FXa binding to CMV (29), the current data provide convincing evidence that both prothrombinase constituents can assemble directly on virus particles.
Activation of FX on Herpesviruses.
After establishing that proPL is available on CMV, HSV-1, and HSV-2, we next determined whether they also have constituents that can facilitate FX activation. To do so, each virus type was incubated with purified FX and either FX-deficient or combined FX- and FVII (the precursor of FVIIa)-deficient plasma and evaluated for clotting times. No detectable FXa activity was generated when the FX/FVII-deficient plasma was used (data not shown). In contrast, significant FXa activity was produced when FVII was present in the plasma (CMV, 25.2 ± 4.5 nM FXa per 108 vp; HSV-1, 74.7 ± 6.6 nM FXa per 108 vp; HSV-2, 185.5 ± 85.4 nM FXa per 108 vp; n = 5). The large difference between these viruses in proPL and FXa-generating activity may provide an explanation for why thrombotic lesions are observed with HSV but not for CMV infection (20, 22).
To confirm that exogenous FX was being converted to FXa in the clotting assay, FXa production was followed electrophoretically by using radioiodinated FX. As shown in Fig. 2, a species was produced in the presence of CMV, HSV-1, or HSV-2 and both FVIIa and Ca2+ that corresponded exactly to the FXa heavy subunit under reducing conditions. No detectable FXa band was visible when either virus, FVIIa, or Ca2+ was absent, which is in agreement with the clotting assays.
Figure 2.
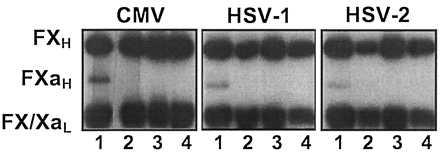
Herpesvirus-dependent proteolytic activation of FX. 125I-FX was incubated with CMV, HSV-1, or HSV-2 or without virus in the presence of combinations of FVIIa and Ca2+. The reaction mixtures were then subjected to reduced SDS/PAGE and the electrophoretic pattern of the 125I-FX under each condition was visualized by autoradiography on Kodak X-Omat film. The position of the FX heavy (FXH), FXa heavy (FXaH), and the FX/Xa light (FX/XaL) subunits are shown. Lanes: 1, 125I-FX, virus, FVIIa, and Ca2+; 2, 125I-FX, FVIIa, and Ca2+; 3, 125I-FX, virus, and Ca2+; 4, 125I-FX, virus, and FVIIa.
The dependence on FVII/VIIa for FXa generation in the clotting and electrophoretic experiments is consistent with the presence of TF on the virus. To further address this possibility, an inhibitory TF-specific mAb was used. In this experiment, purified FX, FVIIa, FVa, and prothrombin were sequentially incubated with virus, and thrombin generation was evaluated as a means to identify FX activation. This experiment (Fig. 3) showed that the virus-dependent generation of FXa was inhibited by anti-TF mAb (CMV, 70%; HSV-1, 40%; HSV-2, 35% inhibition). Higher concentrations of antibody did not increase the extent of inhibition (data not shown). The addition of nonimmune mouse IgG to the positive control (no mAb) also had no effect on FXa generation. Further control experiments showed that this assay was dependent on added Ca2+, FVIIa, FX, FVa, and prothrombin and that no endogenous FX/Xa activity on any of the three viruses was present (data not shown). These combined functional observations suggest a contribution of TF in the activation of FX by CMV, HSV-1, and HSV-2.
Figure 3.
