

Abstract
Activation of striated muscle contraction is a highly cooperative signal transduction process converting calcium binding by troponin C (TnC) into interactions between thin and thick filaments. Once calcium is bound, transduction involves changes in protein interactions along the thin filament. The process is thought to involve three different states of actin-tropomyosin (Tm) resulting from changes in troponin’s (Tn) interaction with actin-Tm: a blocked (B) state preventing myosin interaction, a closed (C) state allowing weak myosin interactions and favored by calcium binding to Tn, and an open or M state allowing strong myosin interactions. This was tested by measuring the apparent rate of Tn dissociation from rigor skeletal myofibrils using labeled Tn exchange. The location and rate of exchange of Tn or its subunits were measured by high-resolution fluorescence microscopy and image analysis. Three different rates of Tn exchange were observed that were dependent on calcium concentration and strong cross-bridge binding that strongly support the three-state model. The rate of Tn dissociation in the non-overlap region was 200-fold faster at pCa 4 (C-state region) than at pCa 9 (B-state region). When Tn contained engineered TnC mutants with weakened regulatory TnI interactions, the apparent exchange rate at pCa 4 in the non-overlap region increased proportionately with TnI–TnC regulatory affinity. This suggests that the mechanism of calcium enhancement of the rate of Tn dissociation is by favoring a TnI–TnC interaction over a TnI–actin–Tm interaction. At pCa 9, the rate of Tn dissociation in the overlap region (M-state region) was 100-fold faster than the non-overlap region (B-state region) suggesting that strong cross-bridges increase the rate of Tn dissociation. At pCa 4, the rate of Tn dissociation was twofold faster in the non-overlap region (C-state region) than the overlap region (M-state region) that likely involved a strong cross-bridge influence on TnT’s interaction with actin-Tm. At sub-maximal calcium (pCa 6.2–5.8), there was a long-range influence of the strong cross-bridge on Tn to enhance its dissociation rate, tens of nanometers from the strong cross-bridge. These observations suggest that the three different states of actin-Tm are associated with three different states of Tn. They also support a model in which strong cross-bridges shift the regulatory equilibrium from a TnI–actin–Tm interaction to a TnC–TnI interaction that likely enhances calcium binding by TnC.
Keywords: troponin, tropomyosin, thin filament, calcium regulation, myosin S1
Introduction
The myofibril is the contractile unit of striated muscle that is made of serially arranged sarcomeres. Within the half sarcomere, there is a region of overlap between the thick and thin filaments where myosin interacts with actin to produce force and shortening. Control of contraction occurs primarily at the thin filament1,2 involving calcium, which binds to the thin filament and results in an uncovering of actin sites for interaction with myosin. Because of the numerous proteins involved, once calcium is bound, there are signal transduction processes (changes in intra and inter-protein interactions) that uncover the actin binding sites. A unique feature of reconstituted thin filaments,3 the myofibril4 and the intact fiber5 is the highly cooperative activation of ATPase activity or force by calcium that involves these signal transduction processes.
The anatomical and biochemical features of the thin filaments are primarily responsible for this highly cooperative behavior. The thin filaments contain actin tropomyosin (Tm) and troponin (Tn).3,6 Tm binds to actin filaments, spanning seven actin monomers. Tn binds to actin-Tm with a stoichiometry of 1Tn:1Tm:7actins. It is the binding of Tn to actin-Tm that is part of the control mechanism of contraction.7,8 Troponin is a heterotrimer made of TnC, the calcium binding subunit, TnI, the actin binding and inhibiting subunit and TnT, the tropomyosin binding subunit.9 Crystal structures of the regulatory domain of Tn show the complex interactions between the subunits.10,11 There are also interactions of TnT with actin12 and TnI with Tm.13 Myosin does not interact directly with TnC but does influence its calcium binding14-17 so there is signal transduction between TnC and myosin.
Exposure of actin sites for myosin interaction occurs by changes in the position of Tm on the actin filament surface.18 Early work suggested a two-state system where Tm sterically blocks the actin sites when calcium is low, while high calcium results in a non-blocking position of Tm.19,20 More recent studies suggest three different states of the thin filament in reference to the average position of Tm on the surface of the actin filament:21 a blocked (B) state where Tm is on the outer domain of the filament and blocks the actin site, a closed (C) state where tropomyosin is mostly along the inner domain of the filament and allows for weak interactions between actin and the motor domain, and an open (M) state where Tm is even further displaced into the inner domain of the filament. The M state allows for force-producing interactions. Both calcium and strong cross-bridges influence the distribution of Tm on the actin filament with it being mostly B state without calcium, mostly C state with calcium and mostly M state with strong cross-bridges. Troponin controls the B to C state transition, since in its absence, Tm is mostly in the C state.21 Troponin is the calcium switch of the thin filament. In the absence of calcium, a domain of TnI binds to actin resulting in Tm being mostly in the B state.22 Regulatory calcium binding to TnC converts a domain of TnC into a high affinity site for this TnI domain,23,24 essentially adding a very nearby TnI domain binding site that competes with actin for the TnI domain. The calcium-saturated TnC site has a higher affinity such that the TnI domain switches from actin to TnC. This is thought to allow Tm to move from the outer domain to the inner domain; a B to C state transition.25 A simple cartoon of Tn subunit interactions on actin-Tm in the three different states is shown in Figure 1.
Figure 1.

Cartoon of thin filament regulation. The states refer to the different nomenclatures used with the upper row being based upon structural studies,21 middle being based upon kinetic and equilibrium binding studies,40 and the lower being based upon ATPase and/or muscle fiber force studies.
