

Abstract
HIV type 1 (HIV-1) specifically uses host cell tRNALys-3 as a primer for reverse transcription. The 3′ 18 nucleotides of this tRNA are complementary to a region on the HIV RNA genome known as the primer binding site (PBS). HIV-1 has a strong preference for maintaining a lysine-specific PBS in vivo, and viral genomes with mutated PBS sequences quickly revert to be complementary to tRNALys-3. To investigate the mechanism for the observed PBS reversion events in vitro, we examined the capability of the nucleocapsid protein (NC) to anneal various tRNA primer sequences onto either complementary or noncomplementary PBSs. We show that NC can anneal different full-length tRNAs onto viral RNA transcripts derived from the HIV-1 MAL or HXB2 isolates, provided that the PBS is complementary to the tRNA used. In contrast, NC promotes specific annealing of only tRNALys-3 onto an RNA template (HXB2) whose PBS sequence has been mutated to be complementary to the 3′ 18 nt of human tRNAPro. Moreover, HIV-1 reverse transcriptase extends this binary complex from the proline-specific PBS. The formation of the noncomplementary binary complex does not occur when a chimeric tRNALys/Pro containing proline-specific D and anticodon domains is used as the primer. Thus, elements outside the acceptor-TΨC domains of tRNALys-3 play an important role in preferential primer use in vitro. Our results support the hypothesis that mutant PBS reversion is a result of tRNALys-3 annealing onto and extension from a PBS that specifies an alternate host cell tRNA.
HIV type 1 (HIV-1) selectively packages host cell tRNALys-3 for use as a replication primer (1–3). In virions, the 18 nt at the 3′ end of tRNALys-3 are believed to be annealed to a complementary sequence located in the 5′ untranslated region (U5) of the viral genome called the primer binding site (PBS) (Fig. 1). Extension of the 3′ hydroxyl at the terminus of the tRNA primer by reverse transcriptase (RT) produces an initial product known as the minus-strand strong-stop DNA [(−)SS cDNA] (Fig. 1, step 1). Synthesis of a full-length double-stranded DNA copy of the viral genome is then completed through a complex series of events that are not yet completely understood (Fig. 1, steps 2–6).
Figure 1.
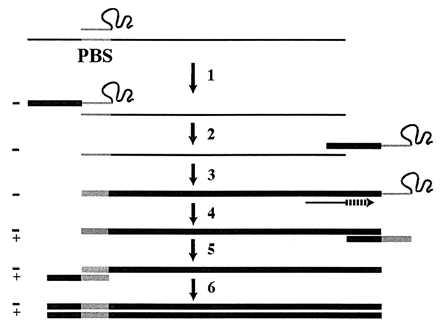
Current model of retroviral replication. (Step 1) After annealing of the tRNA primer to the PBS, RT extends the 3′ end of the tRNA by using the genomic RNA (thin line) as a template to synthesize (−)SS cDNA (thick line) and degrades the RNA template. (Step 2) By virtue of base pair complementarity, the (−)SS cDNA translocates to the 3′ end of the RNA genome. (Step 3) Nearly full-length −cDNA is synthesized, terminating at the PBS. (Step 4) Strong-stop plus-strand cDNA synthesis is initiated from an RNaseH-resistant polypurine tract in the viral RNA. During this step, the 3′ 18 nt of the tRNA primer are copied. (Step 5) Base pair complementarity between the strong-stop plus-strand cDNA and the −cDNA mediates the second strand transfer. (Step 6) Completion of full-length double-stranded proviral cDNA synthesis by RT.
In vivo analyses have shown that HIV-1 mutants with altered PBSs complementary to other tRNA species replicate slowly (4–6) and eventually revert to the wild-type sequence specific for tRNALys-3 (4–8). One mechanism of PBS sequence reversion that has been proposed involves preferential use of tRNALys-3 even when the PBS is complementary to an alternate tRNA (6, 9, 10). According to the current model of retroviral replication, the sequence at the 3′ end of the tRNA primer is copied into the newly transcribed plus-strand strong-stop cDNA to regenerate the PBS (11) (Fig. 1, step 4). Thus, in the event that tRNALys-3 is used to initiate reverse transcription from a noncomplementary PBS, after just one round of replication, 50% of the proviruses would contain a PBS complementary to tRNALys-3. The viruses with a Lys-3-specific PBS have a growth advantage (4, 6, 7) and therefore would quickly predominate. Because abundant packaging of tRNALys-3 into the virion is independent of the PBS sequence (12), it may be reasonable to expect that a mutant PBS would revert to be Lys-specific. However, factors other than tRNALys-3 abundance may also play a role in mutant PBS reversion (13, 14). Moreover, whereas mutant PBS reversion implies that tRNALys-3 was preferentially used, no direct evidence for this mechanism thus far has been presented.
