

Abstract
Just three decades ago the prevailing viewpoint envisaged atherosclerosis as a bland proliferative process. (1) According to that concept, endothelial denuding injury led to platelet aggregation and release of platelet-derived growth factor which would trigger the proliferation of smooth muscle cells in the arterial intima, and form the nidus of the atherosclerotic plaque. This cellular model of atherosclerosis updated Virchow's concepts of atherosclerosis as a response to injury formulated in the mid-nineteenth century. The advent of the cell biological era of atherosclerosis supplanted the simplistic concept of the atheroma as a passive deposition of lipid debris on the artery wall. Beyond the vascular smooth muscle cells long recognized in atherosclerotic lesions, subsequent work identified immune cells and mediators at work in atheromata, implicating inflammatory mechanisms in disease development. (2) The advent of gene-targeting technology enabled the testing of the roles of specific molecules in the development of experimental atherosclerosis in mice. Such data demonstrated a critical role for hypercholesterolemia and also supported the participation of immune mechanisms in the pathogenesis of atherosclerosis. (3)
Multiple independent pathways of evidence now pinpoint inflammation as a key regulatory process that links multiple risk factors for atherosclerosis and its complications with altered arterial biology. This revolution in our thinking about the pathophysiology of atherosclerosis has begun to provide clinical insight and practical tools that may aid patient management. This review provides an update of the role of inflammation in atherogenesis and highlights how translation of these advances in basic science promises to change clinical practice.
Keywords: Atherosclerosis, Inflammation, Heart Disease
Innate and Adaptive Immunity: Twin Arms of the Immune Response Involved in Atherosclerosis
Through evolution, the inflammatory response has grown in complexity and has provided host defenses against infection and injury. Moreover, inflammatory mechanisms also participate in the repair of injured tissues. The primitive arm of inflammation, known as innate immunity, echoes in mammals pathways extant in early eukaryotes. (4) Primitive phagocytic cells, evolutionary precursors of the mammalian monocyte/macrophage (Figure 1), exist in marine invertebrates as recognized by Metchnikoff in the 19th century. (5) The innate immune response mounts rapidly and combats perceived foreign invaders, often with preformed mediators. “Natural antibodies”, certain complement proteins, and families of cell surface receptors recognize microbial products that can elicit an immediate response without requiring “education” of the immune system. The receptors involved in these primordial host defense responses include several families of macrophage scavenger receptors, also implicated in uptake of modified lipoproteins, and a family of Toll-like receptors (TLR) (Figure 2). The TLRs, named after Drosophila genes, belong to the family of pattern recognition receptors that recognize microbial structures and products. These receptors trigger a complex intracellular signaling cascade that stimulates the production of pro-inflammatory cytokines and other inflammatory mediators. The innate immune response, characterized as “fast and blunt”, recognizes a limited diversity of structures on the order of hundreds.
Figure 1. Elements involved in innate immunity.
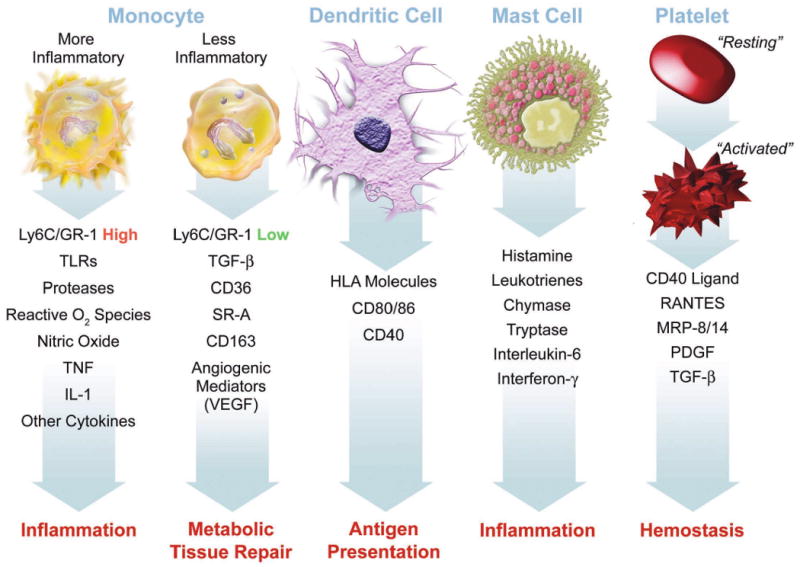
This figure summarizes some of the functions ascribed to various cellular participants in atherosclerosis that may participate in the disease and its complication when dysregulated. Mononuclear phagocytes represent the bulwark of the innate immune defenses in mammals. Monocytes give rise to macrophages, which in the arterial intima form foam cells, the hallmark of the arterial fatty streak. Recent work has focused on heterogeneity of mononuclear phagocytes. We now recognize a pro-inflammatory subset distinct from a less inflammatory population of monocytes. The inflammatory subset expresses high levels of the cell-surface marker Ly6c (also known as GR-1) in the mouse. These inflammatory monocytes express higher levels of Toll-like receptors (TLR), and the other functions indicated, including elaboration of high levels of the cytokines tumor necrosis factor (TNF) and interleukin-1 (IL–1). The less inflammatory subset of monocytes express higher levels of transforming growth factor beta (TGF-beta), the scavenger receptors CD36 and scavenger receptor – A (SR-A), and angiogenic mediators including vascular endothelial growth factor (VEGF). Dendritic cells express human leukocyte antigen (HLA) molecules among the other indicated structures. Dendritic cells present antigens to T cells, linking innate to adaptive immunity. Mast cells elaborate many mediators as shown. Recent data support a causal role for mast cells in mouse atherosclerosis. Platelets also participate in adaptive immunity. When activated, platelets exteriorize CD40 ligand (CD40L or CD154) and release mediators including RANTES (regulated and T cell expressed secreted), myeloid related protein – 8/14 (MRP-8/14), platelet-derived growth factor (PDGF), and TGF-beta.
Figure 2. Cells involved in atherosclerosis express pattern recognition receptors involved in innate immunity.
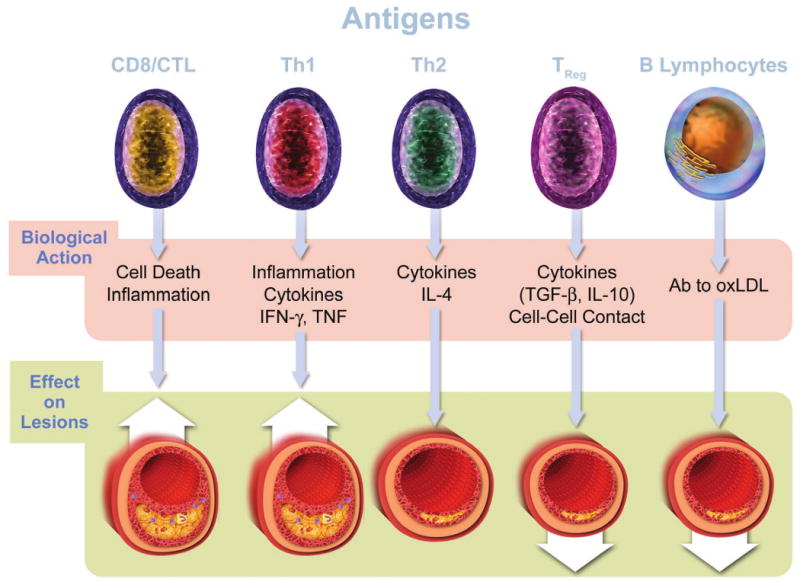
With the cooperation of CD14, TLR4 binds bacterial lipopolysaccharides (LPS) and a variety of other potential instigators of inflammation and atherosclerosis including heat shock proteins (hsp). TLR2 usually exists as a heterodimer with TLR1 or TLR6. TLR2 complexes can bind microbial products as shown and, in addition, apolipoprotein CIII (Apo CIII). Scavenger receptor A binds modified low-density lipoproteins (LDL). CD36 binds oxidatively modified LDL. The receptor for advanced glycation endproducts (RAGE) also decorates many cells involved in atherosclerosis and may function in inflammatory signaling.
