

Abstract
The specific formylation of initiator methionyl-tRNA by methionyl-tRNA formyltransferase (MTF; EC 2.1.2.9) is important for the initiation of protein synthesis in eubacteria and in eukaryotic organelles. The determinants for formylation in the tRNA are clustered mostly in the acceptor stem. As part of studies on the molecular mechanism of recognition of the initiator tRNA by MTF, we report here on the isolation and characterization of suppressor mutations in Escherichia coli MTF, which compensate for the formylation defect of a mutant initiator tRNA, lacking a critical determinant in the acceptor stem. We show that the suppressor mutant in MTF has a glycine-41 to arginine change within a 16-amino acid insertion found in MTF from many sources. A mutant with glycine-41 changed to lysine also acts as a suppressor, whereas mutants with changes to aspartic acid, glutamine, and leucine do not. The kinetic parameters of the purified wild-type and mutant Arg-41 and Lys-41 enzymes, determined by using the wild-type and mutant tRNAs as substrates, show that the Arg-41 and Lys-41 mutant enzymes compensate specifically for the strong negative effect of the acceptor stem mutation on formylation. These and other considerations suggest that the 16-amino acid insertion in MTF plays an important role in the specific recognition of the determinants for formylation in the acceptor stem of the initiator tRNA.
Keywords: protein synthesis initiation, RNA–protein interactions, genetic suppression
Formylation of initiator methionyl-tRNA (Met-tRNA) by methionyl-tRNA formyltransferase (MTF; EC 2.1.2.9) is important for the initiation of protein synthesis in eubacteria and in eukaryotic organelles such as chloroplasts and mitochondria (1–3). The formylation reaction is highly specific. The enzyme formylates the initiator Met-tRNA but not the elongator species of Met-tRNA or any other aminoacyl-tRNA (4, 5). The determinants for formylation are clustered mostly in the acceptor stem (6–8). The gene for the Escherichia coli enzyme has been cloned and sequenced (9). However, there is little information on the overall topology of interaction of the enzyme and the tRNA and on the molecular basis of the specificity of recognition. As a first step in identifying the regions of MTF that come close to the 3′ end of the initiator tRNA, we showed recently that the 3′ end could be crosslinked to a single lysine residue, Lys-206,† of MTF (10).
The crystal structure of MTF has been determined recently (11). The enzyme consists of two domains connected by a linker region. The N-terminal domain is strikingly homologous to the structure of E. coli glycinamide ribonucleotide formyltransferase (GARF), another enzyme which, like MTF, uses N10-formyltetrahydrofolate as a formyl donor in formylation reactions (Fig. 1) (12–14). Included within this domain in GARF from various sources are the amino acid residues Asn-106, His-108, Ser- or Thr-135, and Asp-144, which are thought to participate in the catalytic steps (12, 13, 15). These same amino acids are also present in MTF from various sources, suggesting that the basic catalytic mechanisms for formyl group transfer in these classes of enzymes are similar. A notable difference between MTF and GARF sequences is the insertion in MTF of a 16-amino acid sequence in the loop‡ region between the second β-sheet and the second α-helix from the N terminus of the proteins (11). The insertion sequence is found in all MTFs whose sequences are known to date, and it contains several basic residues (Fig. 2). Another difference between MTF and GARF is the presence in MTF of an approximately 100-amino acid long extension at the C terminus of MTF. This region is structurally homologous to the anticodon binding domain of E. coli lysyl-tRNA synthetase and to the oligonucleotide or oligosaccharide binding domain of other proteins and has been shown to bind tRNA on its own (11). These findings have led to the suggestion that the insertion loop and the C-terminal extension together are involved in recognition of the tRNA by MTF.
Figure 1.
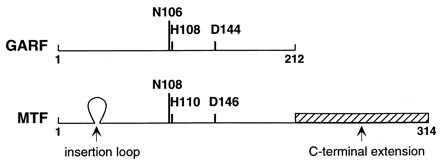
Schematic alignment of GARF and MTF sequences from various sources. The amino acid numbering for MTF begins with serine found at the N terminus of the protein. The amino acids Asn, His, and Asp known to be involved in catalysis in GARF and also found in MTF are indicated. Arrows indicate insertions in MTF compared with GARF, which are thought to be important for tRNA recognition.
Figure 2.

Alignment of MTF sequences highlighting the insertion of a 16-amino acid sequence. Closed circles indicate the amino acids that are highly conserved in at least 8 of the 10 sequences. Open circle indicates a basic amino acid residue conserved in 9 of the 10 sequences. ∗ indicates the site of suppressor mutation in E. coli MTF identified in this work.
For functional studies of mutant initiator tRNAs in E. coli, we previously developed a strategy based on the use of the U35A36 mutant carrying an anticodon sequence change from CAU → CUA (16). The CUA anticodon sequence allows the assessment of the initiator activity of mutant tRNAs in vivo by measuring the levels of chloramphenicol acetyltransferase (CAT) expression from a reporter CAT gene, CATam1.2.5, which has UAG as the initiation codon. Using this assay, we showed that mutations in the acceptor stem, which removed some of the critical determinants important for formylation, essentially abolished activity of the mutant tRNAs in initiation in E. coli (17). As a result, E. coli harboring these mutant tRNAs and the CATam1.2.5 reporter gene are sensitive to chloramphenicol. Here, we report on the use of this system to isolate suppressor mutations in MTF, which can compensate for the formylation defect of the mutant initiator tRNA and, thereby, confer chloramphenicol resistance phenotype on the cells. We show that a Gly-41 to Arg change in MTF, located within the 16-amino acid insertion loop, compensates for the formylation defect of the U35A36/G72G73 mutant initiator tRNA. We describe the purification of this and other site-specific mutants at this position and comparison of their catalytic properties to those of the wild-type enzyme in formylation of the wild-type and the U35A36/G72G73 and the U35A36 mutant initiator tRNAs. The mutant enzymes are 26- to 27-fold more active than the wild-type enzyme toward the U35A36/G72G73 mutant initiator tRNA. In contrast, the mutant enzymes are 3- to 5-fold less active than the wild-type enzyme toward the wild-type and the U35A36 mutant initiator tRNAs. These results provide a direct correlation between phenotypic suppression in vivo and the enhanced activity of the suppressor mutants in vitro on the U35A36/G72G73 mutant initiator tRNA. They also suggest that the 16-amino acid insertion in MTF plays an important role in recognition of the determinants for MTF in the acceptor stem of the initiator tRNA.