In vivo the nucleocapsid protein (NC) is believed to mediate primer/template annealing. It has also been demonstrated that NC can place tRNALys-3 onto the PBS for extension by RT in vitro (15–18). In this report, we describe an in vitro system designed to closely mimic in vivo primer/template annealing using NC and (−)SS cDNA synthesis by RT. Using this system, we carried out experiments using various tRNAs as primers along with complementary and noncomplementary PBS sequences to probe the mechanism of mutant PBS reversion.
MATERIALS AND METHODS
Protein Purification.
The bacterially expressed HIV-1 RT was purified according to the procedure previously reported (19). Plasmid pNCH6, which encodes the gene for HIV-1 NCp15 with poly-histidine at the C terminus, was obtained as a gift from S. Le Grice of Case Western Reserve University. pNCH6 and pREP4 (Qiagen) were cotransformed into protease-deficient Escherichia coli strain BL21(DE3). Purification of the his-tagged NC fusion protein was performed by Ni-chelate affinity chromatography essentially as described (20), except that 10 mM 2-mercaptoethanol and 10 μM ZnCl2 were maintained in all buffered solutions during purification. NC was further purified by chromatographing on a MonoQ FPLC column (Pharmacia) and eluted with an NaCl gradient (0.01–0.25 M) in 25 mM Tris⋅HCl (pH 7.5), 15% glycerol, 75 μM ZnCl2, and 5 mM DTT. T7 RNA polymerase was prepared as previously described (21).
Plasmids.
To prepare unmodified human tRNALys-3 and chimeric tRNALys/Pro, a DNA insert containing a T7 RNA polymerase promoter and the gene for the desired tRNA was made by ligating six chemically synthesized oligodeoxynucleotides together as previously described (22). The insert was ligated into EcoRI/BamHI-digested plasmid pVAL119 (a gift from Jack Horowitz of Iowa State University) (23) to generate plasmids pK-F119 and pK/P-F119. pHIVCG4 (containing the 5′ portion of the HIV-1 MAL isolate genome) and pHIV-PBS (containing the 5′ portion of the HXB2 isolate genome) were gifts from Jean-Luc Darlix of LaboRetro, Institut National de la Santé et de la Recherche Médicale, Lyon, France, and Lawrence Kleiman at the Lady Davis Institute for Medical Research–Jewish General Hospital, Montreal, Canada, respectively. Using overlap-extension PCR (24), the PBS sequences of pHIVCG4 and pHIV-PBS were mutated to a sequence complementary to the 3′ 18 nt of E. coli tRNAPro-3 (pP-PBS) and human tRNAPro (pHIV-hmPPBS), respectively. To incorporate a 4-nt signature tag into pHIV-hmPPBS, the plasmid was cut at a unique AflII site, blunt-ended by Klenow fragment, and religated. The sequences of all the mutant constructs were confirmed by Sanger’s dideoxy sequencing method (25).
RNA Preparation.
FokI-digested pK-F119 and pK/P-F119 were used to prepare human tRNALys-3 and chimeric tRNALys/Pro, respectively, by in vitro transcription (26). E. coli and human tRNAPro were prepared as described (27, 28). In vitro transcription reactions were supplemented with 17 mCi/ml [α-32P]GTP when preparing internally radiolabeled tRNAs. HIV-1 MAL RNA templates were generated by in vitro transcription using RsaI-digested pHIVCG4 and pP-PBS. HIV-1 HXB2 RNA templates were similarly prepared using HaeIII-digested pHIV-PBS and pHIV-hmPPBS. Concentrations of RNA were determined using the following extinction coefficients: tRNA, 60.4 × 104 M−1; HIV MAL RNA template (311 nt), 2.32 × 106 M−1; and HIV HXB2 RNA template (394 nt), 3.56 × 106 M−1. Prior to use, tRNAs were renatured in the presence of MgCl2 as previously described (27). HIV-1 RNA templates were renatured by heating to 85°C for 2.5 min in 10 mM Tris⋅HCl/1 mM EDTA (pH 8.0), followed by incubation at 50°C for 8 min. MgCl2 was then added to 8 mM, and the mixture was incubated at 37°C for 10 min and finally placed on ice.
NC-Mediated Annealing Assays.
NCp15-assisted annealing of [32P]tRNA to the RNA template was performed as described (29), except that MgCl2-renatured tRNA and MgCl2-renatured HIV-RNA template were used. The NC concentration used was either 4 μM (MAL) or 5.1 μM (HXB2) to maintain a total ribonucleotides-to-NC ratio of 8. Primer/template complexes were fractionated from free tRNA by 10% SDS/PAGE (19:1/acrylamide:bisacrylamide) and visualized by autoradiography.
PCR-Based RT Extension Assay.