The adaptive immune response has arisen more recently in evolution (Figure 3). This arm of host defenses, in contrast to the innate immune response, requires “education” of the immune system. Common clinical experience illustrates the lag time in developing an adaptive immune response. For example, an antibody response, or a cellular immune response requires weeks to months following vaccination with an antigen. Also in contrast with the innate immune response, the adaptive arm displays exquisite specificity. Instead of recognizing mere hundreds of molecular patterns, the repertoire of antibodies and T cell receptors can recognize many millions of specific structures.
Figure 3. Cells involved in adaptive immunity.
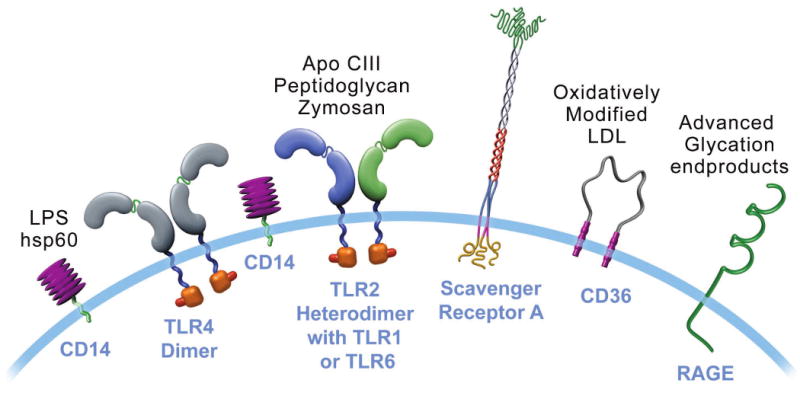
The text describes the functional roles of the five classes of lymphocytes depicted in atherosclerosis. B cells elaborate antibodies (Ab). A specialized subset of B cells (B1 cells) elaborate primarily IgM antibodies, including natural antibodies that recognize constituents of oxidized LDL (oxLDL). The bottom panel of this figure portrays diagrammatically the effect of the various cell types on lesions, based mostly on experiments in mice. Up arrows indicate aggravation of lesion formation. Down arrows indicate reduction in lesion formation. IFN-γ—interferon-gamma; TNF—tumor necrosis factor; IL-4— interleukin-4; TGF-β—transforming growth factor beta; IL-10—interleukin-10; hsp60—heat shock protein 60. This diagram summarizes the “net” effect attributed to the cell type on atherosclerosis primarily on the basis of experiments in mice. In some cases, this figure necessarily oversimplifies the complexity of the data. For example, not all TH2 cell functions and not all antibodies elaborated by B cells may mitigate atherogenesis. (66,67)
The inflammatory response in atherosclerosis involves elements of both the innate and adaptive limbs of immunity.
Innate immunity in atherosclerosis
Considerable evidence supports the early involvement of the monocyte/macrophage, the most prominent cellular component of the innate immune response, during atherogenesis. Observations in human arterial specimens and many experimental models of atherosclerosis have identified monocyte recruitment as an early event in atherogenesis. The recruitment of mononuclear phagocytes involves attachment to activated endothelial cells by leukocyte adhesion molecules. Several protein mediators, specialized cytokines known as chemokines, direct cell migration of monocytes into the intima. Maturation of monocytes into macrophages, their multiplication, and production of many mediators ensues. Previous reviews recount the details of these now well-understood molecular mechanisms that we will not repeat here. (6,7)
Since last reviewed, several new findings regarding monocyte recruitment to atherosclerosis have come to light. First, examination of the kinetics of monocyte recruitment to mouse atherosclerotic lesions suggests that monocyte entry occurs not just during the initial stages of lesion formation, but continues even in established lesions. (8) This observation has implications for targeting monocyte recruitment for atherosclerosis treatment.
Another recent recognition revolves around monocyte heterogeneity in atherosclerosis. (9) Evidence from mouse experiments and in humans suggests a disease-relevant dimorphism of monocytes. (10,11) (12) Hyperlipidemia elicits a profound enrichment of a pro-inflammatory subset of monocytes in the mouse. These pro-inflammatory monocytes, recognized by high levels of a marker known as Ly-6c, or Gr-1, may correspond to a human monocyte subset marked by the presence of P-selectin glycoprotein ligand (PSGL). (13) These pro-inflammatory monocytes home to atherosclerotic lesions, where they propagate the innate immune response by expressing high levels of pro-inflammatory cytokines and other macrophage mediators including matrix metalloproteinases (Figure 1, left).
Recent evidence has also highlighted the potential participation of mast cells in atherosclerosis. Long identified as a minority leukocyte population in the arterial adventitia and atherosclerotic intima, mast cells exhibit numerous functions implicated in atherogenesis. (14) (15) For example, mast cells release vasoactive small molecules such as histamine and leukotrienes, as well as serine proteinases and heparin, a co-factor in growth factor action and angiogenesis. Recent pharmacologic and genetic studies have provided firm evidence for mast cell participation in atherogenesis in mice. (16) (17) As established pharmacologic agents can modulate mast cell functions in humans, these recent observations also have therapeutic implications. The extension of these mouse experiments to the human situation requires further research.
Many links exist between lipoproteins and innate immunity. Modified lipoproteins interact with scavenger receptors (Figure 2), and may thus send proinflammatory signals. Oxidized phospholipids derived from modified lipoproteins may also drive inflammation. A lipoprotein-associated phospholipase (LP-PLA2) currently targeted in clinical trials may generate pro-inflammatory derivatives of oxidatively modified lipoproteins. (18) Recent data show that apolipoprotein CIII, a constituent of certain triglyceride-rich lipoproteins associated with poor clinical outcomes, incite inflammation by binding to TLR2 (Figure 2). (19)
Another area of recent advance in relation to innate immunity in atherosclerosis regards the links between thrombosis and inflammation. Previously considered independent pathways in host defense, current evidence supports considerable crosstalk. (20) For example, prostaglandins produced through the cyclooxygenase pathway control inflammation as well as thrombosis. Therefore, anti-inflammatory cyclooxygenase-2 inhibitors may heighten thrombotic risk. A major protein mediator of coagulation, thrombin, can elicit the expression of pro-inflammatory cytokines from vascular endothelial and smooth muscle cells. Platelets, when activated, can secrete preformed pro-inflammatory cytokines and exteriorize and shed a multipotent pro-inflammatory stimulus, CD40-ligand (CD154). Platelets can also release a pro-inflammatory mediator known as myeloid-related protein 8/14 (MRP-8/14). (21) This heterodimeric molecule serves as a biomarker for adverse cardiovascular events in both apparently well populations, and in survivors of acute coronary syndromes. (22) Current investigations are expanding our knowledge of the inflammatory actions of MRP-8/14. For example MRP-8/14 can bind TLR-4, activating innate immunity through this pattern recognition receptor.(23)(Figure 2) This ligand can also promote endothelial cell apoptosis, a process implicated in plaque thrombosis. (24) These recent observations tighten the link between inflammation and thrombosis, suggesting an intimate interlacing of these two convergent pathways in atherosclerosis.