MATERIALS AND METHODS
Strains and Plasmids.
E. coli CA274 (HfrH lacZam trpEam), E. coli JM109 (18), and E. coli DH5αF′ (19) were used as hosts. Plasmid pACDFMT (resistant to tetracycline) contains the fmt gene for E. coli MTF (20) cloned into the NcoI–EcoRI sites, and the plasmid pRSVCATam1.2.5 (resistant to ampicillin) contains the U35A36/G72G73 mutant initiator tRNA gene, the reporter CATam1.2.5 gene, and the glutaminyl-tRNA synthetase (GlnRS) gene. The plasmid expressing GlnRS as a His6 fusion (pQE70GlnRS) was a gift from the laboratory of Paul Schimmel. The enzyme was purified by using the Talon affinity resin (CLONTECH) as described below for the MTF–His6 fusion protein.
Plasmid for Expression of MTF as a Fusion Protein with His6 Residues.
To facilitate subcloning of the mutants in the coding region of fmt, the PstI site located toward the 5′ end of the gene was altered from CTGCAG to TTGCAA by site-specific mutagenesis without changing the coded sequence, Leu-80–Gln-81. The fmt gene was then amplified by PCR with the plasmid pACDFMT as template and cloned into the BamHI and BglII sites of pQE16 (resistant to ampicillin, Qiagen, Chatsworth, CA) to generate pQE16 FMTp. The derived amino acid sequence of the recombinant fusion protein is MRGS(1SESL---NRLV314)RSHHHHHH in which the amino acid sequence within the parentheses corresponds to MTF.
Mutagenesis of the fmt Gene.
Cells were grown at 37°C in 2× YT medium; the concentrations of antibiotics when used were: ampicillin (Amp), 100 μg/ml; tetracycline (Tet), 15 μg/ml; and chloramphenicol (Cam), 100 μg/ml. N-Methyl-N′-nitro-N-nitrosoguanidine was used to treat E. coli DH5αF′ carrying the plasmid pACDFMT as described (21, 22), and the plasmid DNA was isolated from the mutagenized culture to yield a random pool of mutants (23). Suppressor mutations in the fmt gene were identified from this pool of mutants by transforming E. coli CA274 carrying the pRSVCATam1.2.5/trnfMU35A36/G72G73/GlnRS plasmid. The transformants were grown for 4 hr in the presence of ampicillin and tetracycline and spread on plates containing (in addition) chloramphenicol. Plasmids were isolated from 20 chloramphenicol-resistant colonies, and the pACD plasmid carrying the fmt gene was purified by transformation of E. coli CA274 and selection for Tet+ Amp− phenotype. Following the above screening procedure, four pACD plasmids containing the fmt gene and conferring resistance to chloramphenicol were isolated. The mutation in these was localized to an ≈1-kbp BssHII fragment bearing the fmt gene (including 100 bp upstream but lacking the sequences coding for the C-terminal 20 amino acids) by subcloning and identified by DNA sequencing.
Site-Specific Mutagenesis of the Gene for MTF and Purification of Mutant MTF Proteins.
Mutations at position 41 to a K, L, Q, or D were generated by QuikChange mutagenesis with Pfu DNA polymerase (Stratagene) by using the plasmids pACDFMT or pQE16FMTp. E. coli JM109 was used as host to express the MTF–His6 fusion proteins. The cultures (500 ml) were grown for 4 hr at 37°C in the presence of ampicillin, induced by the addition of isopropyl β-d-thiogalactoside (0.5 mM, final), and the incubation was continued for 2 more hr. Cells were harvested by centrifugation and resuspended in 20 ml of buffer 1 (20 mM Tris⋅HCl, pH 8.0/100 mM NaCl), 1 ml of lysozyme (2 mg/ml in buffer 1), and 0.5 ml of 100 mM phenylmethylsulfonyl fluoride. The suspension was incubated at room temperature for 20 min and freeze thawed once, and 250 μl of DNase I (2.5 mg/ml in 100 mM MgCl2) was added and incubated for a further 20 min. The lysate was clarified by ultracentrifugation (100,000 × g) and applied onto a Talon Sepharose Co2+ metal affinity resin column (24) (CLONTECH, 5.0-ml bed volume) preequilibrated with buffer 1. The column was sequentially washed with buffer 1 (50.0 ml) and buffer 1 + 10 mM imidazole⋅HCl, pH 8.0 (50.0 ml). The bound protein was eluted with buffer 1 + 100 mM imidazole⋅HCl, pH 8.0, and dialyzed against 20 mM imidazole⋅HCl, pH 7.5, 150 mM KCl, 50% glycerol, 10 mM 2-mercaptoethanol. Protein was estimated by the dye binding assay (25) with IgG as standard. Recovery of the protein ranged from 10 to 20 mg. The purity of MTF was monitored by SDS/polyacrylamide gel electrophoresis.