In a reaction volume of 32 μl, 7.2 pmol tRNA and 17.6 pmol HIV-1 RNA template were annealed by NC as described above. The contents of the reaction (80 μl) were then adjusted to 40 mM Tris⋅HCl (pH 7.5), 6 mM MgCl2, 5 mM DTT, 30 mM NaCl, 0.2 mM of each dNTP, 89.6 nM tRNA, and 219 nM HIV RNA template. Samples were preincubated at 37°C for 2 min. Reactions were initiated with either 40 nM or 400 nM RT, as indicated in the figure legends. To select full-length (−)SS cDNA products and to reverse transcribe the extended tRNA primer, the following DNA primers (0.8 pmol) were used: cDNAP1 for MAL templates, 5′-GGGTCTCTCTTGTTAGAC-3′; HXB5P for HXB2 templates, 5′-GGGAGACCGGCAGATC-3′. After a 20-min incubation, the reaction was quenched with 0.5 volume of 0.1 M EDTA (pH 8.0), followed by the addition of SDS (1% wt/vol) and proteinase K (0.4 mg/ml). After incubation at 37°C for 10 min, proteins were removed by phenol-chloroform extraction. Samples were desalted by G-50 Sephadex spin columns (Pharmacia or Sigma), dried, and used in PCR amplification (100 μl) by using cDNAP1 or HXB5P and another primer specific to the 5′ portion of the tRNA used (P-Lys for tRNALys-3 in MAL reactions: 5′-TAATACGACTCACTATAGCCCGGATAGCTCAGT-3′; HMLYS5P and HMPRO5P in HXB2 reactions: 5′-GCCCGGATAGCTCAGTC-3′ and 5′-GGCTCGTTGGTCTAGGG-3′, respectively). PCR was performed using a Perkin–Elmer GeneAmp PCR System 2400. For amplification of the MAL reactions, the first PCR cycle was carried out as follows: 94°C (1 min), 43°C (30 sec), and 72°C (1 min). This was followed by 29 cycles of reactions at 94°C (1 min), 50°C (30 sec), and 72°C (1 min). The amplification conditions for the HXB2 reactions were the same, except the pre-PCR cycle was omitted. DNA samples were cloned into the EcoRV site of LITMUS 29 (New England Biolabs) and sequenced using Sequenase version 2.0 (United States Biochemicals).
RESULTS
Sequences of two of the tRNA primers and the relevant regions of the HIV-1 RNA templates used in this study are shown in Fig. 2. We also prepared E. coli tRNAPro-3 and human tRNAPro (not shown). The chimeric tRNA construct contains human tRNALys-3-specific acceptor-TΨC stem-loop sequences and D-arm and anticodon-arm sequences derived from E. coli tRNAPro-3 (30) (Fig. 2A Right). This construct maintains the important tertiary interactions of a tRNA and is expected to fold into an L-shaped structure. The wild-type HIV-1 MAL and HXB2 RNA templates used in this work have PBSs complementary to human tRNALys-3 and chimeric tRNALys/Pro (Fig. 2B). Mutant templates with PBSs complementary to E. coli tRNAPro-3 (MAL) and human tRNAPro (HXB2) were also prepared (Fig. 2B).
Figure 2.

Sequences of tRNAs (30) and HIV-1 templates (45) used in this study. (A) Sequences of human tRNALys-3 (Left) and chimeric tRNALys/Pro (Right). The chimeric tRNA contains a human tRNALys-3-specific acceptor-TΨC domain (shaded) and D-anticodon stem-loop domains corresponding to E. coli tRNAPro-3. (B) Sequence comparison between the 5′ region of the HIV-1 MAL and HXB2 RNA genomes. The Lys-PBS and the pseudo-PBS are shaded and boxed, respectively. Nucleotides are numbered according to the MAL isolate. The PBSs of the HXB2 and MAL genomes were mutated to be specific for human tRNAPro and E. coli tRNAPro-3, respectively.
NC-Mediated tRNA Annealing to the HIV-1 MAL Template.
We first examined the ability of NC to mediate annealing of different tRNA primers to either complementary or noncomplementary templates derived from the MAL isolate (Fig. 3A). In the absence of NC, none of the three tRNAs tested (human tRNALys-3, chimeric tRNALys/Pro, or E. coli tRNAPro-3) was annealed to a complementary PBS (Fig. 3A, lanes 1, 4, and 7). Upon addition of NC and in the presence of a complementary PBS sequence, two distinct complexes that migrated near the top of the gel were obtained with each of the three tRNAs (Fig. 3A, lanes 2, 5, and 8). Densitometry scans (data not shown) indicated that approximately 18% binary complex formed when tRNALys-3 and tRNALys/Pro were annealed to a noncomplementary Pro-PBS in the presence of NC (Fig. 3A, lanes 3 and 6). In contrast, no binary complex was detected when E. coli tRNAPro-3 was incubated with NC in the presence of a noncomplementary Lys-PBS (Fig. 3A, lane 9).
Figure 3.