Adaptive immunity in atherosclerosis
Accumulating evidence supports a key regulatory role for adaptive immunity in atherosclerosis and its complications. The subject of several recent reviews, this section will highlight recent advances in this area.(3) (25) (26) Interacting with a special subset of mononuclear phagocytes specialized in antigen presentation known as dendritic cells (Figure 1), T lymphocytes encounter antigens and mount a cellular immune response (Figure 3). The dendritic cells populate atherosclerotic plaques and regional draining lymph nodes where they can present antigens to T cells with co-stimulatory molecules that incite this key afferent limb of adaptive immunity. Putative antigens that stimulate T cells in the context of atherosclerosis include certain heat shock proteins, components of plasma lipoproteins, and potentially microbial structures as well. The clone of T cells that recognizes antigen in this context will proliferate to amplify the immune response. Upon renewed exposure to the specific antigen, these T cells produce cytokines, trigger inflammation, and some T cells have mechanisms specialized for killing cells. (Figure 3) This amplification accounts for the delay in the typical adaptive immune response that is slower and much more structurally specific than the “fast and blunt” innate immune response described above.
Various functionally distinct classes of T cells exist. Helper T cells spearhead antigen recognition, and fall into two major functional subtypes known as Th1 and Th2 (Figure 3, left). Th1 responses generally amplify pro-inflammatory pathways by secretion of cytokines such as interferon-gamma. The Th1 response appears to aggravate atherosclerosis. A more recently recognized T cell subset, Th17 cells, may also exert particularly pro-inflammatory actions. Th2 cells elaborate cytokines that may modulate inflammation such as interleukin-4 that can promote humoral immunity (see below). Whereas the role of Th2 in atherosclerosis is controversial,(27-30) some, but not all, evidence suggests that Th2-slanted responses may drive aneurysm formation. (31,32) (33) Humans may have less accentuated polarization of Th1 vs. Th2 cells than inbred mice.
Another T cell subtype, known as regulatory T cells or Treg for short, appears to pay an intriguing modulatory role in atherosclerosis. Treg can dampen inflammatory responses. Genetic manipulations that interfere with Treg functions mediated by transforming growth factor beta (TGF-β) augment atherogenesis in mice, yield lesions with signs of heightened inflammation, and even trigger thrombosis. (34,35) Thus, Treg cells and Th2 vs. Th1 and Th17 cells can counterbalance the pro-atherogenic effects of Th1 cells indicating the yin and yang complexity of cellular immunity.
The types of T cells just described express the surface marker CD4 and recognize antigen presented by dendritic cells and macrophages.(Figure 3) One third of all T cells in human lesions are of a different type that carries the CD8 marker and recognizes antigens bound to HLA molecules on many different cell types, typically viral antigens on infected cells. (Figure 3) When activated, CD8 T cells kill neighbor cells via cell/cell contact. Several mediators produced in lesions can recruit CD8 T cells capable of killing smooth muscle cells and macrophages, processes linked to lesion growth and complication. (36)
All these T cells share the capacity to recognize protein antigens bound to HLA molecules on cell surfaces. The NKT cell, in contrast, reacts towards lipid antigens presented by CD1 molecules on antigen-presenting cells. Once activated, the NKT cell produces proinflammatory cytokines that promotes atherosclerosis. (37)
Humoral immunity in atherosclerosis
B lymphocytes secrete antibodies that like T cells, can recognize many millions if not billions of diverse structures. Convergent lines of experimental evidence suggest that humoral immunity can attenuate rather than promote atherogenesis. For example, splenectomy, ablating an important B cell compartment, aggravates atherosclerosis. (38) Hypercholesterolemic mice develop a strong humoral response directed against epitopes characteristic of oxidatively modified low-density lipoprotein (LDL). (39,40) Immunization of rabbits or mice with oxidized LDL attenuates atherosclerosis. Interestingly, the antibodies elicited in mice in response to oxidized LDL also recognize a pneumococcal antigen. (41) This finding underscores the view that host defenses against infectious agents can overlap with inflammatory pathways involved in atherogenesis. The observation that humoral immunity against oxidized LDL might protect against atherosclerosis has inspired therapeutic explorations of vaccination against oxidized LDL to mitigate this disease.
Clinical Translation of Inflammation Biology: The Role of Biomarkers
Following on the ferment in the basic science laboratory regarding inflammation in atherosclerosis, we have now entered an era of clinical translation of inflammation biology to the clinic. The description of inflammatory pathways above identified several new potential therapeutic avenues. Many existing systemic anti-inflammatory strategies such as glucocorticoids, non-steroidal anti-inflammatory drugs, or anti-cytokine agents exert unwanted actions that render them less than ideal candidates for evaluation as long-term therapeutics for modulation of atherosclerosis. Of many promising more specific anti-inflammatory agents in development for atherosclerosis, none appear sufficiently validated for clinical use at present. In contrast, the use of inflammatory biomarkers to predict risk, to monitor treatments, and to guide therapy has shown substantial potential for clinical applicability.
Biomarkers of inflammation in risk prediction
The contemporary literature now contains numerous reports of the relationship between various biomarkers of inflammation and prospective cardiovascular risk, in apparently well individuals as well as in patients with coronary heart disease or heart failure. The clinical utility of a biomarker for risk prediction depends on practicability, ease, cost, and reproducibility of the measurement, and the ability to add to the predictability of existing biomarkers such as those incorporated in the Framingham algorithm. Many reviews have highlighted this fast moving field. (42) Among the many biomarkers of inflammation proposed for diagnostic use, myeloperoxidase, lipoprotein-associated phospholipase A2 (Lp-PLA2), pentraxin-3, cytokines such as IL-6, proteases such as matrix metalloproteinase 9, and C-reactive protein (CRP) measured by a highly sensitive assay (hs-CRP) have generated considerable attention. For a variety of reasons, CRP has emerged as a leading biomarker of inflammation for clinical application. In well individuals without acute infections or inflammatory diseases (e.g., rheumatoid arthritis), levels of hsCRP remain stable over long periods of time with a year-to-year and decade-to-decade variability comparable to that of cholesterol (43) (44) CRP has considerable chemical stability, requires no special precautions for sampling, and has a relatively long half-life without the diurnal variation that plagues certain other biomarkers.