Measurement of Kinetic Parameters in Formylation of tRNA.
The substrate tRNAs (tRNA2fMet and the U35A36, U35A36/G72G73 mutants) were expressed and purified by gel electrophoresis (8, 26). The assay for formylation used a two-step reaction. tRNA was first quantitatively aminoacylated by using either [35S]methionine and methionyl-tRNA synthetase or [3H]glutamine and GlnRS and then formylated with N10-formyltetrahydrofolate and purified MTF. For the aminoacylation step, the incubation mixture (10 μl) contained 20 mM imidazole⋅HCl, pH 7.5, 0.1 mM Na2EDTA, 2 mM ATP, 150 mM NH4Cl, 10 μg/ml BSA, 100 μM [35S]methionine (specific activity, 6000–10,000 cpm/pmol), various amounts of tRNA (0.3–9.0 μM), and an excess of purified MetRS. Similarly, aminoacylation of the U35A36 and U35A36/G72G73 mutant tRNAs was carried out in the presence of 400 μM [3H]glutamine (specific activity, 500–1000 cpm/pmol) and an excess of GlnRS. After 30 min at 37°C, 0.3 mM N10-formyltetrahydrofolate (2 μl) and appropriately diluted amounts of MTF (3 μl) were added for determination of the initial rate of formylation. Control experiments showed that the aminoacyl ester linkage in the aminoacyl-tRNA was completely stable during the course of the formylation reaction. The reaction was allowed to proceed for 5 min and was terminated by the addition of 15 μl of 0.36 M CuSO4 in 1.1 M Tris⋅HCl (pH 7.3) and incubated further for 5 min. Acid-precipitable radioactivity was measured as described (27). Values of Km and Vmax were determined from Lineweaver–Burk and Eadie–Hofstee or Hanes–Woolf plots.
RESULTS
Isolation of Suppressor Mutations in MTF.
The strategy for isolation of suppressor mutations in MTF is based on the use of the CATam1.2.5 gene carrying the UAG initiation codon as a reporter gene in cells also carrying a mutant initiator tRNA, which can read UAG as an initiation codon but which is inactive in initiation because it is defective in formylation (Fig. 3 Left). The mutant initiator tRNA used for this purpose contains the U35A36 anticodon sequence mutation and the G72G73 mutation in the acceptor stem and the discriminator base, which makes it an extremely poor substrate for MTF (Fig. 4) (16, 17). This mutant tRNA is essentially inactive in initiation even in cells overproducing MTF (20). Because of the anticodon sequence change, the mutant tRNA is aminoacylated with glutamine instead of methionine (28). To ensure that the mutant tRNA was maximally aminoacylated with glutamine, E. coli GlnRS was overproduced by including the gene for GlnRS (29) on the same plasmid as the CATam1.2.5 gene and the mutant tRNA gene (Fig. 3 Left).
Figure 3.
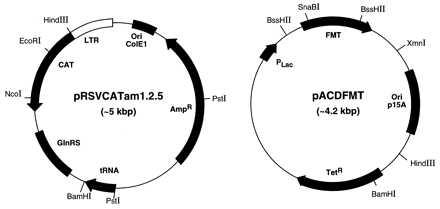
Plasmids used for isolation of suppressor mutations in MTF. pRSVCATam1.2.5 (Left) contains the gene for CATam1.2.5 reporter, the mutant tRNA gene, and the gene for E. coli GlnRS. The pACDFMT plasmid (Right) contains the fmt gene for E. coli MTF and was used for the isolation of mutants in the fmt gene by using N-methyl-N′-nitro-N-nitrosoguanidine mutagenesis (details in text). LTR, long terminal repeat.
Figure 4.
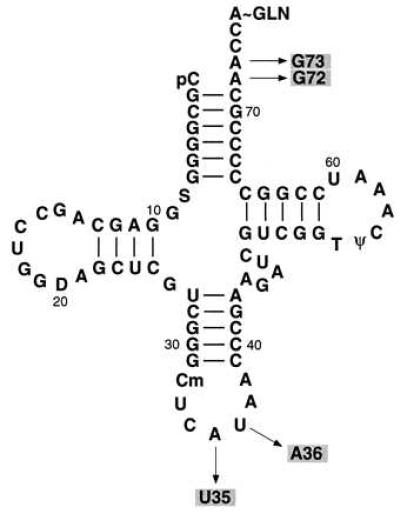
Cloverleaf structure of E. coli initiator tRNA2fMet. Arrows indicate the sites of mutation in the U35A36/G72G73 mutant initiator tRNA used for isolation of the suppressor mutants of MTF.
E. coli DH5α F′ cells carrying the gene for MTF in the pACDFMT plasmid (Fig. 3 Right) were mutagenized with N-methyl-N′-nitro-N-nitrosoguanidine, and plasmid DNA was isolated from the mutagenized culture. The pool of mutagenized pACDFMT plasmid DNA was then used to transform E. coli CA274 carrying the pRSVCATam1.2.5 plasmid (Fig. 3 Left), and the transformants were selected for growth on plates containing ampicillin, tetracycline, and chloramphenicol. Twenty chloramphenicol-resistant colonies were picked for analysis. Purification of the pACDFMT plasmids from these cells followed by retransformation of E. coli CA274 carrying the pRSVCATam1.2.5 plasmid showed that four of these conferred chloramphenicol resistance on the transformants. All four contained the same mutation in the MTF gene (details under Materials and Methods), a GGA → AGA change resulting in a Gly-41 to Arg-41 change in MTF. It is not known whether these mutants are siblings or independent. The G⋅C to A⋅T transition mutation in the MTF gene is consistent with the known preference of N-methyl-N′-nitro-N-nitrosoguanidine-induced mutations (21).