Recent models of the position of Tn on actin-Tm propose different positions of Tn on actin-Tm to accomplish the same function.26,27 Independent of these different models on Tn location, the average Tm position is mostly agreed upon.21,28 Because Tm interacts with seven actin monomers along the thin filament compared to TnI’s one to two, it is likely a critical component in signal transduction along the thin filament. A critical feature of Tm is its elasticity.29,30 There is lateral elasticity that is thought to be involved in allowing Tm to bind to the actin filament.31,32 There is also longitudinal elasticity that is also likely involved in Tm function. An example of Tm’s axial elastic properties is its ability to influence actin’s persistence length. This length is increased twofold by Tm and Tn–Tm in the B state and about 1.3-fold by Tn-Tm in the C state.33 This can be explained if Tm has axial in-elasticity. Another example of its relative in-elasticity is that the binding of one strong cross-bridge displaces Tm into the M state for 10–20 actins adjacent to the cross-bridge in the absence of calcium.34,35 This axial in-elasticity of Tm is one of the features of the thin filament responsible for part of the cooperativity of calcium activation.
Previously, we showed that the distribution of a fluorescently labeled, exogenous myosin motor domain (S1) within rigor myofibrils was modulated by calcium.36 This approach probed the distribution of M-state actin sites within the half sarcomere and suggested that the intrinsic rigor cross-bridges shifted actin –Tm sites into the M state and this was limited to about 14 actins adjacent to the rigor cross-bridge. More refined studies showed that calcium alone did not shift the actin sites into the M state and supported the three-state model.37,38 Based on these observations, Figure 2 shows predictions of the predominant states within the non-overlap and overlap regions of rigor half sarcomeres and the Tn subunit interactions likely responsible for them. The overlap region is the M state. The non-overlap region is the B state without calcium and the C state with calcium. Adding saturating S1 shifts both regions to the M state independent of calcium. A problem with these36-38 and most other studies34,39,40 is that the thin filament state was probed via changes in actin or Tm as a result of strong cross-bridge binding, which in itself, alters the system. Also, previous studies have not probed the state of Tn in the three different states. Tn could have just two states (B and C) while actin-Tm has three, where the M state is myosin-dependent and does not alter Tn’s state on the thin filament.
Figure 2.

Thin filament states within the half sarcomere. Adapted from the work of Zhang et al.38 and the current study.
There are important features of the myofibril that make it an attractive model to study thin filament regulation using high-resolution imaging and image analysis. It has a repeating structure allowing for signal averaging of half sarcomeres. The concentration of Tn sites within a defined width of the half sarcomere is the same in the overlap and non-overlap region, allowing ratiometric image analysis. We know where the last intrinsic rigor cross-bridge is located in the half-arcomere from the phase-contrast image. Lastly, we have hundreds of thin filaments within a cross-section of the half sarcomere that are in lateral register, allowing signal averaging of these regions. Thus, if one region of the thin filament, say, that at the overlap, non-overlap junction, is in a different state, it will likely be detected.
We have taken advantage of the myofibril structure to probe regulation by a relatively simple and subtle method of labeled Tn exchange. Brenner et al.41 developed the technique and showed that the distribution of Tn exchange within the half sarcomere was not uniform. This suggests that the processes involved in Tn exchange may give insight into the mechanism of signal transduction in myofibrils. The exchange process is rate-limited by the rate of intrinsic Tn dissociation, and when labeled Tn is present, the labeled protein becomes incorporated into the myofibril. Observation of the pattern of the fluorescence in the half sarcomere tells where the exchange occurred and observation at different times tells how fast (i.e. the apparent rate of Tn dissociation). Regulation of muscle contraction does not occur by Tn dissociation but by calcium and cross-bridge-dependent rearrangement of Tn on the thin filament. Our hypothesis is that any changes in protein interactions that alter the rate of Tn dissociation may be observed by this method. The calcium and cross-bridge-dependent movement of TnI from actin to TnC may affect Tn’s dissociation from the thin filament. Thus, we further hypothesize that calcium and strong cross-bridges will increase the rate of Tn dissociation.
Results
Troponin dissociates with three different rates in the rigor myofibril
To determine the number of states of Tn in native thin filaments, labeled Tn was exchanged at pCa 9 and 4 in rigor myofibrils (giving three different regions of thin filaments states as shown in Figure 2). Incubation of rigor myofibrils with a fluorescently labeled Tn resulted in incorporation of Tn within the myofibril (Figure 3). At the first time-point, the location of the labeled Tn within the half sarcomere was calcium dependent. At low calcium (pCa 9) there was essentially no exchange in the non-overlap region (B regions) while there was exchange in the overlap region, containing rigor acto-myosin interactions (M regions). At high calcium (pCa 4) the pattern switched in that there was more exchange in the non-overlap (C regions) than the overlap region (M regions). With increasing time of incubation the pattern changed. At pCa 9, there was a very subtle change with the difference between the non-overlap and overlap region becoming slightly less. Observation of the time course at pCa 4 shows that there was a change in the pattern with the amount of exchange in the two regions being similar after 256 min. The different patterns and their change with time suggest that exchange occurred and it occurred with different rates.
Figure 3.