More than a dozen large-scale prospective cohort studies indicate that hsCRP predicts incident myocardial infarction, stroke, and cardiovascular death even after full adjustment for the traditional Framingham covariates (45). Unbiased computational approaches have identified hsCRP as a marker that, along with parental history, sharpens the predictive ability of the traditional Framingham algorithm in women and in men. (46,47) As demonstrated in the Reynolds Risk Scores (www.reynoldsriskscore.org), consideration of hsCRP along with parental history can correctly reclassify many individuals categorized as having intermediate risk according to the traditional Framingham criteria, a risk stratum that accounts for the majority of cardiovascular events. hsCRP may thus serve as a useful adjunct to the Framingham index as a tool to identify individuals at heightened risk for cardiovascular events.
hsCRP as a potential therapeutic goal
Practitioners routinely follow certain biomarkers as a way of monitoring the dosing of cardiovascular therapeutics. We measure LDLC serially when prescribing lipid-lowering agents, blood pressure in anti-hypertensive therapy, and heart rate when titrating beta-adrenergic blocking agents. Given the body of evidence implicating inflammation in atherosclerosis, could an inflammatory biomarker such as hsCRP be used to monitor therapy in a way that would improve clinical effectiveness? A pre-specified analysis of the PROVE-IT (TIMI-22) study, previously discussed in these pages, suggested dual mechanisms of benefit of statin therapy, LDL-lowering, and a direct anti-inflammatory effect independent of LDL-lowering, reflected by reduction of hsCRP. (48) (49) Specifically, within the PROVE IT TIMI-22 trial, clinical outcomes were best among statin-treated participants who not only achieved LDLC levels below 70 mg/dL, but who also achieved hsCRP levels below 2 mg/L.(50) A post hoc analysis of the A to Z trial affirmed this “dual target” concept in survivors of acute coronary syndromes by showing greater benefit following statin initiation among those who achieved lower levels of both LDLC and hsCRP.(51) While these two data sets support the concept of monitoring hsCRP to gauge the intensity of statin therapy, this concept has not undergone prospective testing in outcomes trials, and existing guidelines do not currently recommend this practice.
Targeting therapy using hsCRP
Can the application of biomarkers of inflammation identify individuals that don't meet established treatment criteria who might nonetheless benefit from therapeutic intervention? A post-hoc analysis of the AFCAPS/TexCAPS proved hypothesis-generating in this regard. (52) Stratification of this population of individuals with no established cardiovascular disease into four cells defined by above and below median LDLC, and above and below median hsCRP showed that both groups with high LDLC benefited from therapy as indicated by a number needed to treat below 60. Individuals with LDLC and hs-CRP below median did not benefit from therapy, yielding a number needed to treat of approximately 1,000 to prevent 1 cardiovascular event. The provocative cell in this analysis, the 25% of the individuals in this cohort with below median LDLC but above median hsCRP, yielded clinical benefit indistinguishable from the two high LDL groups. This observation suggested that well individuals with average levels of LDLC, currently below treatment thresholds, might nonetheless benefit from statin therapy if they had concomitant elevations of hsCRP.
The recently reported JUPITER trial tested this hypothesis prospectively. JUPITER enrolled 17,802 individuals without manifest cardiovascular disease, with LDLC levels below 130 mg/dL, but with hsCRP levels greater than 2 mg/dL; all study participants were randomly allocated to rosuvastatin 20 mg daily or to placebo and were then followed for incident vascular events. On the advice of its Independent Data Monitoring Board, JUPITER was stopped early due to a 44 percent reduction in the trial primary endpoint of all vascular events (P<0.00001), a 54 percent reduction in myocardial infarction (P=0.0002), a 48 percent reduction in stroke (P=0.002), a 46 percent reduction in the need for arterial revascularization (P<0.0001), and a 20 percent reduction in all-cause mortality (Figure 4). All pre-specified subgroups within the trial significantly benefitted from rosuvastatin including those traditionally considered to be at low risk such as those with Framingham Risk Scores less than 10 percent, those without metabolic syndrome, women, and those with elevated levels of hsCRP but no other major ATP-III risk factor (figure 5).
Figure 4. Cumulative incidence of cardiovascular events in the JUPITER trial, according to study group.
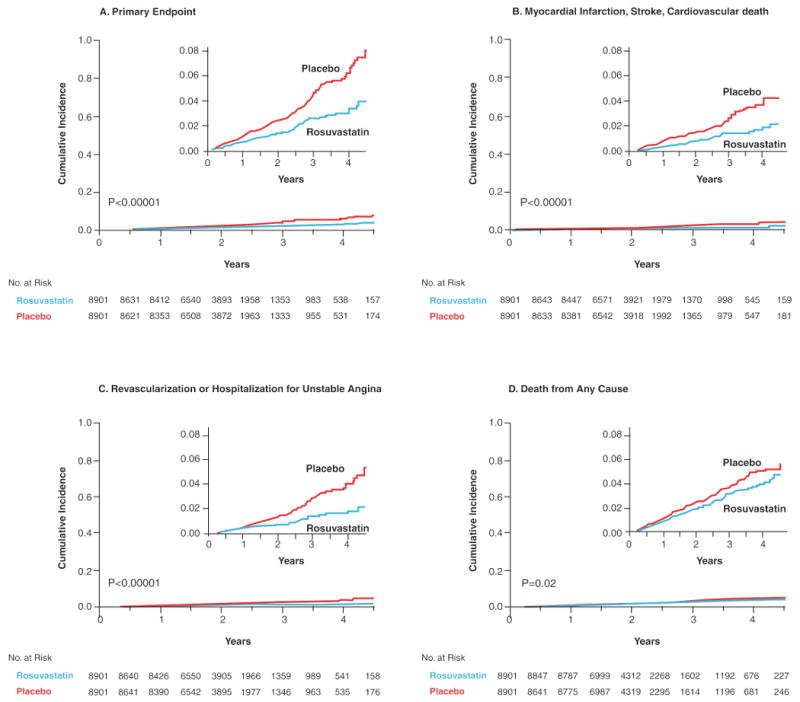
Panel A shows the cumulative incidence of the primary endpoint (nonfatal myocardial infarction, nonfatal stroke, arterial revascularization, hospitalization for unstable angina, or confirmed death from a cardiovascular cause). Panel B shows the cumulative incidence of nonfatal myocardial infarction, nonfatal stroke, or confirmed death from a cardiovascular cause. Panel C shows cumulative incidence for arterial revascularization or hospitalization for unstable angina. Panel D shows the cumulative incidence of death from any cause. [Adopted from Ridker PM, Danielson E, Fonseca FAH, Genest J, Gottto AM, Kastelein JJP, Koenig W, Libby P, Lorenzatti AJ, MacFadyen JG, Nordestgaard BG, Shepherd J, Willerson JT, Glynn RJ for the JUPITER Study Group. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008;359:2195-207.]
Figure 5. Effects of rosuvastatin on the primary trial endpoint, according to baseline characteristics of the JUPITER cohort.