The chloramphenicol resistance phenotype of E. coli CA274 carrying the pRSVCATam1.2.5 and the mutant pACDFMT plasmids is also reflected in the increase in CAT activity in extracts of these cells. CAT activity in extracts of four independent transformants carrying the mutant MTF gene on plasmids increased by factors of 5.5–8.8 compared with those carrying the wild-type MTF on plasmids (data not shown).
Allele Specificity of Suppression.
The G41R suppressor mutant of MTF did not compensate for the formylation defect of another mutant tRNA, which is also inactive in initiation because it is an extremely poor substrate for formylation. The mutant (Mi:2/5) tRNA, derived from E. coli elongator methionine tRNA (30, 31), has the features in the acceptor stem important for formylation but is an extremely poor substrate for formylation because it is aminoacylated with lysine.
Generation of Gly-41 to Asp, Gln, Leu, and Lys Mutants in MTF by Site-Specific Mutagenesis.
To understand the mechanism by which the G41R mutant compensates for the formylation defect of the U35A36/G72G73 mutant initiator tRNA, other mutants were generated at this site by site-specific mutagenesis. The mutant MTF genes were introduced into E. coli CA274 carrying the pRSVCATam1.2.5 plasmid, and the transformants were screened for phenotypic resistance to chloramphenicol. The G41K mutant also rescued the formylation defect of the U35A36/G72G73 tRNA, whereas the G41D, G41L, and G41Q mutants did not. The G41D, G41L, and G41Q mutant MTFs were stably overproduced in E. coli CA274 to the same levels as wild-type MTF. These and other results (described below on the purified proteins) indicate that the presence of a positive charge instead of Gly at position 41 is necessary for suppression of the formylation defect of the U35A36/G72G73 mutant initiator tRNA.
Purification and Catalytic Properties of the Mutant Enzymes.
For detailed analysis of the properties of the mutant enzymes, it is necessary to obtain them in homogenous form and free of any contamination from wild-type MTF. This was achieved by expressing the wild-type and the mutant MTFs as His-tagged proteins and purifying them by chromatography on Co2+ chelate columns (24). The proteins thus obtained are essentially homogeneous (Fig. 5). Interestingly, the G41R and G41K mutants are somewhat sensitive to proteolysis and are cleaved to a small extent between amino acids 41 and 42 to yield a fragment (Fig. 5, indicated by an asterisk), deleted of amino acids 1–41 as determined by N-terminal sequence analysis of the fragment (data not shown).
Figure 5.
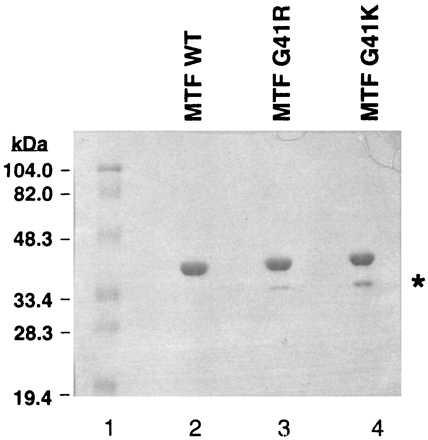
SDS/10% polyacrylamide gel electrophoresis of purified wild-type (WT) and the G41R and G41K suppressor mutants of MTF. The gel was stained with Coomassie blue R-250. ∗ indicates a truncated fragment of MTF.
The His-tagged MTFs contain an extension of four amino acids at the N terminus and eight at the C terminus (see Materials and Methods). Therefore, before their use for measurement of catalytic properties, we studied the effect of introducing the His-tag on the activity of MTF toward the wild-type initiator tRNA. Addition of the His-tag had a negligible effect—at the most a 1.5- to 2-fold effect—on the activity of wild-type MTF (data not shown).
Fig. 6 compares the relative activities in formylation of the wild-type MTF and the G41R and G41K suppressor mutants of MTF on the wild-type and the U35A36 and U35A36/G72G73 mutant initiator tRNAs. The U35A36 mutant initiator tRNA was also used as a substrate in these studies to examine whether any increased activity of the suppressor mutants of MTF on the U35A36/G72G73 mutant initiator tRNA could be because of their preference for the U35A36 anticodon sequence mutation or because of a preference for glutamine over methionine attached to the tRNA. The U35A36 mutant tRNA behaved similarly to the wild-type initiator tRNA in these assays and differently from the U35A36/G72G73 mutant tRNA. Both mutant enzymes showed slightly reduced rates (3- to 5-fold) of formylation of the wild-type and the U35A36 mutant initiator tRNA compared with the wild-type MTF (Fig. 6 A and C). In contrast, both the mutant enzymes showed significantly higher rates (26- to 27-fold) of formylation of the U35A36/G72G73 mutant initiator tRNA compared with the wild-type MTF (Fig. 6B). These results demonstrate: (i) that the phenotypic rescue of the activity of the U35A36/G72G73 mutant initiator tRNA in initiation by the suppressor mutations in MTF is because of an increased activity of the mutant MTFs on this tRNA and not because of a general nonspecific increase in rates of formylation by the mutant enzymes (32) and (ii) that the increased activity of the G41R and G41K enzymes is because of their compensation—specifically of the strong negative effect of the G72G73 mutation in the initiator tRNA on formylation.
Figure 6.
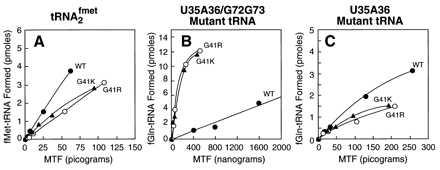
Activities in formylation of wild-type, G41R, and G41K mutant MTFs on wild-type initiator tRNA (A) and the U35A36/G72G73 (B) and U35A36 (C) mutant initiator tRNAs. Details of the assay are described under Materials and Methods.