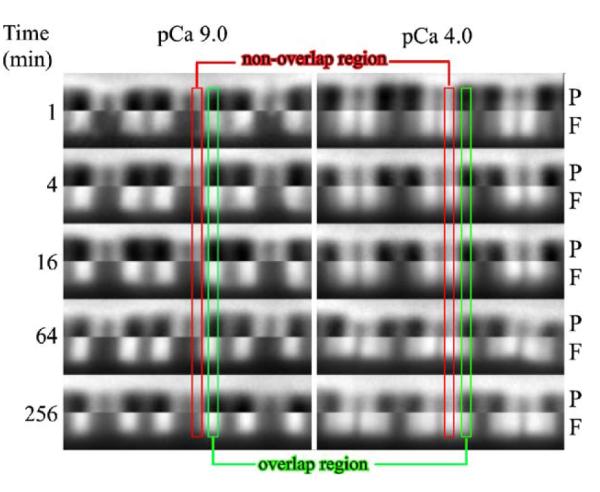
Influence of calcium on labeled Tn exchange pattern in rigor myofibrils. Fluorescently labeled Tn was incubated with myofibrils for different times at pCa 9 or 4 then imaged. The myofibril region of interest (both phase contrast (P) and fluorescent (F)) were cropped and spliced together with the phase contrast being the upper half and the fluorescence image (showing relative amount within a myofibril) being the lower half. Part of the non-overlap region is highlighted within the red rectangle and part of the overlap region is noted by the green rectangle outline. At pCa 9, exchange was in the overlap region that contained rigor cross-bridges and there was a slight increase in the amount in the non-overlap region by 256 min. At pCa 4, exchange was faster in the non-overlap than overlap region at early time-points and the extent of exchange in each region was nearly equal by 256 min.
When myofibrils were incubated with saturating levels of S1, giving M regions in both the non-overlap and overlap region, there was no difference in the pattern as a function of time along the full length of the contractile thin filament independent of pCa (images not shown). Additionally, when exchange was done for 60 min at pCa 9 with MgATP, to dissociate the rigor acto-myosin linkages (B regions in both non-overlap and overlap regions), there was no difference in the pattern between the two regions and the signal was very low (data not shown). This result suggests that there were very few “empty” troponin sites along the thin filament, since these sites would have quickly become occupied by labeled Tn and give a significant signal. These results suggest that there were three different Tn exchange rates with the rank order of rates being C>M>>B regions. These experiments do not directly address which troponin subunit interactions within the thin filament were influenced by calcium and strong cross-bridges nor do they give quantitative estimates on the magnitude of the difference in the rates of Tn dissociation.
Strong cross-bridges enhance TnIT dissociation but slow TnT dissociation from myofibrillar thin filaments
To determine the role of Tn subunits in Tn exchange, labeled Tn was exchanged into TnIT and TnT myofibrils. A hypothesis is that calcium will not influence TnIT or TnT dissociation rates, since TnC is absent and strong cross-bridges will influence dissociation rates independent of TnC. TnIT or TnT was first exchanged for TnCIT by incubating myofibrils at pCa 4. This favored dissociation of intrinsic TnCIT and ensured a uniform distribution of the TnIT or TnT along the full length of the thin filament. The ATPase activity and labeled Tn exchange patterns after 8 min incubation at pCa 9 and 4 were then determined. Exchange of TnIT for Tn resulted in a loss of calcium activation of ATPase activity while exchange of TnT for Tn resulted in a loss of calcium regulation (Figure 4(a)). Comparison of the pattern of Tn exchange between TnCIT and TnIT myofibrils shows that without TnC, the exchange was almost solely in the overlap region, independent of calcium (Figure 4(b)). Comparison of the pattern between TnCIT and TnT myofibrils shows that there was less exchange in the non-overlap than overlap region at pCa 9 for TnCIT compared to TnT myofibrils, whereas this was not as obvious at pCa 4 (Figure 4(b)). When exchange was done with saturating S1 there was little difference in the patterns between TnCIT, TnIT and TnT myofibrils, it was uniform along the full length of the contractile thin filament (images not shown).
Figure 4.

Influence of Tn subunits on myofibrillar ATPase activity and labeled Tn exchange pattern. Intrinsic Tn (TnCIT) was exchanged out with either TnIT or TnT at pCa 4. ATPase activity was measured at pCa 9 and pCa 4 and expressed relative to TnCIT myofibril pCa 4 activity (a). Labeled Tn was exchanged into these myofibrils as for Figure 2 for 8 min (b). Images are presented as in Figure 2. Exchange of TnIT for TnCIT resulted in complete loss of calcium activation of ATPase activity while exchange of TnT for TnCIT resulted in complete loss of calcium regulation of ATPase activity. There was limited exchange in the non-overlap region for TnIT myofibrils showing that TnC was needed for the calcium dependency of Tn exchange (C state). For TnT myofibrils, non-overlap region exchange (C state) was fast, independent of calcium showing that TnI was needed for B state and TnIC was needed for calcium dependency of switching between the B and C state. Another feature of TnT myofibrils was that exchange was slower in the overlap than non-overlap, suggesting that strong cross-bridges (M state) slowed TnT dissociation.
Troponin dissociation rates are slow and influenced by calcium, strong cross-bridges and Tn subunit composition
To obtain quantitative estimates of the rate of Tn dissociation, labeled Tn was exchanged in TnCIT and TnT myofibrils followed by image analysis. The working hypothesis is that, as Tn exchange reaches equilibrium, the fluorescence intensity in the non-overlap region should equal that of the overlap region, since there is an equivalent amount of Tn in each region. A consequence of the main hypothesis is that since TnI is involved in slowing Tn’s dissociation, TnT dissociation will be faster than Tn. Over the 1024 min time-course, the pattern of exchange was time dependent for all myofibrils (Figure 5(a)) with most giving a near equal intensity in the non-overlap and overlap region by 1024 min. The exception was TnCIT myofibrils at pCa 9 where the non-overlap region was just starting to show Tn exchange by 1024 min. For all others (TnCIT at pCa 4 and TnT at pCa 9 or 4), the earliest time point showed the greatest difference in the amount of exchange between the non-overlap region compared to the overlap. There were subtle differences when comparing the non-overlap to overlap region within this group with the rank order of the magnitude of the difference being TnT-pCa 9 > TnCIT-pCa 4 > TnT-pCa 4.