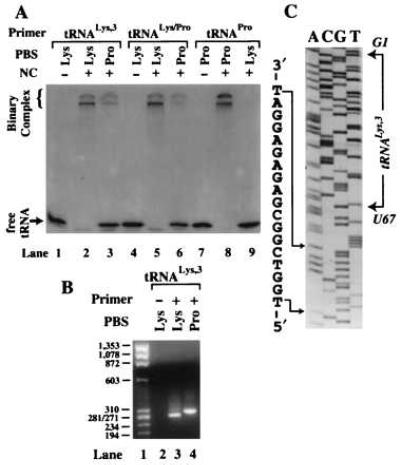
NC-mediated annealing and RT extension using the HIV-1 MAL template. (A) Polyacrylamide gel showing the results of NC-mediated annealing of 32P-labeled tRNA primers to the 311-nt template. The − and + signs represent the absence and presence of NC in the reactions. The identity of each primer/template combination used is indicated above the lanes. (B) Agarose gel (2%) showing PCR-amplified primer extension products. The tRNA primers were extended by either 40 nM (lane 3) or 400 nM (lanes 2 and 4) RT. Lane 1, DNA size marker; the length indicated is in base pairs. Lanes 2–4, extension was carried out in the absence (−) or presence (+) of tRNALys-3 primer using a template complementary to either human tRNALys-3 (Lys) or E. coli tRNAPro-3 (Pro). (C) DNA sequence of the PCR product obtained in lane 4 (B). Nucleotides corresponding to the E. coli Pro-PBS are explicitly shown. The choice of sequencing primer resulted in obtaining the sense sequence of the RNA template and the antisense sequence of tRNALys-3. The position of nucleotides G1 and U67 of tRNALys-3 are shown in italics to indicate that they are complementary to the sequence shown in the gel.
To determine the exact location on the MAL template to which tRNALys-3 was annealed by NC, the complementary and noncomplementary binary complexes were extended by HIV-1 RT. The fully extended products were detected by PCR (Fig. 3B). A PCR-based assay for (−)SS cDNA was used to facilitate cloning and sequencing of the extended products. A direct assay involving RT extension of 32P-labeled tRNA would not allow us to readily establish the site of annealing and extension. When tRNALys-3 and the complementary RNA template were used, the major PCR product was approximately 270 bp in length (Fig. 3B, lane 3). Sequence analysis of this product showed that priming was initiated from the Lys-PBS, as expected (data not shown). A slightly longer product (approximately 300 bp in length) was obtained on extension of tRNALys-3 annealed to an RNA template containing a Pro-PBS (Fig. 3B, lane 4). Sequencing of this product showed that (−)SS cDNA synthesis was initiated at nucleotide 210 of the MAL template from an internal position of the tRNA primer (U67) (Fig. 3C). Nucleotides 58–67 of tRNALys-3 are perfectly complementary to nucleotides 211–220 of the HIV-1 MAL isolate (31). Our results show that on mutation of the authentic PBS to a sequence complementary to tRNAPro-3, tRNALys-3 is annealed by NC to this alternate or “pseudo-PBS” sequence (Fig. 2B).
NC-Mediated tRNA Annealing to the HIV-1 HXB2 Template.
Sequence alignments reveal that the pseudo-PBS is in a region of the MAL isolate (nucleotides 211–233) that is absent in all other HIV-1 isolates sequenced to date (31). Therefore, we decided to test NC-mediated annealing using an RNA template derived from the HIV-1 HXB2 isolate. This isolate lacks the pseudo-PBS sequence and is also commonly used in in vivo studies (4, 5, 7–9, 14). Using this construct, we mutated the PBS to be complementary to the 3′ 18 nt of human tRNAPro (Fig. 2B). The capability of NC to anneal tRNALys-3, human tRNAPro, and the chimeric tRNALys/Pro to both complementary and noncomplementary PBS sequences was assessed. As expected, all three tRNAs were efficiently annealed to a complementary HXB2 template (Fig. 4, lanes 2, 5, and 8). The extent of annealing determined by densitometry was again very similar (≈90%) in all three cases (data not shown). Interestingly, only tRNALys-3 could be annealed to a template containing a noncomplementary Pro-PBS (Fig. 4, lane 3). Densitometry indicated that 34% (average of three independent trials) binary complex formed in this case. Human tRNAPro and the chimeric tRNALys/Pro failed to anneal to the noncomplementary PBS sequences (<1%) (Fig. 4, lanes 6 and 9). Significantly, the chimeric tRNA did not anneal to the Pro-PBS, even though it has the same percent complementarity to the Pro-PBS as tRNALys-3. This result suggests that sequences outside the acceptor-TΨC stem-loop domain of tRNALys-3 may help mediate noncomplementary binary complex formation.
Figure 4.
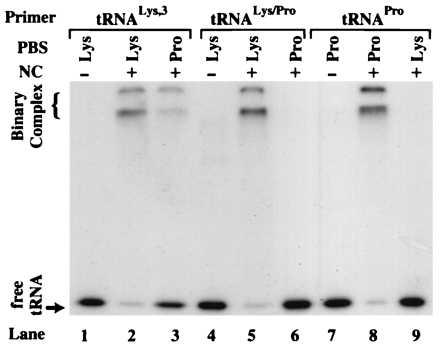
NC-mediated annealing using the HIV-1 HXB2 template. Polyacrylamide gel showing the results of NC-mediated annealing of 32P-labeled tRNA primers to the 394-nt template. The identity of each primer/template combination used is indicated above the lanes. The − and + signs represent the absence and presence of NC in the reactions.