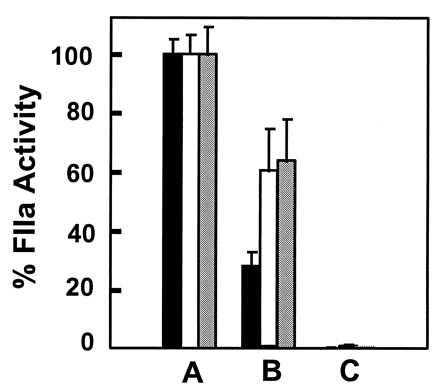
Inhibition of herpesvirus-dependent FX activation with anti-TF mAb. (Bars A) CMV, HSV-1, and HSV-2 were preincubated for 2 hr in buffer and then with FX, FVIIa, and Ca2+ for 5 min at room temperature. FVa and prothrombin were then added and the reaction mixture incubated for a further 25 min. Thrombin generation was measured by using the chromogenic substrate S-2238 and calculated as 100% activity. (Bars B) As for bars A but viruses were preincubated for 2 hr with an inhibitory anti-TF mAb (American Diagnostica, product 4508). (Bars C) As for bars A but no virus (n = 9). Bars: solid, CMV; open, HSV-1; shaded, HSV-2.
To provide direct evidence that the TF activity is associated with the virus, flow cytometry was conducted. As depicted by the histograms in Fig. 4A, 94% of CMV, 88% of HSV-1, and 84% of HSV-2 particles were indeed recognized by the anti-TF mAb. These data demonstrate the presence of TF antigen on virus particles. Substantiating evidence was obtained by immunogold electron microscopy (Fig. 4B), in which the electron-dense gold particles identify specific interactions between the anti-TF mAb and each virus type. As a negative control, no gold staining was observed when an identical amount of anti-actin mAb was substituted for the primary anti-TF mAb.
Figure 4.
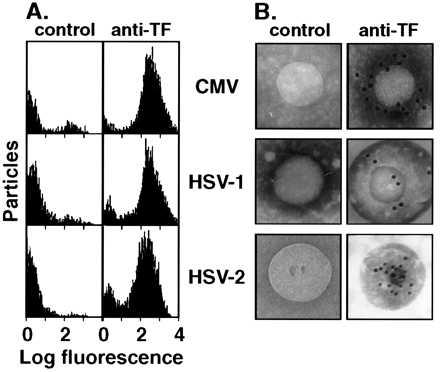
Demonstration of TF on herpesviruses. (A) Flow cytometry to detect the binding of a TF-specific mAb (anti-TF) or nonimmune IgG (control) to viral particles using goat anti-mouse IgG conjugated to phycoerythrin as a secondary antibody. (B) Immunogold electron microscopy to detect the binding of TF-specific mAb (anti-TF) or nonimmune IgG (control) to viral particles, with goat anti-mouse IgG conjugated to gold as the secondary antibody.
DISCUSSION
Many types of virus, including herpesviruses, are covered by a membrane that functions in early stages of infection by contributing components necessary for host cell entry (38) and for evasion of the immune system (39). This envelope contains proteins that are encoded by both the virus and host genomes, with a phospholipid component derived solely from the host cell. Under normal resting conditions, cells actively maintain an asymmetric phospholipid distribution with proPL located on the inner side of the lipid bilayer (5, 40). Thus, membrane sidedness acts as an important hemostatic barrier that limits the exposure of proPL to sites of vascular damage, where it is “flipped” to the surface. In a previous study (29), we demonstrated that the CMV envelope has proPL on its surface and consequently circumvents an important cellular hemostatic control. Through the use of clotting and chromogenic assays, flow cytometry, and electron microscopy, the current study extends this finding to include both HSV-1 and HSV-2 as having accessible proPL.
Although the three viruses that we evaluated in this report are from the herpesvirus family, it is conceivable that accessible proPL is a ubiquitous characteristic of enveloped viruses. In fact it has been shown that HIV type 1 and type 2, vesicular stomatitis virus, extracellular autographa californica nuclear polyhedrosis virus, and HSV have a higher proportion of PS in their envelope than the host cell membrane (41–44). There is further evidence in the literature that identifies a role for PS and PS-binding proteins in hepatitis B, influenza A and B, hemorrhagic septicemia virus, sendai virus, vesicular stomatitis virus, rubella virus, sindbus virus, vaccinia virus, and CMV infection (44–51). Therefore, our observation that CMV, HSV-1, and HSV-2 can assemble prothrombinase on their surface may be a consequence of a distinct function for proPL in the infection process. In this regard, we have identified a specific role for proPL as an anchor for the PS-binding protein annexin II, which is a CMV receptor (30, 52, 53). However, no analogous function has yet been identified for either HSV-1 or HSV-2.