Measurement of Steady-State Kinetic Parameters in Formylation of Wild-Type and Mutant Initiator tRNAs by Using Wild-Type and Mutant MTFs.
Table 1 compares the kinetic parameters in formylation of the wild-type and mutant initiator tRNAs by the wild-type and the mutant enzymes. The G41R and G41K enzymes behave essentially in an identical manner in all respects. With the wild-type and the U35A36 mutant initiator tRNAs as substrate, the kcat/Km for the G41R and G41K mutants is 3- to 5-fold lower compared with that for the wild-type enzyme. This is partly because of a slight increase in Km and a slight decrease in kcat. With the U35A36/G72G73 mutant tRNA as substrate, the kcat/Km for the G41R and G41K enzymes is 26- to 27-fold higher than for the wild-type enzyme. This is exclusively because of an effect on kcat, suggesting that the effect is because of stabilization of the transition state during the formylation reaction. Although the G41R and G41K mutants compensate for the strong formylation defect of the U35A36/G72G73 tRNA, the compensation is only partial. The kcat/Km in formylation of the U35A36/G72G73 tRNA by the G41R or the G41K enzymes is still approximately 2,000-fold lower than formylation of the wild-type tRNA by the same enzymes (Table 1).
Table 1.
Steady-state kinetic parameters in formylation using wild-type and mutant MTFs and wild-type and mutant initiator tRNAs
| tRNA used | Enzyme | Km, μM | kcat, s−1 | kcat/Km, μM−1⋅s−1 | Relative ratio |
|---|---|---|---|---|---|
| Wild type | WT | 0.56 ± 0.14 | 41.52 ± 13.85 | 72.48 | 1.00 |
| G41R | 0.66 ± 0.16 | 11.40 ± 0.159 | 18.49 | 0.25 | |
| G41K | 1.02 ± 0.03 | 14.60 ± 0.053 | 14.28 | 0.20 | |
| U35A36 | WT | 2.90 ± 1.35 | 8.85 ± 2.29 | 3.13 | 1.00 |
| G41R | 2.30 ± 1.12 | 2.78 ± 0.94 | 1.21 | 0.38 | |
| G41K | 2.01 ± 0.50 | 2.95 ± 0.56 | 1.64 | 0.52 | |
| U35A36/G72G73 | WT | 5.70 ± 0.89 | (1.48 ± 0.41) × 10−3 | 2.67 × 10−4 | 1.00 |
| G41R | 5.27 ± 1.18 | (3.45 ± 0.71) × 10−2 | 6.93 × 10−3 | 25.95 | |
| G41K | 5.19 ± 0.07 | (3.71 ± 0.28) × 10−2 | 7.14 × 10−3 | 26.69 |
Kinetic parameters were measured by using Lineweaver–Burk and Eadie–Hofstee or Hanes–Woolf plots. These gave basically the same numbers. The kinetic parameters listed are the average of four to six separate measurements. The tRNA concentrations used were 0.2–2 μM for wild-type (WT) tRNA and 0.5–6 μM for the U35A36 and the U35A36/G72G73 mutant tRNAs. The enzyme concentrations used varied from 0.025 to 0.106 nM for the wild-type tRNA, 0.34 to 0.65 nM for the U35A36 tRNA, and 0.12 to 1.55 μM for the U35A36/G72G73 tRNA. For calculation of relative ratio of kcat/Km, the kcat/Km of wild-type MTF for each tRNA is taken as 1.
DISCUSSION
The determinants for the specific recognition of the initiator tRNA by MTF are clustered mostly at the end of the acceptor stem (6–8). In attempts to identify the amino acid residues of MTF that interact with these determinants, we have isolated and identified suppressor mutations in MTF that compensate for the formylation defect of the U35A36/G72G73 mutant initiator tRNA. Suppression is allele-specific and requires the presence of a positively charged amino acid at position 41. The G41R and G41K suppressor mutants are 26- to 27-fold more active on the U35A36/G72G73 mutant initiator tRNA than the wild-type MTF. In contrast, these enzymes are 3- to 5-fold less active than the wild-type enzyme on the wild-type initiator tRNA. Besides providing direct in vitro evidence for the phenotypic suppression seen in vivo, these results show that the phenotypic suppression is not because of a general nonspecific increase in activity of the suppressor mutants. Also, the finding that the suppressor mutants are only 3- to 5-fold less active than the wild-type enzyme on the wild-type tRNA suggests that the suppression observed is not because of a disruption of normal tRNA recognition leading to a general loss in selectivity of the enzyme for the tRNA (33).
The mutant tRNA used for selection of the suppressor mutations in MTF has changes in the anticodon sequence (U35A36) and in the acceptor stem (G72G73). Whereas the G72G73 mutation is undoubtedly responsible for the formylation defect of the tRNA, the U35A36 mutation alone makes the tRNA a poor substrate (approximately 20-fold, Table 1) for MTF. Therefore, it was possible that the suppressor mutants compensate for the effect of the U35A36 mutation or for the fact that this mutant tRNA is aminoacylated with glutamine. Our finding that the U35A36 mutant tRNA, also aminoacylated with glutamine, behaves identically to the wild-type tRNA in its properties as a substrate for the wild-type and the G41R and G41K suppressor mutants of MTF (Fig. 6, A and C) rules out this possibility and shows that the suppressor mutations compensate specifically for the strong negative effect of the G72G73 mutation in the acceptor stem of the tRNA on formylation. A direct test of this with just the G72G73 mutant tRNA as a substrate is, unfortunately, not possible because this tRNA is an extremely poor substrate for methionyl-tRNA synthetase and cannot be aminoacylated with methionine to any appreciable extent (8).