Figure 5.

The rate of Tn dissociation from different states of the thin filament. Troponin T was exchanged for TnCIT as described in Figure 4. Labeled Tn was exchanged into TnCIT and TnT myofibrils as described in Figure 2. Only images from the fluorescence are shown and demonstrate pattern not amount (a). Imaging was done using fixed exposure time (note the noisy signal for the slowest exchanging sample (pCa 9, TnCIT at 1 min). Localization of non-overlap and overlap region is as in Figure 2. The time-dependent change in the pattern is obvious for all myofibrils. Sarcomere intensity increased with time (not shown) while the ratio approached one from either below (pCa 9, TnCIT) or above (pCa 9, TnT; pCa 4, TnCIT and TnT) one. The intensity ratio was fitted to [(1-exp (kno*t))/(1-exp(kol*t))] + C, where kno is the dissociation rate in the non-overlap region; kol is the dissociation rate in the overlap region and C is a constant. Fitted values are given in Table 1.
The total sarcomere fluorescence intensity increased ca sixfold from 1–1024 min of exchange (data not shown) but the data were quite noisy. Also, the increase in total intensity was the result of at least two different rates of Tn dissociation. Thus, fitting the change in total fluorescence intensity as a function of time to obtain the rate of Tn dissociation was not done. To extract rate data from the images, the intensity was measured within a sub-region of the non-overlap and a sub-region of the overlap region for six contiguous half sarcomeres. This gives the non-overlap/overlap intensity, which is a measure of the distribution or intensity pattern. A similar approach was used for equilibrium binding studies on labeled S1 binding to myofibrils but the variable was total fluorescent S137,38 instead of time. A plot of the ratio as a function of time allows for extraction of the rate of change of the ratio that is dependent upon the rate of Tn dissociation in each region. The data were fit by the equation, intensity ratio = [(1-exp(kno*t))/(1-exp(kol*t))] + C, where kno is the dissociation rate in the non-overlap region; kol is the dissociation rate in the overlap region and C is a constant that should =0 if there is an equivalent amount of exchange in each region at infinite time. The results from this fitting are shown in Figure 5(b) and the fitted values are given in Table 1. The fit for TnCIT data from myofibrils at pCa 9 is not shown as the exchange in the non-overlap region was less than half complete by the 1024 min. Estimates for this rate (B-state regions) were from fitting the ratio after subtracting 0.32 (intensity ratio at 1 min) from the ratio values and fixing the kol at 28.9×10−3 min−1. Globally, the dissociation rates were faster for TnT myofibrils than TnCIT myofibrils. For TnCIT myofibrils the relative rates were ca 200:100:1 (C:M:B). For TnT myofibrils the dissociation rate was 1.1 to 2.2 faster in the non-overlap region (C regions) than overlap (M regions) and there appeared to be a modest influence of calcium on TnT dissociation rate. The rates were kinetically very slow with values of 150×10−3 min−1 (0.0024 s−1) or slower.
Table 1.
Apparent rate of troponin dissociation from the different regions within the half sarcomere
| Myofibril | B region (×10−3 min) | C region (×10−3/min) | M region (×10−3/min) | Plateau | R2 |
|---|---|---|---|---|---|
| pCa 9 TnCITa | 0.32±0.03 | – | 28.9b | 0.01245±0.009 | 0.928 |
| pCa 9 TnT | – | 147.4±19 | 66.5±7 | 0.07905±0.019 | 0.991 |
| pCa 4 TnCIT | – | 52.8±3.6 | 28.9±1.7 | 0.256±0.009 | 0.987 |
| pCa 4 TnT | – | 125.0±84 | 107.6±71 | 0.3102±0.019 | 0.727 |
Best-fit value±SE.
Fit after correcting data by subtracting 0.32 (ca 1 min intensity ratio) from data points to correct for the signal from the overlap region measured in the non-overlap region.
From pCa 4 TnCIT fitting.
To graphically display the intra-sarcomeric fluorescence intensity and its change with conditions, the average intensity (four pixels tall) as a function of position in the sarcomere (44 pixels wide) was plotted. The intensity profiles are from images presented in Figure 5(a) using TnCIT myofibrils at pCa 9 and 4 (Figure 6(a)) and TnT myofibrils at pCa 9 and 4 (Figure 6(b)). For TnCIT myofibrils at pCa 9, the peaks are in the middle of the overlap region while for pCa 4, they are in the middle of the non-overlap region. With increased time at pCa 9, there was an increase in the relative intensity in the non-overlap region. Conversely, at pCa 4, there was an increase in the intensity in the overlap region. For TnT myofibrils, there was a subtle influence of calcium and a more complex influence of time of exchange on the profile. At pCa 9, the peaks are in the middle of the non-overlap region at 4 min, but in the middle of the half sarcomere (non-overlap/overlap junction) by 1024 min. At pCa 4, the peaks are in the middle of the non-overlap region at 4 min, but are in the non-overlap region nearer the Z-line by 1024 min. Comparison of pCa 9 and pCa 4 at the same time shows there are subtle differences in the profiles at both 4 and 1024 min. Note that the theoretical curve for a uniform distribution of labeled Tn along the length of the contractile thin filament would result in a broad peak in the middle of the half-sarcomere, much like that for the TnT sarcomere at pCa 9 and 1024 min of exchange. Thus, the images, intensity ratio data, and intensity profiles suggest that there are three different rates of Tn dissociation with a rank order of C>M>>B.