To determine the location on the HXB2 RNA template to which tRNALys-3 was annealed, the NC-annealed tRNA primer/RNA template binary complexes were extended by RT. The results of PCR amplification of the fully extended products are shown in Fig. 5A. In the case of tRNALys-3 annealed to a Pro-PBS, the length of the DNA fragment produced was identical to the product obtained from extension with the complementary binary complex (Fig. 5A, lanes 3 and 4). DNA sequencing of the extension products revealed that (−)SS cDNA synthesis was initiated at the nucleotide immediately 5′ to the PBS in both cases (Fig. 5B, sets 1 and 2). These data confirm that tRNALys-3 was annealed to and extended from the Pro-PBS (Fig. 5B, set 2). As expected, when tRNAPro was used as the primer in the presence of a Pro-PBS, a 270-bp extension product was also obtained (Fig. 5A, lane 6, and 5B, set 3). However, no PCR products of this length were detected when tRNAPro was used in the presence of a Lys-PBS (Fig. 5A, lane 7).
Figure 5.
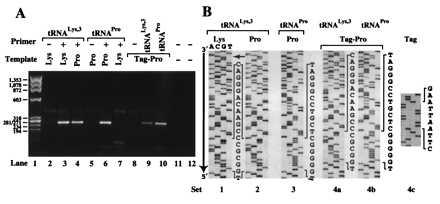
RT extension of tRNAs NC-annealed to complementary and noncomplementary PBS sequences using the HIV-1 HXB2 template. (A) Agarose gel (2%) showing PCR-amplified primer extension products. The tRNA primers were extended by 400 nM RT. The presence (+) or absence (−) of a tRNA primer in the reactions is indicated. PCR-negative controls were carried out with the DNA primers specific for the tRNALys-3-primed product (lanes 11) or the tRNAPro-primed product (lane 12). The RNA templates used contain a PBS complementary to either human tRNALys-3 (Lys) or human tRNAPro (Pro). In addition to a Pro-PBS, the Tag-Pro template contains a 4-nt signature tag near the 5′ end. (B) DNA sequences of PCR products obtained in A. Sets 1–4b, the portion of each ladder shown includes the nucleotide sequence of the tRNA primer and the PBS (nucleotides explicitly shown). The choice of sequencing primer resulted in obtaining the sense and antisense sequence of the RNA template and tRNA primer, respectively. The small arrow indicates the 5′ end of the tRNA sequence. Set 4c, sequence information to confirm the presence of the signature tag in the PCR product obtained from RT extension of tRNALys-3 annealed onto the Pro-PBS (A, lane 9). The inserted sequence (5′-AATT-3′) is italicized.
To rule out the possibility of wild-type RNA template contamination when the Pro-PBS template was used, a 4-nt signature tag was inserted 131 nt 5′ to the Pro-PBS. This tag sequence should not be present in the PCR product if tRNALys-3 was annealed to a contaminating wild-type Lys-PBS template. Once again, the major PCR product resulting from extension using the tagged Pro-PBS template and either tRNALys-3 or tRNAPro was ≈270 nt long (Fig. 5A, lanes 9 and 10). DNA sequencing of the products revealed that the tag sequence was present in both cases (Fig. 5B, set 4c; data not shown) and that tRNALys-3 was extended from the Pro-PBS (Fig. 5B, set 4a). Taken together, these data show that NC can anneal tRNALys-3 to a noncomplementary Pro-PBS and form a binary complex that is extended by RT.
DISCUSSION
By using RNA templates derived from both the HIV-1 MAL and HXB2 genomes, we show that NC can mediate annealing of different tRNAs onto the RNA templates, provided that the PBS is complementary to the 3′ 18 nt of the tRNA used (Figs. 3A and 4). The presence of the noncognate anticodon and D domains apparently does not interfere with complementary tRNA/template annealing by NC, in agreement with previous reports that NC binds and unwinds tRNAs in a nonspecific manner (32, 33).