The chemical nature of the interaction between Arg-41 or Lys-41 of MTF and the acceptor stem of the mutant tRNA is not known. The U35A36/G72G73 mutant initiator tRNA is defective in formylation because it is lacking one of the most critical determinants for formylation. Extensive mutagenic studies on the tRNA have shown that nucleotides 1 and 72 at the end of the acceptor stem must be unpaired for formylation. The nature of the bases 1 and 72 themselves appears not to be important (7, 8, 34). The three-dimensional structure of the wild-type initiator tRNA (35) and comparative NMR structural analyses of acceptor stem microhelices corresponding to tRNAs, which are either substrates for MTF or not (36), suggest that MTF prefers a tRNA structure in which bases 1 and 72 are unpaired and the 3′ end of the tRNA is folded back toward the 5′ end of the tRNA. The U35A36/G72G73 mutant initiator tRNA has a C1⋅G72 base pair. In addition, it has G73 in the discriminator position, which makes it an even worse substrate for MTF, presumably because the mutant tRNA adopts a structure quite different from that required for formylation (37). Therefore, suppression of the formylation defect of the U35A36/G72G73 mutant tRNA by the Arg-41 and Lys-41 mutant enzymes requires that these enzymes facilitate melting of the C1⋅G72 base pair and/or compensate for the effect of G73 so that the amino acid attached to the 3′ end of the tRNA can fit into the catalytic pocket of MTF. Further evidence for the role of Arg-41 mutant enzyme in facilitating melting of the C1⋅G72 base pair comes from the finding that the Arg-41 enzyme also suppresses the formylation defect of the U35A36/G72 mutant initiator tRNA (data not shown). This mutant tRNA is a poor substrate for formylation only because it has a C1⋅G72 base pair at the end of the acceptor stem.
The approximately 26- to 27-fold increase in kcat/Km (this difference is exclusively because of the effect on kcat) of the G41R and G41K enzymes over the wild-type enzyme with the U35A36/G72G73 mutant initiator tRNA as substrate (Table 1) corresponds to a stabilization of the transition state by 2 kcal/mol (1 kcal = 4.18 kJ) (ΔG = RTln[(kcat/Km)wild type/(kcat/Km)mutant]) (38). This means that the interaction of the mutant enzymes with the U35A36/G72G73 mutant initiator tRNA in the transition state is worth 2 kcal/mol more than that of the wild-type MTF. This difference is higher than the energy of one uncharged hydrogen bond (39). The finding that the Gln-41 mutant is inactive as a suppressor is consistent with this. It is possible that the G41R and G41K enzymes help disrupt the C1⋅G72 base pair and increase kcat by forming a hydrogen bond or ionic bond with one or more of the phosphates at the end of the acceptor stem. Additionally, the suppressor mutants could stabilize the transition state by interacting directly with G72 or G73 through hydrogen bond formation with O6 and/or N7 of the guanine residues.
Because a single mutation within the 16-amino acid insertion in MTF suppresses the very strong negative effect of the G72G73 mutation on the tRNA, this region of MTF most likely interacts directly with some of the critical determinants for formylation in the acceptor stem of the tRNA (23, 40). Although this conclusion is based on the isolation and analysis of suppressor mutations at a single position within the 16-amino acid insertion, several other observations from our laboratory and from that of Blanquet and co-workers (see below) support this conclusion. (i) The 16-amino acid insertion is found in all of the MTFs of known sequence, including that in yeast mitochondria, where the insertion sequence is longer. The sequence within the 16 amino acids is quite basic and has several highly conserved residues (Fig. 2). (ii) Mutations within this region greatly affect the activity of the mutant enzymes. For example, mutation of Arg-42, the conserved amino acid adjacent to the site of suppressor mutation (Fig. 2), to Leu or Ala lowers kcat/Km of the enzyme by more than 1000-fold (V.R., S.G., and U.L.R., unpublished observations; Y. Mechulam, M. Panvert, E. Schmitt, and S. Blanquet, personal communication). (iii) The crystal structure of MTF shows that the 16-amino acid insertion loop is proximal to the catalytic site of MTF and is in a position to interact with the acceptor stem of the tRNA (11). (iv) The loop is mostly unstructured and uniquely susceptible to cleavage by proteases, and the cleavage product is inactive in formylation (D. Mangroo, S.G., and U.L.R., unpublished results; ref. 11; also see Fig. 5). However, in the presence of fMet-tRNA, the product of MTF, the loop is protected against proteolytic cleavage (11). It is possible that in the presence of the cognate tRNA, the mobile loop adopts a more rigid structure similar to the situation in E. coli tyrosyl-tRNA synthetase, in which two mobile loops come together in the transition state during tyrosyl-AMP formation in an induced-fit mechanism (41).
The conclusion that the amino acids in the 16-amino acid insertion loop of MTF interact specifically with determinants for formylation in the acceptor stem of the initiator tRNA does not mean that this is the only region of MTF that contacts the determinants in the acceptor stem. MTF also contains a C-terminal extension over GARF (Fig. 1). This C-terminal extension can bind to tRNA on its own, and crosslinking experiments have shown that Lys-206, which precedes this part of MTF comes close to the 3′-terminal A of the tRNA (10, 11). It is possible that the C-terminal extension also interacts with the determinants in the acceptor stem. However, binding of the C-terminal fragment of MTF, which contains several basic and aromatic residues on the surface of the enzyme, to tRNA is nonspecific with respect to the tRNA substrate (11). This, combined with the fact that mutations of most of these basic residues individually have only small effects on enzyme activity (Y.L., S.G., and U.L.R., unpublished results; Y. Mechulam, M. Panvert, E. Schmitt and S. Blanquet, personal communication), suggests that the C-terminal extension may contribute mostly nonspecific binding energy, whereas the amino acid residues in the insertion loop provide the necessary specificity for tRNA.