Figure 6.

Intra-sarcomeric relative intensity profiles for TnCIT and TnT myofibrils. The column average (4×44 pixel ROI) was obtained from pCa 9 (red) and 4 (blue), TnCIT (a) and TnT (b) myofibrils at 4 (broken line) and 1024 (continuous line) min from the images in Figure 5(a). For each myofibril, the intensity was normalized to the peak intensity within each sarcomere. Arrowheads mark the intensity peaks for pCa 9 while arrows mark the intensity peaks for pCa 4. For pCa 9, TnCIT myofibrils, there was no change in the position of the peak but there was a change in the intensity in the non-overlap region with increased time. For pCa 4, TnCIT myofibrils, the peak was in the non-overlap region at both 4 and 1024 min and there was an increase in the intensity in the overlap region with increased time. With TnT myofibrils, there was a change in the position of the peak with increased time. At pCa 9, the peak moved from the non-overlap to the A-I band junction while at pCa 4, it moved from the middle of the non-overlap region to nearer the Z-line.
The TnC–TnI regulatory domain affinity determines the calcium-dependent TnCIT dissociation rate
The above experiments suggest that when the regulatory domain of TnI is bound to the thin filament (either in TnCIT without calcium or TnIT thin filaments), Tn exchange is slow. An hypothesis is that if the affinity of the regulatory interaction between TnC and TnI at saturating calcium is altered, then the rate of Tn dissociation would be altered. To test this, engineered TnCs, with biochemically characterized TnI–TnC regulatory domain Kd42 values were incorporated into myofibrils followed by labeled Tn exchange. The TnCs had a 16-fold range of TnI affinity at saturating calcium. Figure 7 shows that a weaker regulatory interaction (higher Kd value) between TnI and TnC resulted in slower dissociation rates of Tn in the non-overlap region. There was no difference in the pattern between the different TnCs when exchange was done at pCa 9 (images not shown). The pCa 4 ATPase activity also graded with TnI–TnC regulatory domain affinity (data not shown). These observations suggest that the influence of saturating calcium on the dissociation of Tn is mediated through TnC–TnI regulatory domain interactions.
Figure 7.
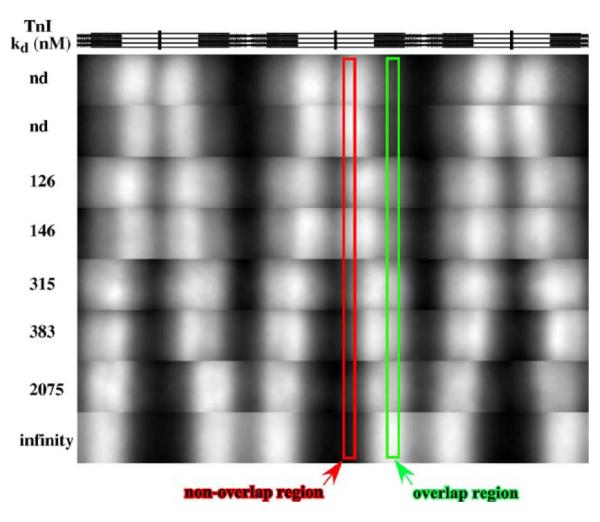
The C-state rates of Tn dissociation are determined by regulatory TnC–TnI interactions. TnIT was exchanged for native Tn, TnCs were added to 0.2 mg/ml TnIT myofibrils at 2 μM, incubated for 30 min, then labeled Tn exchange done at pCa 4 by mixing with an equal volume of 2 μM labeled Tn. Exchange was done for 16 min and samples were processed as for Figure 2. Images were cropped within the footprint of the fluorescence signal and then converted to 8 bit to maximize visualization of subtle differences. Annotations are as for Figure 2. A stick diagram of sarcomere anatomy is shown at the top as a map for the regions within the sarcomere. The nd samples are rabbit skeletal and wild-type chicken TnC, respectively, while 126, 146, 315, 383, and 2075 nM are F78Q TnCF29W, TnCF29W, M46Q TnCF29W, M82Q TnCF29W, and I62Q TnCF29W, respectively. Infinity is no TnC added (TnIT myofibril). The rate of Tn dissociation, in the presence of calcium (nominal C state), grades with TnC–TnI regulatory domain affinity. Note the regions of enhanced Tn exchange in the region just proximal to the overlap region for nearly all mutant TnCs.
Strong cross-bridges enhance calcium-dependent Tn dissociation in a cooperative manner
Observation of the patterns in Figure 7 suggests that the Tn’s dissociation rate was slightly greater in the C-state region, just proximal to the overlap region (i.e. those Tns close to, but not in actin-Tm units with rigor cross-bridges). We hypothesized that this is the result of signal transduction from the cross-bridge to Tn, several actins toward the Z-line, along the thin filament. To test this, labeled Tn was exchanged into myofibrils at different calcium levels. There are two potential outcomes for this experiment: (1) the dissociation rate of Tn in the non-overlap region will grade with [calcium] and be uniform within this region; or (2) the dissociation rate of Tn will grade with [calcium] but not be uniform within the non-overlap region, the dissociation rate will be faster the closer the Tn is to the overlap region. Outcome (1) suggests no long-range influence of the strong cross-bridge on Tn, whereas outcome (2) does. The resulting images from this experiment are shown in Figure 8. At 16 min, the rate of Tn dissociation in the non-overlap region was not uniform as the pCa was increased. Rather, there was a faster dissociation rate for those Tns closest to but not in the overlap region in the pCa range in which cooperative activation of myofibril ATPase activity was observed (ATPase data not shown). These experiments suggest that the rate of Tn dissociation was fastest at the overlap/non-overlap junction and decreased the closer the Tn was to the Z-line at sub-maximal calcium.