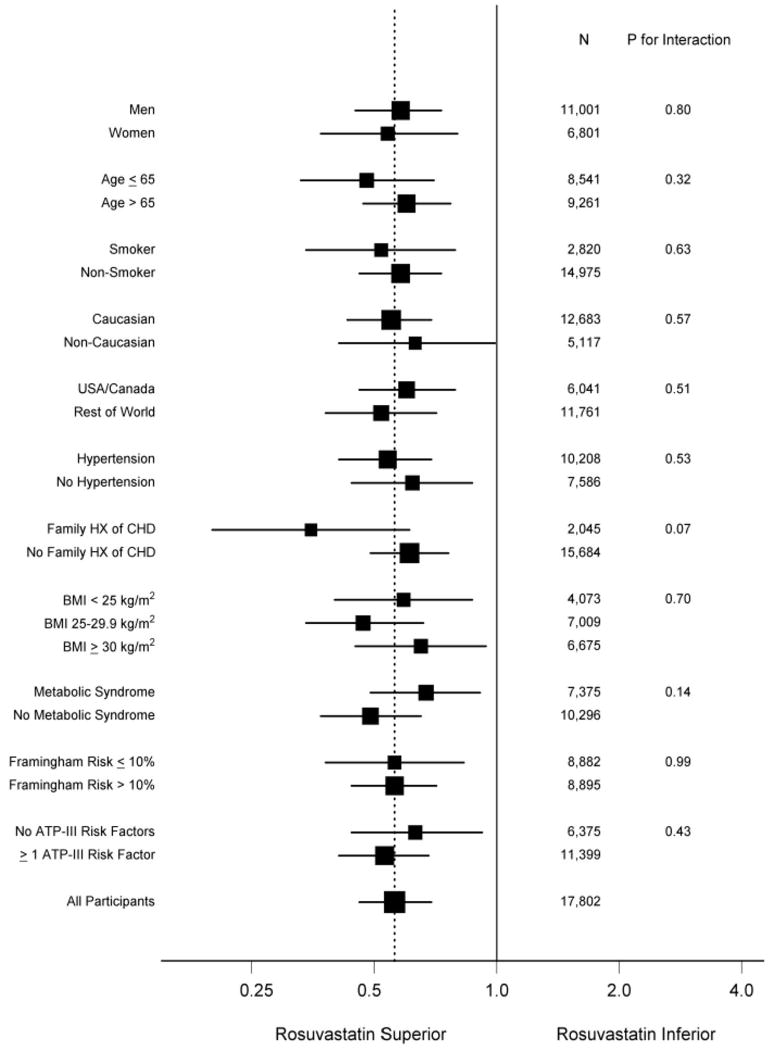
[Adopted from Ridker PM, Danielson E, Fonseca FAH, Genest J, Gottto AM, Kastelein JJP, Koenig W, Libby P, Lorenzatti AJ, MacFadyen JG, Nordestgaard BG, Shepherd J, Willerson JT, Glynn RJ for the JUPITER Study Group. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008;359:2195-207.]
The treated group in JUPITER enjoyed substantive reductions in both absolute and relative risk. Despite excluding all individuals with hyperlipidemia (LDL>130, actual median LDL at entry was 108 mg/dL), the placebo event rate in JUPITER exceeded that of AFCAPS/TexCAPS, indicating that those with a heightened inflammatory burden disclosed by elevated hsCRP have high vascular risk even when cholesterol levels lie within a range considered acceptable by current guidelines. With regard to cost-effectiveness, the commonly accepted metric of Number Needed to Treat at 5 years (5-yr NNT) is 25 in JUPITER for the primary study endpoint and 32 for the “hard endpoint” of myocardial infarction, stroke, or death. These NNT values compare favorably to the 5-yr NNT values of 50 that have been reported in prior primary prevention trials of statin therapy in the setting of overt hyperlipidmia. They also compare very favorably to the treatment of hypertension (5-yr NNT 80 to 160) or to aspirin prophylaxis (5-year NNT 250-300). Prespecified analyses within the JUPITER database affirm that maximum treatment benefit occurs with reduction of both LDLC and hsCRP. (53) This finding has clinical relevance since, in JUPITER, the median on-treatment LDLC was only 55 mg/dL (and 25 percent of the trial had LDLC less than 45 mg/dL), yet optimum benefits not only when LDLC levels reached these very low targets, but when hsCRP levels also fell greatly.
The Future of Inflammation in Atherosclerosis
Targeting inflammation in atherosclerosis: Beyond statins
As described above a growing body of evidence supports the use of statins as an anti-inflammatory intervention in atherosclerosis due to both LDL-lowering and direct anti-inflammatory actions. Progress in understanding the basic biology of inflammation in atherosclerosis has identified potential novel strategies for modulating inflammation in atherosclerosis. No large-scale clinical trial has yet established that an anti-inflammatory intervention that does not alter lipid levels can improve cardiovascular outcomes. Although certain established systemic anti-inflammatory therapies such as corticosteroids or non-steroidal anti-inflammatory agents do not appear promising as anti-atherosclerotic interventions, other agents warrant consideration in this regard. Clinical trials currently underway are exploring the potential of inhibiting lipoprotein-associated phospholipase A2 as an anti-inflammatory therapy, although the first hypothesis testing trial for this agent failed to meet either of its pre-specified primary endpoints. (53,54) Various protein therapeutic strategies such as anti-integrin or anti-cytokine therapies have received consideration for therapeutic application. Therapeutic vaccination with lipoprotein peptides is also being considered for clinical evaluation (55). All of these potential direct anti-inflammatory modalities will require extensive clinical evaluation and direct testing in randomized trials before adoption and practice.
Imaging of inflammation in atherosclerosis
Traditional cardiovascular imaging has focused on anatomy. Magnetic resonance and nuclear imaging techniques can approach aspects of cardiac function such as perfusion and viability. The identification of molecular mediators of inflammation that operate during atherogenesis has generated considerable interest in harnessing them as targets for imaging. Examples of tempting targets in this regard include adhesion molecules such as vascular cell adhesion molecule-1 (VCAM-1), monocyte/macrophage functions such as phagocytosis tracked with microparticulate markers, glucose uptake as monitored by fluorodeoxyglucose, microvessels identified by integrin-directed agents, modified LDL accumulating in lesions, and proteinases implicated in vascular remodeling and plaque destabilization. (56-59) A growing experimental literature has demonstrated the feasibility of many of these targeted imaging strategies. Few if any of these modalities appear near ready for clinical application however. Even those currently feasible in clinical practice, such as 18F-fluorodeoxyglucose imaging, will require considerable clinical validation before adoption in clinical practice. (60,61)
Genetics of inflammation in atherosclerosis
Progress in genetics and genomics, and enormous technical strides in genotyping have heightened interest in defining genetic biomarkers of cardiovascular risk that may open new perspectives in personalized medicine in the future. The computational analysis of various biomarkers alluded to previously identified family history of cardiovascular disease in a parent at age equal to or less than 60 (≤ 60) along with hs-CRP added to the traditional Framingham variables in predicting cardiovascular risk. (46,47) This observation suggests the importance of genetic factors as contributors to cardiovascular risk prediction not completely captured by the Framingham algorithm.
An initial wave of enthusiasm stimulated multiple studies of individual single nucleotide polymorphisms (SNPs) or in a more sophisticated approach haplotypes.(62) The advent of genome-wide association screens (GWAS) has proven quite fruitful. (63) (64) The concordant identification of a region on chromosome 9 as associated with cardiovascular disease in several independent large genetic studies has reinforced future potential of genetics in identifying risk predictors and potential therapeutic targets. (63) Identification by GWAS of “sentinel” members of pathways known to participate in atherosclerosis enhances confidence in the validity of this approach, yet many questions remain unanswered. (65) The functional genomic work required to unravel the biological pathways revealed by GWAS will require considerable investigative effort in years to come. Inevitably, the pursuit of genetic factors identified by GWAS will identify participants in inflammatory pathways that will broaden our understanding and mastery of inflammation in atherosclerosis.