Finally, comparison of MTF and GARF sequences from a variety of sources suggests that MTF has a modular structure in which polypeptide sequences necessary for tRNA binding and discrimination have been inserted into an ancestral enzyme, which could bind N10-formyltetrahydrofolate and transfer the formyl group to simple acceptors. In this respect, it is similar to aminoacyl-tRNA synthetases, which also interact with tRNAs in a highly specific manner. In aminoacyl-tRNA synthetases, large polypeptide sequences are thought to have been inserted during evolution into a catalytic core, which had the ability to form aminoacyl-AMP (42–45). Depending on the aminoacyl-tRNA synthetase and source of the enzyme, these polypeptide sequences have been inserted into the N terminus, the C terminus, and/or within the catalytic core. These insertions confer on the aminoacyl-tRNA synthetase the properties of binding and aminoacylation of a specific tRNA, editing of misacylated tRNAs, and oligomerization of the protein. Domains of proteins involved in recognition of the acceptor stem region, the anticodon region, and other parts of the tRNA molecule are thought to have been inserted at different times during evolution of the translational machinery (42). A striking example of tRNA specificity conferred by these insertions comes from the work of Auld and Schimmel (46). These authors have shown that the ability of two closely related aminoacyl-tRNA synthetases such as isoleucyl-tRNA synthetase and methionyl-tRNA synthetase to discriminate among the corresponding tRNA anticodons, which differ by a single nucleotide, resides in a 10-amino acid loop sequence that is part of a larger insertion in these enzymes.
Acknowledgments
We thank Dr. Paul Schimmel for comments and suggestions on the manuscript and Annmarie McInnis for patience, cheerfulness, and care in the preparation of this manuscript. We thank Dr. Dev Mangroo for initiation of work on isolation of suppressor mutants of MTF. This work was supported by National Institutes of Health Grant GM17151.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: MTF, methionyl-tRNA formyltransferase; GARF, glycinamide ribonucleotide formyltransferase; CAT, chloramphenicol acetyltransferase; GlnRS, glutaminyl-tRNA synthetase.
The amino acid numbering of MTF is different from that used previously by us (10). It has been changed to conform with the numbering system used by Schmitt et al. (11) in their description of the crystal structure of MTF. This numbering system (11) is based on the sequence of the protein, which like most proteins lacks the N-terminal methionine encoded in the gene.
The E. coli GARF also contains a loop between the second β-strand and the second α-helix. However, the loop contains only 3 amino acids instead of the 16 in the E. coli and other MTFs shown on Fig. 2. Yeast mitochondrial MTF has a longer insertion of 37 amino acids.
References
- 1.Kozak M. Microbiol Rev. 1983;47:1–45. doi: 10.1128/mr.47.1.1-45.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gualerzi C O, Pon C L. Biochemistry. 1990;29:5881–5889. doi: 10.1021/bi00477a001. [DOI] [PubMed] [Google Scholar]
- 3.RajBhandary U L. J Bacteriol. 1994;176:547–552. doi: 10.1128/jb.176.3.547-552.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marcker K, Sanger F. J Mol Biol. 1964;8:835–840. doi: 10.1016/s0022-2836(64)80164-9. [DOI] [PubMed] [Google Scholar]
- 5.Dickerman H W, Steers E, Jr, Redfield B G, Weissbach H. J Biol Chem. 1967;242:1522–1525. [PubMed] [Google Scholar]
- 6.Lee C P, Seong B L, RajBhandary U L. J Biol Chem. 1991;266:18012–18017. [PubMed] [Google Scholar]
- 7.Guillon J-M, Meinnel T, Mechulam Y, Lazennec C, Blanquet S, Fayat G. J Mol Biol. 1992;224:359–367. doi: 10.1016/0022-2836(92)91000-f. [DOI] [PubMed] [Google Scholar]
- 8.Lee C-P, Dyson M R, Mandal N, Varshney U, Bahramian B, RajBhandary U L. Proc Natl Acad Sci USA. 1992;89:9262–9266. doi: 10.1073/pnas.89.19.9262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guillon J M, Mechulam Y, Schmitter J M, Blanquet S, Fayat G. J Bacteriol. 1992;174:4294–4301. doi: 10.1128/jb.174.13.4294-4301.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gite S, RajBhandary U L. J Biol Chem. 1997;272:5305–5312. doi: 10.1074/jbc.272.8.5305. [DOI] [PubMed] [Google Scholar]
- 11.Schmitt E, Blanquet S, Mechulam Y. EMBO J. 1996;15:4749–4758. [PMC free article] [PubMed] [Google Scholar]
- 12.Almassy R J, Janson C A, Kan C-C, Hostomska Z. Proc Natl Acad Sci USA. 1992;89:6114–6118. doi: 10.1073/pnas.89.13.6114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klein C, Chen P, Arevalo J J, Stura E A, Marolewski A, Warren M S, Benkovic S J, Wilson I. J Mol Biol. 1995;249:153–175. doi: 10.1006/jmbi.1995.0286. [DOI] [PubMed] [Google Scholar]