Figure 8.

The C-state rate of Tn dissociation is cooperative with calcium and strong cross-bridges. Labeled Tn exchange was done as for Figure 2, and presented as in Figure 6. The non-overlap region is noted by a red rectangle outline. The rate of Tn dissociation, at sub-maximal calcium (pCa 6.1 to 5.8, the region of cooperative activation of ATPase activity) was not uniform along the length of the non-overlap thin filament at 16 min. A region of enhanced exchange occurred just proximal to the overlap region, and depending on pCa, slowed from the proximal overlap region towards the Z-line. After extensive exchange (1024 min), the intensity was mostly equal in the non-overlap and overlap region except for pCa 9. A subtle sub-region of extra Tn exchange was apparent at pCa 4, just proximal to the Z-line. This was also evident for TnCIT and TnT myofibrils at pCa 4 (Figure 5(a)).
Discussion
Current models on thin filament regulation propose an equilibrium between three different states in relation to the position of Tm on the surface of actin.21,40 Shifting the proportion of actin-Tm between the different states involves Tn and the strong cross-bridge. No studies have determined if there are three different states of Tn that correspond to the proposed three different states of actin-Tm. Independent of any detailed analysis, the images from our studies clearly demonstrate that there are three different states of Tn, at least as determined by labeled Tn exchange pattern (Figures 3, 4, 5, 6, 7 and 8). A simple kinetic pathway for Tn dissociation from actin-Tm is shown in Figure 9. More refined analysis of the images leads to several additional interpretations.
Figure 9.
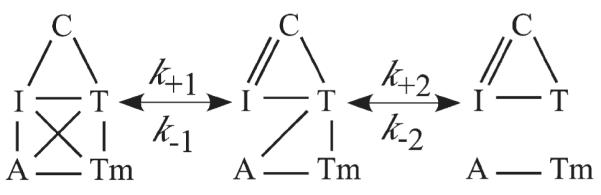
Pathway for Tn dissociation. Protein interactions are noted by lines connecting the letters with A, Tm, T, I, C, being actin, tropomyosin, TnT, TnI and TnC. Steps (1) or (2) can limit rate of Tn dissociation while step (1) is involved in regulation. Step (1) is dissociation of the regulatory domain of TnI from actin-Tm and its binding to the regulatory domain of TnC. Step (2) is TnT dissociation from actin-Tm. Step (1) limits the B-state region the rate of Tn dissociation while step (2) limits C- and M-state region dissociation rate. Calcium and strong cross-bridges influence step (1) while strong cross-bridges also influence step (2).
The first, and most simple interpretation is that calcium influences the apparent rate of Tn dissociation. If it is assumed that the Tn association rate is very rapid (105−108 M−1s−1) compared to the dissociation rate for the three different states of actin-Tm, then calcium weakens the binding of Tn to actin-Tm. This was proposed by Potter & Gergely8 over 30 years ago. Assuming an on-rate of 1×107, M−1s−1, and the estimates for the off-rate from Table 1 of 8.8×10−4 s−1 and 5×10−6 s−1 gives association constants of 1×1010 and 2×1012 M−1 for C and B states, respectively. Globally, these values are within three orders of magnitude of those determined by equilibrium linkage analysis of cardiac Tn binding to actin-Tm.43 However, attempts to quantify the affinity of Tn for actin-Tm by biochemical experiments observed small to no influence of calcium on the measured affinity.44,43 Other biochemical experiments using conformational45 and FRET46,47 probes suggest that TnI conformation changes or that it moves further from actin upon addition of calcium to reconstituted actin-Tm–Tn. This could result in weaker Tn binding to actin-Tm.
Our approach was rather simple, instead of focusing on the equilibrium binding constant, we focused on the dissociation rate. Also, our analytical method is most sensitive to small differences in dissociation rates (Figure 5). We observe that calcium enhances Tn dissociation about 200-fold (C state/B state). Further testing of the hypothesis that this is the result of the switching of the TnI regulatory domain between actin-Tm and TnC was done using Tn subunit-deficient myofibrils. The results show that the C-region rate was dependent upon calcium-saturated TnC (Figure 3). We further tested this hypothesis using engineered TnCs that bind TnI with different affinities at saturating calcium, with observations that strongly support the hypothesis (Figure 7). And lastly, we tested this hypothesis by varying free calcium concentration (Figure 8). Fundamentally, addition of calcium switches part of TnI from actin-Tm to TnC, an hypothesis presented many years ago and strongly supported by our studies. In terms of the kinetic pathway (Figure 9), at pCa 9, step (1) determines the rate of Tn dissociation whereas at pCa 4, step (2) determines the rate in the non-overlap region.