To initiate thrombin production, proPL and a mechanism to convert FX to FXa must become available to the circulating plasma coagulation proteins. Under normal physiological conditions, the first FXa molecules are made by the TF/FVIIa tenase (54). Both HSV-1 and CMV have been demonstrated to enhance TF expression on host endothelial cells (12, 55). Similarly, experiments using UV-inactivated viruses revealed an increase in TF activity on the cultured cells we used to propagate viruses (data not shown). Therefore, we speculated that TF produced in response to infection may be routed to the virus particle when the envelope is formed. We now report that all three viruses can participate in the activation of FX, as concluded from clotting, electrophoretic, and chromogenic data. For each, the activation of FX was dependent on Ca2+ and FVIIa, which suggested the presence of a TF-like constituent on the virus surface. By using a mAb raised to TF, we demonstrated by flow cytometry and electron microscopy that TF antigen is present on virions. Furthermore, we found that a different TF mAb inhibited the virus-dependent generation of FXa.
The data presented herein cumulatively demonstrate that CMV, HSV-1, and HSV-2 possess the molecular machinery necessary to initiate thrombin generation on their envelope surface. This activity involves endogenous proPL, to facilitate coagulation enzyme-cofactor-substrate complex assembly, and a species functionally and antigenically identical to TF, which participates in FX activation. Because neither the CMV nor HSV genome encode a TF homologue, it must be acquired along with proPL from the host cell during virus envelope formation. Additional components on the virus may contribute to thrombin production. One example is the HSV-1-encoded glycoprotein C that has been shown to accelerate FX activation when expressed on the surface of an infected endothelial cell (15). The purified virus is known to have glycoprotein C on its surface (39). Whether it can function like the cellular form within a tenase has not yet been determined but provides a possible explanation for the incomplete inhibition of FXa generation observed by using the anti-TF mAb. The fact that we did not observe direct FXa generation by virus alone in our electrophoresis experiment suggests that the activity not inhibited by the TF antibody may be dependent on FVIIa.
The ability of viruses to convert resting cells from an anticoagulant to a procoagulant state is not unique to herpesviruses. Measles virus, murine hepatitis virus, and avian hemangioma retrovirus also cause host cells to become thrombogenic (49, 56, 57). It is therefore compelling to consider that virus infection in general may manifest certain procoagulant effects. Because thrombin is a potent cell modulator, viruses may have evolved to directly produce thrombin as a way to signal host cells during infection. Herpesviruses and thrombin have been shown to trigger similar intra- and extracellular events that are attributed to G protein-coupled receptor perturbation (58, 59). Repercussions of endothelial cell signaling by herpesviruses or thrombin include increases in tissue factor expression (14, 55), platelet binding (11), and monocyte adhesion (60). These characteristics give rise to a procoagulant and proinflammatory phenotype, which are contributing factors in atherogenesis (12, 61, 62). Our current data suggest that the production of thrombin on virus attached to the host cell surface may, therefore, be an initiating event in the vascular pathology mediated by herpesviruses.
Acknowledgments
We thank Garry Kessler for protein purification, Karen Poirier for technical assistance, Nadine Barrette for figure preparation, Steven Doyle (Microscopy Imaging Laboratory, University of Toronto) for assistance with electron microscopy, and Drs. Pierre Neuenschwander and James Morrissey for helpful discussions regarding the antigenic detection of tissue factor. This project was supported by the Canadian Red Cross Society R&D Grant HO1O925 (E.P. and J.F.W.). J.F.W. was supported by a Bayer/Canadian Red Cross Society/Medical Research Council Scholarship.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: proPL, procoagulant phospholipid; HSV, herpes simplex virus; CMV, cytomegalovirus; FX, factor X; FVII, factor VII; TF, tissue factor; FV, factor V; AnV, annexin V; PS, phosphatidylserine; vp, virus particle(s).