The results of previous in vivo studies suggested that the mutant PBS reversion events are likely to occur via tRNALys-3 placement onto a noncomplementary PBS and subsequent extension by RT (5–7, 9, 10). We wished to provide direct evidence for the specific annealing of tRNALys-3 onto a noncomplementary PBS and to establish an in vitro system that would allow us to elucidate the molecular determinants for this interaction. In our initial studies using the HIV-1 MAL isolate, we found that in the presence of a noncomplementary PBS, NC prefers to anneal tRNALys-3 to a region 3′ to the PBS. Moreover, RT extends the primer from an internal nucleotide (U67) located in the acceptor stem of tRNALys-3 (Fig. 6 Left). Although the exact mechanism by which this occurs has not been investigated, we imagine that an uncharacterized ribonuclease degrades the 3′ 9 nt of the tRNA to allow extension from an internal position. This is similar to the known mechanism of initiation of reverse transcription in the yeast Ty5 and Drosophila copia retrotransposon elements, which also involves DNA synthesis primed from an internal nucleotide (2 nt after the anticodon loop) of a tRNAiMet primer (34, 35). Recently, Lu et al. mutated the acceptor stem of tRNALys-3 so that nucleotides 66–72 were no longer complementary to the Lys-PBS but were instead complementary to a conserved sequence in the TAR region of the HIV-1 genome (NL4–3 isolate) (36). These researchers observed that the mutated tRNALys-3 was redirected to prime from the alternate site in the TAR region in vitro. Their result is consistent with our observation that tRNALys-3 can be annealed to and extended from an alternate site in the MAL isolate when a perfectly complementary PBS is not available.
Figure 6.
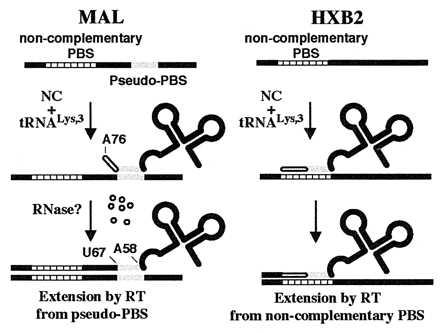
Scheme summarizing results of NC-mediated tRNA annealing onto noncomplementary PBSs by using template sequences derived from the HIV-1 MAL (Left) and HXB2 (Right) genomes.
The priming we observe from a pseudo-PBS located downstream of the authentic PBS, however, does not explain the mutant PBS sequence reversion events observed in vivo by others. Because the pseudo-PBS present in the MAL RNA genome is not found in the vast majority of HIV-1 isolates sequenced to date, we next tested a 394-nt RNA template corresponding to the 5′ end of the HIV-1 HXB2 isolate. In the presence of an HXB2 template containing a noncomplementary PBS, NC is able to anneal only tRNALys-3 to the Pro-PBS (Fig. 4). DNA sequencing following RT-PCR showed that tRNALys-3 was indeed annealed to and extended from the noncomplementary Pro-PBS (Fig. 6 Right). This result is in agreement with recent studies showing that NC can promote annealing between mutated sequences representing (−)SS cDNA and 3′ viral RNA sequences (37) and that RT can extend mispaired binary complexes (37, 38). However, we also show that tRNAPro cannot be annealed to the Lys-PBS by NC. This could be because of the decrease in potential base pair complementarity between tRNAPro and the Lys-PBS (10 Watson–Crick base pairs) compared with that between tRNALys-3 and the Pro-PBS (10 Watson–Crick and three G:U wobble base pairs). However, NC also failed to anneal the chimeric tRNALys/Pro onto the Pro-PBS. Because tRNALys-3 and tRNALys/Pro have the same extent of base pair complementarity between the 3′ 18 nt of the tRNAs and the Pro-PBS, our results suggest that tRNALys-3 placement onto the noncomplementary PBS is mediated by certain sequence and/or structural elements located in the D and/or anticodon domain of tRNALys-3. We cannot rule out the possibility that altered folding of the chimeric tRNALys/Pro may affect noncomplementary annealing by NC. However, we feel this is unlikely, because tRNALys/Pro and tRNALys-3 were annealed by NC to the complementary PBS to similar extents (Fig. 4).
Domains outside the 3′ 18 nt of tRNALys-3 previously have been shown to play a role in primer function. For example, using natural tRNALys-3 heat-annealed to a wild-type HIV-1 RNA template (MAL isolate), Ehresmann and coworkers determined that the highly modified U-rich anticodon loop of tRNALys-3 interacts with an A-rich loop on the RNA genome located upstream of the PBS (39–43). Disruption of this loop–loop interaction appears to affect viral replication during (−)SS cDNA synthesis, either at the initiation step (44) or by modulating the transition from initiation to elongation mode of RT (42). Deletion of the A loop does not affect in vitro tRNALys-3 annealing onto the complementary PBS either by heat (44) or by NC (18). Our observation that tRNALys/Pro was annealed as efficiently as tRNALys-3 to a Lys-PBS supports this notion (Fig. 4). On the other hand, the loop–loop interaction appears to play a role in tRNALys-3 annealing when the PBS is mutated. For example, changing the PBS alone to be specific to another tRNA does not prevent PBS reversion to a sequence complementary to tRNALys-3 in vivo (4–8). However, in two cases (tRNAHis and tRNAMet) when both the PBS and the A loop were mutated to be complementary to the 3′ 18 nt and the anticodon loop of the alternate tRNA, PBS reversion to a sequence complementary to tRNALys-3 did not occur (13, 14). Taken together, these data suggest that an A-rich loop sequence on the RNA genome interacts with the anticodon loop of tRNALys-3 and that this interaction may facilitate placement of the tRNA onto a noncomplementary PBS. In vitro selection experiments designed to elucidate whether specific nucleotides within the tRNALys-3 anticodon loop are indeed required for NC-mediated annealing to both complementary and noncomplementary PBS sequences are underway.