The second interpretation is that strong cross-bridges favor dissociation of TnI from actin-Tm. FRET studies suggest that the distance between TnI and actin increases upon strong cross-bridge binding,46,47 in agreement with our observations. This is shown by the rate of Tn dissociation being 100-fold faster in M-state regions than B state regions (Figure 5). Further support for this was obtained using TnIT myofibrils (Figure 4). We lost the calcium influence upon non-overlap Tn dissociation as we see no difference in the pattern of Tn exchange at high and low calcium with the overlap region being much faster than the non-overlap. In terms of the pathway, step (2) limits the rate Tn dissociation at pCa 9 in the overlap region while step (1) limits rate of Tn dissociation in the non-overlap region. An important question is how does the strong cross-bridge favor dissociation of TnI from actin-Tm? It could be by direct competition for the same site on actin or it could be through an influence of the strong cross-bridge upon Tm position that disfavors a TnI–actin interaction, thus favoring Tn dissociation.
The third interpretation is that strong cross-bridges favor dissociation of TnI from actin-Tm at actin sites distant from the rigor cross-bridge (Figures 7 and 8). A cartoon of this influence is shown in Figure 10 that is an extension of our earlier cartoon on the influence of the strong cross-bridge on switching actin-Tm sites into M state (see Figure 8 of Swartz et al.36). Biochemical studies suggest this in that the apparent number of actin-Tm sites exposed by one strong cross-bridge is greater than 7.34 Physiological studies suggest a similar exposure of M state actin-Tm sites.48 Structural studies done by us several years ago suggested this36 and later, electron microscopy studies on reconstituted actin-Tm–Tn confirmed our observations.18 Most of these studies used the strong cross-bridge to influence the thin filament state, but the strong cross-bridge itself alters the system. Furthermore, these studies did not determine if Tn was influenced. The current study investigated the influence of a strong cross-bridge on Tn, but distant from the strong cross-bridge. When we modulated the affinity of TnC for TnI (step (1)), either by using TnC mutants (Figure 7), or the physiological method, calcium (Figure 8), we observed an influence of the strong cross-bridge on Tn dissociation that was more than seven actins from the strong cross-bridge.
Figure 10.

Cartoon of the signal transduction pathway from the strong cross-bridge to Tn. Adapted from Swartz et al.36 Actin is represented by circles, Tm the contiguous red line, Tn the blue oblong structure, and the motor domain the dark brown, tadpole-like structure. Strong cross-bridges displace Tm into the M state. Because of Tm’s relative in-elasticity, this displacement is sensed by Tn, to favor TnI (arm-like structure projecting from Tn) dissociation from actin and binding to TnC, which favors calcium binding by TnC.
The observed patterns are very similar to those observed with fluorescent S1 binding experiments.36,37 Both this and our previous work suggest that Tm is displaced from the B-state position to M-(previous work) or C-state position (this work). Considering that dissociation of part of TnI from actin-Tm is likely required for C-region rates of Tn dissociation, the enhanced dissociation rate in the region of the thin filaments proximal to the overlap region results from the strong cross-bridge favoring TnI dissociation from actin-Tm and binding of this part of TnI to TnC. This binding of TnI to TnC then favors calcium binding by TnC,49,50 and propagates this effect down the thin filament. The proposed displacement occurs at a distance from the strong cross-bridge via a displacement of Tm (see Figure 10), not by direct displacement of TnI by the strong cross-bridge. Note that this propagation occurred at both ends of the thin filament if the sarcomere length was at about 2.2 μm (images not shown). Thus, this is an anatomical pathway for how the strong cross-bridge enhances calcium binding by TnC, part of the pathway for cooperative signal transduction. There could be calcium–calcium coupling between Tns along the thin filament to influence calcium binding by downstream TnCs that is independent of strong cross-bridges. This has been inferred from biochemical51 and physiological studies.48 Employing de-convolution methods or modeling of the convoluted half sarcomere intensity profile (see below) are required to address this using our approach.
There are recent models of the position of Tn on actin-Tm that propose different positions of Tn on actin-Tm to accomplish the same function.26,27 Both used electron microscopy/image reconstruction and single particle analysis, followed by docking of the Tn core domain crystal structures into the density envelope utilizing biochemical data on residue distances and interaction sites between actin and TnI. Both propose an additional mass on the reconstituted thin filament in the absence of calcium that is thought to be the regulatory domain of TnI. One model26 has Tn on the azimuthally adjacent actin monomers of the Tm it regulates with the TnI regulatory domain contacting the azimuthall adjacent actin. They propose that the TnI regulatory domain electrostatically alters the actin surface to maintain Tm in the B state. The other model27 has Tn on the same axial actin monomers as the Tm it regulates and is similar (but with much more detail) to classic models on Tn position.8 They propose that the Tn core domain and the TnI extension from it form a C-clamp configuration around Tm and actin to hold Tm in the B state. Our studies do not rule out either of these models. Both suggest the presence of a domain of TnI in B state filaments but not in C state filaments, and this supports the idea that the regulatory domain of TnI dissociates from actin when actin-Tm switches from the B to C state. Either model can also explain the long-range influence of the strong cross-bridge on TnI’s interaction with actin.