References
- 1.Stubbs M T, Bode W. Trends Biochem Sci. 1995;20:23–28. doi: 10.1016/s0968-0004(00)88945-8. [DOI] [PubMed] [Google Scholar]
- 2.Ruf W, Rehemtulla A, Edgington T S. J Biol Chem. 1991;266:2158–2166. [PubMed] [Google Scholar]
- 3.Broze G J. Semin Hematol. 1992;29:159–169. [PubMed] [Google Scholar]
- 4.Bevers E M, Comfurius P, van Rijn J L, Hemker H C, Zwaal R F. Eur J Biochem. 1982;122:429–436. doi: 10.1111/j.1432-1033.1982.tb05898.x. [DOI] [PubMed] [Google Scholar]
- 5.Bevers E M, Comfurius P, Zwaal R F. Biochim Biophys Acta. 1983;736:57–66. doi: 10.1016/0005-2736(83)90169-4. [DOI] [PubMed] [Google Scholar]
- 6.Mann K G, Nesheim M E, Church W R, Haley P, Krishnaswamy S. Blood. 1990;76:1–16. [PubMed] [Google Scholar]
- 7.Krishnaswamy S, Nesheim M E, Pryzdial E L G, Mann K G. Methods Enzymol. 1994;222:260–280. doi: 10.1016/0076-6879(93)22018-b. [DOI] [PubMed] [Google Scholar]
- 8.van Dam-Mieras M C E, Bruggeman C A, Muller A D, Debie W H M, Zwaal R F A. Thromb Res. 1987;47:69–75. doi: 10.1016/0049-3848(87)90241-6. [DOI] [PubMed] [Google Scholar]
- 9.van Dam-Mieras M C E, Muller A D, van Hinsbergh V W M, Mullers W J H A, Bomans P H H, Bruggeman C A. Thromb Haemostasis. 1992;68:364–370. [PubMed] [Google Scholar]
- 10.Etingin O R, Silverstein R L, Friedman H M, Hajjar D P. Cell. 1990;61:657–662. doi: 10.1016/0092-8674(90)90477-v. [DOI] [PubMed] [Google Scholar]
- 11.Visser M R, Tracy P B, Vercellotti G M, Goodman J L, White J G, Jacob H S. Proc Natl Acad Sci USA. 1988;85:8227–8230. doi: 10.1073/pnas.85.21.8227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vercellotti G M. Blood Cells. 1990;16:209–216. [PubMed] [Google Scholar]
- 13.Bruggeman C A, Debie W H M, Muller A D, Schutte B, van Dam-Mieras M C E. Thromb Haemostasis. 1988;59:264–268. [PubMed] [Google Scholar]
- 14.Key N S, Bach R R, Vercellotti G M, Moldow C F. Lab Invest. 1993;68:645–651. [PubMed] [Google Scholar]
- 15.Altieri D C, Etingin O R, Fair D S, Brunck T K, Geltosky J E, Hajjar D P, Edgington T S. Science. 1992;254:1200–1202. doi: 10.1126/science.1957171. [DOI] [PubMed] [Google Scholar]
- 16.Speir E, Modali R, Huang E S, Leon M B, Shawl F, Finkel T, Epstein S E. Science. 1994;265:391–394. doi: 10.1126/science.8023160. [DOI] [PubMed] [Google Scholar]
- 17.Zhou Y F, Guetta E, Yu Z X, Finkel T, Epstein S E. J Clin Invest. 1996;98:2129–2138. doi: 10.1172/JCI119019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nieto F J, Adam E, Sorlie P, Farzadegan H, Melnick J L, Comstock G W, Szklo M. Circulation. 1996;94:922–927. doi: 10.1161/01.cir.94.5.922. [DOI] [PubMed] [Google Scholar]
- 19.Zhou Y F, Leon M B, Waclawiw M A, Popma J J, Yu Z X, Finkel T, Epstein S E. New Eng J Med. 1996;335:624–630. doi: 10.1056/NEJM199608293350903. [DOI] [PubMed] [Google Scholar]