Acknowledgments
We thank Drs. Jean-Luc Darlix, Jack Horowitz, Lawrence Kleiman, and Stuart Le Grice for kindly providing plasmids used in this study. We gratefully acknowledge Drs. Christine Debouck and F. William Studier for the E. coli strains containing recombinant HIV-1 RT and T7 RNA polymerase, respectively. We also thank Dr. Paul Siliciano for helpful comments on the manuscript. This work was supported by the University of Minnesota McKnight-Land Grant Professorship Program and Grant NP-953 from the American Cancer Society.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: NC, nucleocapsid protein; PBS, primer binding site; RT, reverse transcriptase; (−)SS cDNA, minus-strand strong-stop DNA.
References
- 1.Kleiman L, Caudry S, Boulerice F, Wainberg M A, Parniak M A. Biochem Biophys Res Commun. 1991;174:1272–1280. doi: 10.1016/0006-291x(91)91559-u. [DOI] [PubMed] [Google Scholar]
- 2.Jiang M, Mak J, Wainberg M A, Parniak M A, Cohen E, Kleiman L. Biochem Biophys Res Commun. 1992;185:1005–1015. doi: 10.1016/0006-291x(92)91727-8. [DOI] [PubMed] [Google Scholar]
- 3.Huang Y, Mak J, Cao Q, Li Z, Wainberg M A, Kleiman L. J Virol. 1994;68:7676–7683. doi: 10.1128/jvi.68.12.7676-7683.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li X, Mak J, Arts E J, Gu Z, Kleiman L, Wainberg M A, Parniak M A. J Virol. 1994;68:6198–6206. doi: 10.1128/jvi.68.10.6198-6206.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wakefield J K, Rhim H, Morrow C D. J Virol. 1994;68:1605–1614. doi: 10.1128/jvi.68.3.1605-1614.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Das A T, Klaver B, Berkhout B. J Virol. 1995;69:3090–3097. doi: 10.1128/jvi.69.5.3090-3097.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wakefield J K, Wolf A G, Morrow C D. J Virol. 1995;69:6021–6029. doi: 10.1128/jvi.69.10.6021-6029.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kang S-M, Wakefield J K, Morrow C D. Virology. 1996;222:401–414. doi: 10.1006/viro.1996.0437. [DOI] [PubMed] [Google Scholar]
- 9.Rhim H, Park J, Morrow C D. J Virol. 1991;65:4555–4564. doi: 10.1128/jvi.65.9.4555-4564.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Das A T, Koken S E C, Essink B B O, van Wamel J L B, Berkhout B. FEBS Lett. 1994;341:49–53. doi: 10.1016/0014-5793(94)80238-6. [DOI] [PubMed] [Google Scholar]
- 11.Gilboa E, Mitra S W, Goff S, Baltimore D. Cell. 1979;18:93–100. doi: 10.1016/0092-8674(79)90357-x. [DOI] [PubMed] [Google Scholar]
- 12.Jiang M, Mak J, Ladha A, Cohen E, Klein M, Rovinski B, Kleiman L. J Virol. 1993;67:3246–3253. doi: 10.1128/jvi.67.6.3246-3253.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kang S-M, Zhang Z, Morrow C D. J Virol. 1997;71:207–217. doi: 10.1128/jvi.71.1.207-217.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wakefield J K, Kang S-M, Morrow C D. J Virol. 1996;70:966–975. doi: 10.1128/jvi.70.2.966-975.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barat C, Lullien V, Schatz O, Keith G, Nugeyre M T, Grüninger-Leitch F, Barré-Sinoussi F, Le Grice S F, Darlix J-L. EMBO J. 1989;8:3279–3285. doi: 10.1002/j.1460-2075.1989.tb08488.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Rocquigny H, Gabus C, Vincent A, Fournié-Zaluski M-C, Roques B, Darlix J-L. Proc Natl Acad Sci USA. 1992;89:6472–6476. doi: 10.1073/pnas.89.14.6472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weiss S, König B, Morikawa Y, Jones I. Gene. 1992;121:203–212. doi: 10.1016/0378-1119(92)90123-7. [DOI] [PubMed] [Google Scholar]
- 18.Li X, Quan Y, Arts E J, Li Z, Preston B D, de Rocquigny H, Roques B P, Darlix J-L, Kleiman L, Parniak M A, Wainberg M A. J Virol. 1996;70:4996–5004. doi: 10.1128/jvi.70.8.4996-5004.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mizrahi V, Lazarus G M, Miles L M, Meyers C A, Debouck C. Arch Biochem Biophys. 1989;273:347–358. doi: 10.1016/0003-9861(89)90493-1. [DOI] [PubMed] [Google Scholar]