Conclusion
Since our last reviews on these topics, evidence for the involvement of the immune and inflammatory responses in atherogenesis has only intensified. This review has focused on recent advances in this area. We stand on the threshold of an era when clinical inflammation of inflammation biology will prove clinically useful and transformative of clinical practice. This example of translational medicine indicates how clinical challenges have inspired laboratory research that revolutionized our concepts of the pathogenesis of atherosclerosis over the last two decades. The rapid clinical application of these advances in basic science to clinical cardiovascular medicine promises to provide important new tools for diagnosis, monitoring, and management of patients with or at risk for cardiovascular disease in the near future.
Figure 6. Hazard ratios for incident cardiovascular events in the JUPITER trial according to achieved concentrations of LDL cholesterol and high-sensitivity C-reactive protein (hsCRP) after initiation of rosuvastatin therapy.
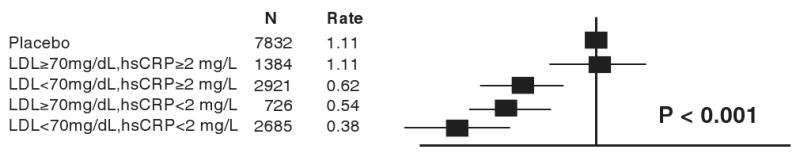
Data were adjusted for age, baseline LDL and HDL cholesterol, baseline hsCRP, blood pressure, gender, body mass index, smoking status, and parental history of premature coronary heart disease. Event rates are per 100 person-years. [Adopted from Ridker PM, Danielson E, Fonseca FAH, Genest J, Gotto AM, Kastelein JJP, Koenig W, Libby P, Lorenzatti AJ, MacFadyen JG, Nordestgaard BG, Shepherd J, Willerson JT, Glynn RJ, on behalf of the JUPITER Trial Study Group. Reduction in C-reactive protein and LDL cholesterol and cardiovascular event rates after initiation of rosuvastatin: a prospective study of the JUPITER trial. Lancet 2009;373:1175-1182.]
Acknowledgments
Funding: This work was supported by grants from the Fondation Leducq (P.L., G.H., P.R.), the Swedish Research Council (G.H.), the Swedish Heart-Lung Foundation (G.H.), and the D.W. Reynolds Foundation (P.L., P.R.).
Footnotes
From the Leducq Transatlantic Network on Atherothrombosis
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Dr. Libby is an unpaid consultant to AstraZeneca. Dr. Ridker reports having received research funding support from multiple not-for-profit entities including the National Heart, Lung, and Blood Institute, the National Cancer Institute, the American Heart Association, the Doris Duke Charitable Foundation, the Leducq Foundation, the Donald W. Reynolds Foundation, and the James and Polly Annenberg La Vea Charitable Trusts. Dr. Ridker also reports having received investigatore-initiated research support from multiple for-profit entities including AstraZeneca, Novartis, Pharmacia, Roche, Sanofi-Aventis, and Abbott, as well as non-financial research support from Amgen. Dr. Ridker is listed as a co-inventor on patents held by the Brigham and Women's Hospital that relate to the use of inflammatory biomarkers in cardiovascular disease that have been licensed to Siemens and AstraZeneca, and has served as a research consultant to Schering-Plough, Sanofi-Aventis, AstraZeneca, Isis, Dade, Merck, Novartis, and Vascular Biogenics. Dr. Hansson has no disclosures to report.
References
- 1.Ross R, Glomset JA. The pathogenesis of atherosclerosis I. New England Journal of Medicine. 1976;295:369–377. doi: 10.1056/NEJM197608122950707. [DOI] [PubMed] [Google Scholar]
- 2.Libby P, Hansson GK. Involvement of the immune system in human atherogenesis: Current knowledge and unanswered questions. Lab. Invest. 1991;64:5–15. [PubMed] [Google Scholar]
- 3.Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol. 2006;6:508–519. doi: 10.1038/nri1882. [DOI] [PubMed] [Google Scholar]
- 4.Hansson GK, Libby P, Schonbeck U, Yan ZQ. Innate and adaptive immunity in the pathogenesis of atherosclerosis. Circ Res. 2002;91:281–91. doi: 10.1161/01.res.0000029784.15893.10. [DOI] [PubMed] [Google Scholar]
- 5.Karnovsky ML. Metchnikoff in Messina: a century of studies on phagocytosis. N Engl J Med. 1981;304:1178–80. doi: 10.1056/NEJM198105073041922. [DOI] [PubMed] [Google Scholar]
- 6.Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. 2006;354:610–21. doi: 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- 7.Viola A, Luster AD. Chemokines and their receptors: drug targets in immunity and inflammation. Annu Rev Pharmacol Toxicol. 2008;48:171–97. doi: 10.1146/annurev.pharmtox.48.121806.154841. [DOI] [PubMed] [Google Scholar]
- 8.Swirski FK, Pittet MJ, Kircher MF, et al. Monocyte accumulation in mouse atherogenesis is progressive and proportional to extent of disease. Proc Natl Acad Sci U S A. 2006 doi: 10.1073/pnas.0604260103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Libby P, Nahrendorf M, Pittet MJ, Swirski FK. Diversity of denizens of the atherosclerotic plaque: not all monocytes are created equal. Circulation. 2008;117:3168–3170. doi: 10.1161/CIRCULATIONAHA.108.783068. [DOI] [PubMed] [Google Scholar]
- 10.Swirski FK, Libby P, Aikawa E, et al. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117:195–205. doi: 10.1172/JCI29950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tacke F, Alvarez D, Kaplan TJ, et al. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. 2007;117:185–94. doi: 10.1172/JCI28549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 13.An G, Wang H, Tang R, et al. P-selectin glycoprotein ligand-1 is highly expressed on Ly-6Chi monocytes and a major determinant for Ly-6Chi monocyte recruitment to sites of atherosclerosis in mice. Circulation. 2008;117:3227–37. doi: 10.1161/CIRCULATIONAHA.108.771048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kovanen PT. Mast cells: multipotent local effector cells in atherothrombosis. Immunol Rev. 2007;217:105–22. doi: 10.1111/j.1600-065X.2007.00515.x. [DOI] [PubMed] [Google Scholar]
- 15.Libby P, Shi GP. Mast cells as mediators and modulators of atherogenesis. Circulation. 2007;115:2471–3. doi: 10.1161/CIRCULATIONAHA.107.698480. [DOI] [PubMed] [Google Scholar]
- 16.Sun J, Sukhova GK, Wolters PJ, et al. Mast cells promote atherosclerosis by releasing proinflammatory cytokines. Nat Med. 2007;13:719–24. doi: 10.1038/nm1601. [DOI] [PubMed] [Google Scholar]
- 17.Bot I, de Jager SC, Zernecke A, et al. Perivascular mast cells promote atherogenesis and induce plaque destabilization in apolipoprotein E-deficient mice. Circulation. 2007;115:2516–25. doi: 10.1161/CIRCULATIONAHA.106.660472. [DOI] [PubMed] [Google Scholar]