- 14.Buchanan J M. In: Amino Acids, Fermentations and Nucleic Acids: A Symposium. Snell E E, editor. Palo Alto, CA: Annual Reviews; 1982. pp. 57–92. [Google Scholar]
- 15.Warren M S, Marolewski A E, Benkovic S J. Biochemistry. 1996;35:8855–8862. doi: 10.1021/bi9528715. [DOI] [PubMed] [Google Scholar]
- 16.Varshney U, RajBhandary U L. Proc Natl Acad Sci USA. 1990;87:1586–1590. doi: 10.1073/pnas.87.4.1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Varshney U, Lee C-P, Seong B L, RajBhandary U L. J Biol Chem. 1991;266:18018–18024. [PubMed] [Google Scholar]
- 18.Yanisch-Perron C, Viera J, Messing J. Gene. 1985;33:103–119. doi: 10.1016/0378-1119(85)90120-9. [DOI] [PubMed] [Google Scholar]
- 19.Woodcock D M, Crowther P J, Doherty J, Jefferson S, DeCruz E, Noyer-Weidner M, Smith S S, Michael M Z, Graham M W. Nucleic Acids Res. 1989;17:3469–3478. doi: 10.1093/nar/17.9.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mangroo D, RajBhandary U L. J Biol Chem. 1995;270:12203–12209. doi: 10.1074/jbc.270.20.12203. [DOI] [PubMed] [Google Scholar]
- 21.Miller J H. A Short Course in Bacterial Genetics. Plainview, NY: Cold Spring Harbor Lab. Press; 1992. pp. 293–297. [Google Scholar]
- 22.Ramesh V, Varshney U, RajBhandary U L. RNA. 1997;3:1220–1232. [PMC free article] [PubMed] [Google Scholar]
- 23.Meinnel T, Mechulam Y, Le Corre D, Panvert M, Blanquet S, Fayat G. Proc Natl Acad Sci USA. 1991;88:291–295. doi: 10.1073/pnas.88.1.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hochuli E, Döbeli H, Schacher A. J Chromatogr. 1987;411:177–184. doi: 10.1016/s0021-9673(00)93969-4. [DOI] [PubMed] [Google Scholar]
- 25.Bradford M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 26.Mandal N, RajBhandary U L. J Bacteriol. 1992;174:7827–7830. doi: 10.1128/jb.174.23.7827-7830.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.RajBhandary U L, Ghosh H P. J Biol Chem. 1969;244:1104–1113. [PubMed] [Google Scholar]
- 28.Schulman L H, Pelka H. Biochemistry. 1985;24:7309–7314. doi: 10.1021/bi00346a043. [DOI] [PubMed] [Google Scholar]
- 29.Yamao F, Inokuchi H, Cheung A, Ozeki H, Söll D. J Biol Chem. 1982;257:11639–11643. [PubMed] [Google Scholar]
- 30.Varshney U, Lee C P, RajBhandary U L. Proc Natl Acad Sci USA. 1993;90:2305–2309. doi: 10.1073/pnas.90.6.2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li S, Kumar N V, Varshney U, RajBhandary U L. J Biol Chem. 1996;271:1022–1028. doi: 10.1074/jbc.271.2.1022. [DOI] [PubMed] [Google Scholar]
- 32.Rogers M J, Adachi T, Inokuchi H, Söll D. Proc Natl Acad Sci USA. 1994;91:291–295. doi: 10.1073/pnas.91.1.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sherman J M, Thomann H-U, Söll D. J Mol Biol. 1996;256:818–828. doi: 10.1006/jmbi.1996.0128. [DOI] [PubMed] [Google Scholar]
- 34.Lee C P, Mandal N, Dyson M R, RajBhandary U L. Proc Natl Acad Sci USA. 1993;90:7149–7152. doi: 10.1073/pnas.90.15.7149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Woo N H, Roe B, Rich A. Nature (London) 1980;286:346–351. doi: 10.1038/286346a0. [DOI] [PubMed] [Google Scholar]
- 36.Puglisi E V, Puglisi J D, Williamson J R, RajBhandary U L. Proc Natl Acad Sci USA. 1994;91:11467–11471. doi: 10.1073/pnas.91.24.11467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dyson M, Mandal N, RajBhandary U L. Biochimie. 1993;75:1051–1060. doi: 10.1016/0300-9084(93)90004-c. [DOI] [PubMed] [Google Scholar]
- 38.Wilkinson A J, Fersht A R, Blow D M, Winter G. Biochemistry. 1983;22:3581–3586. doi: 10.1021/bi00284a007. [DOI] [PubMed] [Google Scholar]
- 39.Fersht A R. Trends Biochem Sci. 1987;12:301–304. [Google Scholar]
- 40.Miller W T, Hou Y-M, Schimmel P. Biochemistry. 1991;30:2635–2641. doi: 10.1021/bi00224a011. [DOI] [PubMed] [Google Scholar]
- 41.Fersht A R, Knill-Jones J W, Bedouelle H, Winter G. Biochemistry. 1988;27:1581–1587. doi: 10.1021/bi00405a028. [DOI] [PubMed] [Google Scholar]
- 42.Schimmel P, de Pouplana L R. Cell. 1995;81:983–986. doi: 10.1016/s0092-8674(05)80002-9. [DOI] [PubMed] [Google Scholar]
- 43.Cusack S. Nat Struct Biol. 1995;2:824–831. doi: 10.1038/nsb1095-824. [DOI] [PubMed] [Google Scholar]
- 44.Moras D, Arnez J G. Trends Biochem Sci. 1997;22:211–216. doi: 10.1016/s0968-0004(97)01052-9. [DOI] [PubMed] [Google Scholar]
- 45.Lin L, Hale S P, Schimmel P. Nature (London) 1996;384:33–34. doi: 10.1038/384033b0. [DOI] [PubMed] [Google Scholar]
- 46.Auld D S, Schimmel P. Science. 1995;267:1994–1996. doi: 10.1126/science.7701322. [DOI] [PubMed] [Google Scholar]