- 20.Tsuchihashi Z, Brown P O. J Virol. 1994;68:5863–5870. doi: 10.1128/jvi.68.9.5863-5870.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grodberg J, Dunn J J. J Bacteriol. 1988;170:1245–1253. doi: 10.1128/jb.170.3.1245-1253.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sampson J R, Uhlenbeck O C. Proc Natl Acad Sci USA. 1988;85:1033–1037. doi: 10.1073/pnas.85.4.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu M, Horowitz J. BioTechniques. 1993;15:264–266. [PubMed] [Google Scholar]
- 24.Horton R M, Pease L R. In: Directed Mutagenesis. McPherson M J, editor. New York: IRL; 1991. pp. 217–247. [Google Scholar]
- 25.Sanger F, Nicklen S, Coulson A R. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Milligan J R, Groebe D R, Witherell G W, Uhlenbeck O C. Nucleic Acids Res. 1987;15:8783–8798. doi: 10.1093/nar/15.21.8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu H, Musier-Forsyth K. Biochemistry. 1994;33:12708–12714. doi: 10.1021/bi00208a023. [DOI] [PubMed] [Google Scholar]
- 28.Heacock D, Forsyth C J, Shiba K, Musier-Forsyth K. Bioorg Chem. 1996;24:273–289. [Google Scholar]
- 29.Lapadat-Tapolsky M, Pernelle C, Borie C, Darlix J-L. Nucleic Acids Res. 1995;23:2434–2441. doi: 10.1093/nar/23.13.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Steinberg S, Misch A, Sprinzl M. Nucleic Acids Res. 1993;21:3011–3015. doi: 10.1093/nar/21.13.3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baudin F, Marquet R, Isel C, Darlix J-L, Ehresmann B, Ehresmann C. J Mol Biol. 1993;229:382–397. doi: 10.1006/jmbi.1993.1041. [DOI] [PubMed] [Google Scholar]
- 32.Mély Y, de Rocquigny H, Sorinas-Jimeno M, Keith G, Roques B P, Marquet R, Gérard D. J Biol Chem. 1995;270:1650–1656. doi: 10.1074/jbc.270.4.1650. [DOI] [PubMed] [Google Scholar]
- 33.Khan R, Giedroc D P. J Biol Chem. 1992;267:6689–6695. [PubMed] [Google Scholar]
- 34.Kikuchi Y, Ando Y, Shiba T. Nature (London) 1986;323:824–826. doi: 10.1038/323824a0. [DOI] [PubMed] [Google Scholar]
- 35.Voytas D F, Boeke J D. Nature (London) 1992;358:717. doi: 10.1038/358717a0. [DOI] [PubMed] [Google Scholar]
- 36.Lu Y, Planelles V, Li X, Palaniappans C, Day B, Challita-Eid P, Amado R, Stephens D, Kohn D B, Bakker A, Fay P, Bambara R A, Rosebblatt J D. J Biol Chem. 1997;272:14523–14531. doi: 10.1074/jbc.272.23.14523. [DOI] [PubMed] [Google Scholar]
- 37.Lapadat-Tapolsky M, Gabus C, Rau M, Darlix J-L. J Mol Biol. 1997;268:250–260. doi: 10.1006/jmbi.1997.0978. [DOI] [PubMed] [Google Scholar]
- 38.Das A T, Berkhout B. Nucleic Acids Res. 1995;23:1319–1326. doi: 10.1093/nar/23.8.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Isel C, Marquet R, Keith G, Ehresmann C, Ehresmann B. J Biol Chem. 1993;268:25269–25272. [PubMed] [Google Scholar]
- 40.Isel C, Ehresmann C, Keith G, Ehresmann B, Marquet R. J Mol Biol. 1995;247:236–250. doi: 10.1006/jmbi.1994.0136. [DOI] [PubMed] [Google Scholar]
- 41.Arts E J, Ghosh M, Jacques P S, Ehresmann B, Le Grice S F J. J Biol Chem. 1996;271:9054–9061. doi: 10.1074/jbc.271.15.9054. [DOI] [PubMed] [Google Scholar]
- 42.Isel C, Lanchy J-M, Le Grice S F J, Ehresmann C, Ehresmann B, Marquet R. EMBO J. 1996;15:917–924. [PMC free article] [PubMed] [Google Scholar]
- 43.Skripkin E, Isel C, Marquet R, Ehresmann B, Ehresmann C. Nucleic Acids Res. 1996;24:509–514. doi: 10.1093/nar/24.3.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liang C, Li X, Rong L, Inouye P, Quan Y, Kleiman L, Wainberg M. J Virol. 1997;71:5750–5757. doi: 10.1128/jvi.71.8.5750-5757.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Myers G, Berzofsky J A, Korber B, Smith R F, Pavlakis G N. In: Human Retroviruses and AIDS, 1991: A Compilation and Analysis of Nucleic Acid and Amino Acid Sequences. Myers G, et al., editors. Los Alamos, NM: Los Alamos National Laboratory; 1991. [Google Scholar]