- 20.McSorley J, Shapiro L, Brownstein M H, Hsu K C. Int J Dermatol. 1974;13:69–75. doi: 10.1111/j.1365-4362.1974.tb01769.x. [DOI] [PubMed] [Google Scholar]
- 21.Lever W F, Schaumberg-Lever G. Histopathology. Philadelphia: Lippincott; 1974. p. 360. [Google Scholar]
- 22.Phinney P R, Fligiel S, Bryson Y J, Porter D D. Arch Pathol Lab Med. 1982;106:64–67. [PubMed] [Google Scholar]
- 23.Benditt E P, Barrett T, McDougall J K. Proc Natl Acad Sci USA. 1983;80:6386–6389. doi: 10.1073/pnas.80.20.6386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yamashiroya H M, Gosh L, Yang R, Robertson A. Am J Pathol. 1988;130:71–79. [PMC free article] [PubMed] [Google Scholar]
- 25.Melnick J, Adam E, DeBakey M E. J Am Med Assoc. 1990;263:2204–2207. [PubMed] [Google Scholar]
- 26.Fabricant C G, Fabricant J, Minick C R, Litrenta M M. Fed Proc. 1983;42:2476–2479. [PubMed] [Google Scholar]
- 27.Span A H M, Grauls G, Bosman F, Vanboven C P A, Bruggeman C A. Atherosclerosis. 1992;93:41–52. doi: 10.1016/0021-9150(92)90198-p. [DOI] [PubMed] [Google Scholar]
- 28.Virgin, H. W., Weck, K. E., Dal Canto, A. J., Gould, J. D., Latreille, P. & Speck, S. H. (1997) Am. Soc. Virol. July 19-23, Bozeman, Montana, p. 178 (abstr.).
- 29.Pryzdial E L G, Wright J F. Blood. 1994;84:3749–3757. [PubMed] [Google Scholar]
- 30.Wright J F, Kurosky A, Pryzdial E L G, Wasi S. J Virol. 1995;69:4784–4791. doi: 10.1128/jvi.69.8.4784-4791.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pryzdial E L G, Bajzar L, Nesheim M E. J Biol Chem. 1995;270:17871–17877. doi: 10.1074/jbc.270.30.17871. [DOI] [PubMed] [Google Scholar]
- 32.Koopman G, Reutelingsperger C P M, Kuijten G A M, Keehnen R M J, Pals S T, van Oers M J H. Blood. 1994;84:1415–1420. [PubMed] [Google Scholar]
- 33.Tait J F, Gibson D, Fujikawa K. J Biol Chem. 1989;264:7944–7949. [PubMed] [Google Scholar]
- 34.Laemmli U K. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 35.Tait J F, Gibson D. Arch Biochem Biophys. 1992;298:187–191. doi: 10.1016/0003-9861(92)90111-9. [DOI] [PubMed] [Google Scholar]
- 36.Meers P, Mealy T. Biochemistry. 1993;32:11711–11721. doi: 10.1021/bi00094a030. [DOI] [PubMed] [Google Scholar]
- 37.Swairjo M A, Seaton B A. Annu Rev Biophys Biomol Struct. 1994;23:193–213. doi: 10.1146/annurev.bb.23.060194.001205. [DOI] [PubMed] [Google Scholar]
- 38.Roizman B, Whitley R J, Lopez C. The Human Herpesviruses. New York: Raven; 1993. [Google Scholar]
- 39.Friedman H M, Wang L, Fishman N O, Lambris J D, Eisenberg R J, Cohen G H, Lubinski J. J Virol. 1996;70:4253–4260. doi: 10.1128/jvi.70.7.4253-4260.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zwaal R F A, Comfurius P, Bevers E M. Biochem Soc Trans. 1993;21:248–253. doi: 10.1042/bst0210248. [DOI] [PubMed] [Google Scholar]