- 18.Serruys PW, Garcia-Garcia HM, Buszman P, et al. Effects of the direct lipoprotein-associated phospholipase A(2) inhibitor darapladib on human coronary atherosclerotic plaque. Circulation. 2008;118:1172–82. doi: 10.1161/CIRCULATIONAHA.108.771899. [DOI] [PubMed] [Google Scholar]
- 19.Kawakami A, Osaka M, Aikawa M, et al. Toll-like 2 receptor mediates apolipoprotein CIII-induced monocyte activation. Circ Res. 2008;103:1402–1409. doi: 10.1161/CIRCRESAHA.108.178426. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20.Croce K, Libby P. Intertwining of thrombosis and inflammation in atherosclerosis. Curr Opin Hematol. 2007;14:55–61. doi: 10.1097/00062752-200701000-00011. [DOI] [PubMed] [Google Scholar]
- 21.Healy AM, Pickard MD, Pradhan AD, et al. Platelet expression profiling and clinical validation of myeloid-related protein-14 as a novel determinant of cardiovascular events. Circulation. 2006;113:2278–84. doi: 10.1161/CIRCULATIONAHA.105.607333. [DOI] [PubMed] [Google Scholar]
- 22.Morrow DA, Wang Y, Croce K, et al. Myeloid-related protein 8/14 and the risk of cardiovascular death or myocardial infarction after an acute coronary syndrome in the Pravastatin or Atorvastatin Evaluation and Infection Therapy: Thrombolysis in Myocardial Infarction (PROVE IT-TIMI 22) trial. Am Heart J. 2008;155:49–55. doi: 10.1016/j.ahj.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vogl T, Tenbrock K, Ludwig S, et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med. 2007;13:1042–9. doi: 10.1038/nm1638. [DOI] [PubMed] [Google Scholar]
- 24.Viemann D, Barczyk K, Vogl T, et al. MRP8/MRP14 impairs endothelial integrity and induces a caspase-dependent and -independent cell death program. Blood. 2007;109:2453–60. doi: 10.1182/blood-2006-08-040444. [DOI] [PubMed] [Google Scholar]
- 25.Mallat Z, Taleb S, Ait-Oufella H, Tedgui A. The role of adaptive T cell immunity in atherosclerosis. J Lipid Res. 2008 doi: 10.1194/jlr.R800092-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weyand CM, Younge BR, Goronzy JJ. T cells in arteritis and atherosclerosis. Curr Opin Lipidol. 2008;19:469–77. doi: 10.1097/mol.0b013e32830bfdc2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davenport P, Tipping PG. The role of interleukin-4 and interleukin-12 in the progression of atherosclerosis in apolipoprotein E-deficient mice. Am J Pathol. 2003;163:1117–1125. doi: 10.1016/S0002-9440(10)63471-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Binder CJ, Hartvigsen K, Chang MK, et al. IL-5 links adaptive and natural immunity specific for epitopes of oxidized LDL and protects from atherosclerosis. J Clin Invest. 2004;114:427–437. doi: 10.1172/JCI20479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fredrikson GN, Andersson L, Soderberg I, et al. Atheroprotective immunization with MDA-modified apo B-100 peptide sequences is associated with activation of the Th2 specific antibody expression. Autoimmunity. 2005;38:171–179. doi: 10.1080/08916930500050525. [DOI] [PubMed] [Google Scholar]
- 30.van Wanrooij EJ, van Puijvelde GH, de Vos P, Yagita H, van Berkel TJ, Kuiper J. Interruption of the Tnfrsf4/Tnfsf4 (OX40/OX40L) pathway attenuates atherogenesis in low-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2007;27:204–210. doi: 10.1161/01.ATV.0000251007.07648.81. [DOI] [PubMed] [Google Scholar]
- 31.Schonbeck U, Sukhova GK, Gerdes N, Libby P. T(H)2 predominant immune responses prevail in human abdominal aortic aneurysm. Am J Pathol. 2002;161:499–506. doi: 10.1016/S0002-9440(10)64206-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shimizu K, Shichiri M, Libby P, Lee RT, Mitchell RN. Th2-predominant inflammation and blockade of IFN-gamma signaling induce aneurysms in allografted aortas. J Clin Invest. 2004;114:300–8. doi: 10.1172/JCI19855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tang PC, Yakimov AO, Teesdale MA, et al. Transmural inflammation by interferon-gamma-producing T cells correlates with outward vascular remodeling and intimal expansion of ascending thoracic aortic aneurysms. Faseb J. 2005;19:1528–30. doi: 10.1096/fj.05-3671fje. [DOI] [PubMed] [Google Scholar]
- 34.Robertson AK, Rudling M, Zhou X, Gorelik L, Flavell RA, Hansson GK. Disruption of TGF-beta signaling in T cells accelerates atherosclerosis. J Clin Invest. 2003;112:1342–50. doi: 10.1172/JCI18607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ait-Oufella H, Salomon BL, Potteaux S, et al. Natural regulatory T cells control the development of atherosclerosis in mice. Nat Med. 2006;12:178–180. doi: 10.1038/nm1343. [DOI] [PubMed] [Google Scholar]
- 36.Ludewig B, Freigang S, Jaggi M, et al. Linking immune-mediated arterial inflammation and cholesterol-induced atherosclerosis in a transgenic mouse model. Proc Natl Acad Sci U S A. 2000;97:12752–7. doi: 10.1073/pnas.220427097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tupin E, Nicoletti A, Elhage R, Rudling M, Ljunggren HG, Hansson GK. CD1d-dependent Activation of NKT Cells Aggravates Atherosclerosis. J Experi Med. 2004;199:417–422. doi: 10.1084/jem.20030997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Caligiuri G, Nicoletti A, Poirier B, Hansson GK. Protective immunity against atherosclerosis carried by B cells of hypercholesterolemic mice. J Clin Invest. 2002;109:745–53. doi: 10.1172/JCI07272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hartvigsen K, Chou MY, Hansen LF, et al. The role of innate immunity in atherogenesis. J Lipid Res. 2008 doi: 10.1194/jlr.R800100-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chou MY, Hartvigsen K, Hansen LF, et al. Oxidation-specific epitopes are important targets of innate immunity. J Intern Med. 2008;263:479–88. doi: 10.1111/j.1365-2796.2008.01968.x. [DOI] [PubMed] [Google Scholar]
- 41.Binder CJ, Shaw PX, Chang MK, et al. The role of natural antibodies in atherogenesis. J Lipid Res. 2005;46:1353–63. doi: 10.1194/jlr.R500005-JLR200. [DOI] [PubMed] [Google Scholar]
- 42.Vasan RS. Biomarkers of cardiovascular disease: molecular basis and practical considerations. Circulation. 2006;113:2335–2362. doi: 10.1161/CIRCULATIONAHA.104.482570. [DOI] [PubMed] [Google Scholar]
- 43.Danesh J, Wheeler JG, Hirschfield GM, et al. C-reactive protein and other circulating markers of inflammation in the prediction of coronary heart disease. N Engl J Med. 2004;350:1387–97. doi: 10.1056/NEJMoa032804. [DOI] [PubMed] [Google Scholar]