- 41.Aloia R C, Tian H, Jensen F C. Proc Natl Acad Sci USA. 1993;90:5181–5185. doi: 10.1073/pnas.90.11.5181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Gendren I L, Brandimarti R, Torrisis M R, Campadelli G, van Meer G. Virology. 1994;200:831–836. doi: 10.1006/viro.1994.1252. [DOI] [PubMed] [Google Scholar]
- 43.Braunagel S C, Summers M D. Virology. 1994;202:315–328. doi: 10.1006/viro.1994.1348. [DOI] [PubMed] [Google Scholar]
- 44.Luan P, Yang L, Glaser M. Biochemistry. 1995;34:9874–9883. doi: 10.1021/bi00031a008. [DOI] [PubMed] [Google Scholar]
- 45.Neurath A R, Strick N. Virology. 1994;204:475–477. doi: 10.1006/viro.1994.1558. [DOI] [PubMed] [Google Scholar]
- 46.Otto M, Gunther A, Fan H, Rick O, Huang R T C. FEBS Lett. 1994;356:125–129. doi: 10.1016/0014-5793(94)01241-5. [DOI] [PubMed] [Google Scholar]
- 47.Chejanovsky N, Zakai N, Amselem S, Barenholz Y, Loyter A. Biochemistry. 1986;25:4810–4817. doi: 10.1021/bi00365a014. [DOI] [PubMed] [Google Scholar]
- 48.Estepa A, Coll J M. Virology. 1996;216:60–70. doi: 10.1006/viro.1996.0034. [DOI] [PubMed] [Google Scholar]
- 49.Mastromarino P, Cioe L, Rieti S, Orsi N. Med Microbiol Immunol. 1990;179:105–114. doi: 10.1007/BF00198531. [DOI] [PubMed] [Google Scholar]
- 50.Kuge O, Akamatsu Y, Nishijima M. Biochem Biophys Acta. 1989;986:61–69. doi: 10.1016/0005-2736(89)90272-1. [DOI] [PubMed] [Google Scholar]
- 51.Oie M. Virology. 1985;142:299–306. doi: 10.1016/0042-6822(85)90338-1. [DOI] [PubMed] [Google Scholar]
- 52.Wright J F, Kurosky A, Wasi S. Biochem Biophys Res Commun. 1994;198:983–989. doi: 10.1006/bbrc.1994.1140. [DOI] [PubMed] [Google Scholar]
- 53.Pryzdial, E. L. G., Raynor, C. M., Sutherland, M. R. & Wright, J. F. (1997) Am. Soc. Virol. July 19-23, Bozeman, Montana, p. 230 (abstr.).
- 54.Martin D M, Boys C W, Ruf W. FASEB J. 1995;9:852–859. doi: 10.1096/fasebj.9.10.7615155. [DOI] [PubMed] [Google Scholar]
- 55.Vercellotti, G. M., Kovacs, A., Key, N. S., Dandelet, L. A. & Jacob, H. S. (1997) Thromb. Haemostasis (Suppl.), 203 (abstr.).
- 56.Fung L S, Neil G, Leibowitz J, Cole E H, Chung S, Crow A, Levy G A. J Biol Chem. 1991;266:1789–1795. [PubMed] [Google Scholar]
- 57.Resnick-Roguel N, Eldor A, Burstein H, Hy-Am E, Vlodavsky I, Panet A, Blajchman M A, Kotler M. J Virol. 1990;64:4029–4032. doi: 10.1128/jvi.64.8.4029-4032.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vu T-K H, Hung D T, Wheaton V I, Coughlin S R. Cell. 1991;64:1057–1068. doi: 10.1016/0092-8674(91)90261-v. [DOI] [PubMed] [Google Scholar]
- 59.Zentner J, Albrecht T, Heuser D. Funct Neurol. 1991;6:411–412. [PubMed] [Google Scholar]
- 60.Etingin O R, Silverstein R L, Hajjar D P. Proc Natl Acad Sci USA. 1991;88:7200–7203. doi: 10.1073/pnas.88.16.7200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Melnick J L, Schattner A. Isr J Med Sci. 1992;28:463–465. [PubMed] [Google Scholar]
- 62.Hajjar D P. Am J Pathol. 1991;139:1195–1211. [PMC free article] [PubMed] [Google Scholar]