- 44.Glynn RJ, MacFadyen JG, Ridker PM. Tracking of high-sensitivity C-reactive protein after an initially elevated concentration: the JUPITER Study. Clin Chem. 2009;55:305–312. doi: 10.1373/clinchem.2008.120642. [DOI] [PubMed] [Google Scholar]
- 45.Ridker PM. C-reactive protein and the prediction of cardiovascular events among those at intermediate risk: moving an inflammatory hypothesis toward consensus. J Am Coll Cardiol. 2007;49:2129–38. doi: 10.1016/j.jacc.2007.02.052. [DOI] [PubMed] [Google Scholar]
- 46.Ridker PM, Buring JE, Rifai N, Cook NR. Development and validation of improved algorithms for the assessment of global cardiovascular risk in women: the Reynolds Risk Score. Jama. 2007;297:611–9. doi: 10.1001/jama.297.6.611. [DOI] [PubMed] [Google Scholar]
- 47.Ridker PM, Paynter NP, Rifai N, Gaziano JM, Cook NR. C-reactive protein and parental history improve global cardiovascular risk prediction: the Reynolds Risk Score for men. Circulation. 2008;118:2243–51. doi: 10.1161/CIRCULATIONAHA.108.814251. 4p following 2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ridker PM, Cannon CP, Morrow D, et al. C-reactive protein levels and outcomes after statin therapy. N Engl J Med. 2005;352:20–8. doi: 10.1056/NEJMoa042378. [DOI] [PubMed] [Google Scholar]
- 49.Libby P, Ridker PM. Inflammation and atherothrombosis: from population biology and bench research to clinical practice. J Am Coll Cardiol. 2006;48:A33–46. [Google Scholar]
- 50.Ridker PM, Morrow DA, Rose LM, Rifai N, Cannon CP, Braunwald E. Relative efficacy of atorvastatin 80 mg and pravastatin 40 mg in achieving the dual goals of low-density lipoprotein cholesterol <70 mg/dl and C-reactive protein <2 mg/l: an analysis of the PROVE-IT TIMI-22 Trial. J Am Coll Cardiol. 2005;45:1644–1648. doi: 10.1016/j.jacc.2005.02.080. [DOI] [PubMed] [Google Scholar]
- 51.Morrow DA, de Lemos JA, Sabatine MS, et al. Clinical relevance of C-reactive protein during follow-up of patients with acute coronary syndromes in the Aggrastat-to-Zocor Trial. Circulation. 2006;114:281–8. doi: 10.1161/CIRCULATIONAHA.106.628909. [DOI] [PubMed] [Google Scholar]
- 52.Ridker PM, Rifai N, Clearfield M, et al. Measurement of C-reactive protein for the targeting of statin therapy in the primary prevention of acute coronary events. N Engl J Med. 2001;344:1959–65. doi: 10.1056/NEJM200106283442601. [DOI] [PubMed] [Google Scholar]
- 53.Ridker PM, Danielson E, Fonseca FAH, et al. Reduction in C-reactive protein and LDL cholesterol and cardiovascular event rates after initiation of rosuvastatin: a prospective study of the JUPITER trial. Lancet. 2009;373:1175–1182. doi: 10.1016/S0140-6736(09)60447-5. [DOI] [PubMed] [Google Scholar]
- 54.Boekholdt SM, de Winter RJ, Kastelein JJ. Inhibition of lipoprotein-associated phospholipase activity by darapladib: shifting gears in cardiovascular drug development: are antiinflammatory drugs the next frontier? Circulation. 2008;118:1120–2. doi: 10.1161/CIRCULATIONAHA.108.795195. [DOI] [PubMed] [Google Scholar]
- 55.Hansson GK, Nilsson J. Introduction: Atherosclerosis as inflammation: a controversial concept becomes accepted. J Int Med. 2008;263:462–463. doi: 10.1111/j.1365-2796.2008.01959.x. [DOI] [PubMed] [Google Scholar]
- 56.Kaul S, Lindner JR. Visualizing coronary atherosclerosis in vivo: thinking big, imaging small. J Am Coll Cardiol. 2004;43:461–3. doi: 10.1016/j.jacc.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 57.Wickline SA, Neubauer AM, Winter P, Caruthers S, Lanza G. Applications of nanotechnology to atherosclerosis, thrombosis, and vascular biology. Arterioscler Thromb Vasc Biol. 2006;26:435–41. doi: 10.1161/01.ATV.0000201069.47550.8b. [DOI] [PubMed] [Google Scholar]
- 58.Jaffer FA, Vinegoni C, John MC, et al. Real-time catheter molecular sensing of inflammation in proteolytically active atherosclerosis. Circulation. 2008;118:1802–1809. doi: 10.1161/CIRCULATIONAHA.108.785881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kaufmann BA, Sanders JM, Davis C, et al. Molecular imaging of inflammation in atherosclerosis with targeted ultrasound detection of vascular cell adhesion molecule-1. Circulation. 2007;116:276–84. doi: 10.1161/CIRCULATIONAHA.106.684738. [DOI] [PubMed] [Google Scholar]
- 60.Tahara N, Kai H, Nakaura H, et al. The prevalence of inflammation in carotid atherosclerosis: analysis with fluorodeoxyglucose-positron emission tomography. Eur Heart J. 2007;28:2243–8. doi: 10.1093/eurheartj/ehm245. [DOI] [PubMed] [Google Scholar]
- 61.Rudd JH, Myers KS, Bansilal S, et al. Atherosclerosis inflammation imaging with 18F-FDG PET: carotid, iliac, and femoral uptake reproducibility, quantification methods, and recommendations. J Nucl Med. 2008;49:871–8. doi: 10.2967/jnumed.107.050294. [DOI] [PubMed] [Google Scholar]
- 62.Miller DT, Ridker PM, Libby P, Kwiatkowski DJ. Atherosclerosis: the path from genomics to therapeutics. J Am Coll Cardiol. 2007;49:1589–99. doi: 10.1016/j.jacc.2006.12.045. [DOI] [PubMed] [Google Scholar]
- 63.Schunkert H, Gotz A, Braund P, et al. Repeated replication and a prospective meta-analysis of the association between chromosome 9p21.3 and coronary artery disease. Circulation. 2008;117:1675–84. doi: 10.1161/CIRCULATIONAHA.107.730614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ridker PM, Chasman DI, Zee RY, et al. Rationale, design, and methodology of the Women's Genome Health Study: a genome-wide association study of more than 25,000 initially healthy american women. Clin Chem. 2008;54:249–55. doi: 10.1373/clinchem.2007.099366. [DOI] [PubMed] [Google Scholar]
- 65.Ridker PM, Pare G, Parker A, et al. Loci related to metabolic-syndrome pathways including LEPR,HNF1A, IL6R, and GCKR associate with plasma C-reactive protein: the Women's Genome Health Study. Am J Hum Genet. 2008;82:1185–92. doi: 10.1016/j.ajhg.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Furukawa Y, Becker G, Stinn JL, Shimizu K, Libby P, Mitchell RN. Interleukin-10 (IL-10) augments allograft arterial disease: paradoxical effects of IL-10 in vivo. Am J Pathol. 1999;155:1929–39. doi: 10.1016/S0002-9440(10)65512-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Caligiuri G, Rudling M, Ollivier V, et al. Interleukin-10 deficiency increases atherosclerosis, thrombosis, and low-density lipoproteins in apolipoprotein E knockout mice. Mol Med. 2003;9:10–17. [PMC free article] [PubMed] [Google Scholar]