The fourth interpretation is that the strong cross-bridge may influence TnT (Figures 3 and 5). An influence of the strong cross-bridge on TnT position on the reconstituted thin filament was observed recently,52 in agreement with our observations. A caveat with our observations is that TnT interactions with actin-Tm may be different when in near quaternary (actin-Tm–TnT) or quaternary (actin-Tm–TnCIT) form.53,54 The structures for the Tn core domain show a coiled-coil formed by TnI and TnT that will not be present in TnT’s tertiary structure.10,11 The quaternary structure may position domains of TnT differently than in the tertiary structure and thus interact differently with actin-Tm. Surprisingly, we can readily reconstitute regulation of ATPase activity and troponin exchange pattern by adding back TnCI to TnT myofibrils, and do so using near stoichiomtric levels of TnCI (data not shown). Similarly, we can readily reconstitute inhibition of ATPase activity and Tn exchange rate by adding TnI to TnT myofibrils, again, at near stoichiometric levels (data not shown). Lastly, if TnT did not bind to the thin filament with reasonable affinity, it would not displace Tn very well and if it did, Tn exchange would be very fast relative to the time resolution of our methods. The estimated off rates for TnT are at most threefold faster than Tn (C state), suggesting a strong interaction between TnT and actin-Tm. These observations suggest that TnT has native tertiary structure in the TnT myofibril, but there could be quaternary structure missing that could alter the interactions.
The proposed strong cross-bridge influence on TnT can explain why the M region Tn dissociation is slower than the C region for native Tn at pCa 4 (Figures 3 and 5). The extensive regulatory interactions between TnC and TnI make it likely that the apparent affinity of TnI10,11 for actin-Tm is decreased, and considering that the strong cross-bridge may compete with the regulatory domain of TnI for a similar site on actin,22 it is even less likely that there are TnI interactions with actin-Tm. Within this framework, one would predict that the M-region rate of Tn dissociation would be faster than the C region if the Tn off-rate was solely mediated by TnI. The observation is that it is slower and thus the most likely mechanism for this is a strong cross-bridge influence on TnT. Further support for this is that the magnitude of the strong –cross-bridge affect on TnT is within twofold of what is observed for TnCIT at pCa 4 (Figure 5(b), y-intercepts). Thus, we infer that the strong cross-bridge influences TnT whether alone (near quaternary structure) or in Tn (complete quaternary structure). A possibility is that enhanced TnT binding to actin-Tm favors the M state position of Tm and thus activation of contraction. Biochemical studies suggest that the Tm binding domain of TnT modulates thin filament activation.53,54 This has important implications for TnT function, as there are cardiomyopathies associated with TnT mutations and altered contractile function, but the molecular pathway of signal transduction that is altered by these mutants is not easily explained. At present, we do not know which part of TnT, either its Tm binding domain or TnCI binding domain, or both, are influenced by the strong cross-bridge. Further studies are required to address this issue and if TnT functions differently when in tertiary form compared to quaternary form.
There are additional subtleties that can be interpreted from our images. These include an apparent influence of calcium on TnT dissociation (Figures 5(a) and 6(b)) and possibly “extra” Tn sites in the proximal Z-line region that are calcium dependent (Figures 5(a), 6(a) and (b), and 7). Recent work suggests that there are weak calcium binding sites in the Tm binding domain of skeletal TnT isoforms55 and this may be involved in the apparent influence of calcium on TnT exchange. In terms of extra Tn sites, careful observation of images (Figures 5(a), 6(a) and (b), and 7) shows that there is additional intensity in the region of the non-overlap region just proximal to the Z-line (most obvious in Figure 6(b); pCa 4). The width of this region is less than that of the non-overlap region. This Tn exchange near the Z-line is calcium-dependent as we do not observe it at pCa 9 in either TnCIT and especially in TnT myofibrils. This region has slower apparent exchange kinetics compared to C state regions. These extra sites likely explain why the ratio does not reach one for TnCIT or TnT myofibrils at pCa 4 (see Figure 5(b) and Table 1), since the non-overlap region of interest (ROI) intensity is likely the combination of the signal from “normal” and “extra” Tn exchanged in. Further studies are needed to address these issues. While this corrupts our current data analysis, we are confident in the magnitude of our estimates for B, C, and M regions. Future work can correct this problem by using longer thin filament myofibrils56 and moving the non-overlap ROI for ratio measurement, more distant from the Z-line.
Relating the current work to other studies that estimate the population of thin filament states is of interest. Biochemical studies suggest that at pCa 9, the fractional distribution of states is 0.7 B, 0.25 C and 0.05 M while at pCa 4 it is 0.0 B, 0.8 C and 0.2 M.39,40 Our work addresses the influence of calcium on the B to C equilibrium and does not estimate directly the proportion of states nor did we modulate the amount of strong cross-bridges to influence M-state occupancy. We can, however, infer that at pCa 9, the myofibril Tn state has a much higher fractional occupancy in B state. If it was 0.25 C, we would expect much faster exchange rates than we observe. This suggests, that in our system at pCa 9, the thin filament is more off, or B state, than in the reconstituted system.
There is a very important caveat when making this relationship: we investigated the state of Tn at its actin-Tm site, not the actin-Tm sites between the Tns along the thin filament. As alluded to in the Introduction, the state of Tn need not be directly coupled to that of the actin-Tm sites between the Tns for the system to work. At the extreme of tight coupling, requiring an in-elastic or rigid Tm, the state of Tn and actin-Tm would be directly coupled. This would result in the full length of a thin filament being in the same state (both Tn and actin-Tm). This is obviously not the case as shown here and our earlier work.36-38 At the extreme of very loose coupling, involving a highly elastic or non-rigid Tm, Tn could be in the B state but the actin-Tms between the Tns can be in any of the three states. This would result in only one to two of seven actins being regulated, likely not enough to regulate contraction. This highly elastic Tm model would result in there being no propagation of the strong cross-bridge effect. We do not observe this in the current and previous studies.36-38 Thus, we infer that it is somewhere between these extremes.
Modeling and measurement of Tm’s axial elasticity on the actin filament is of importance because it is one of the critical proteins involved in signal transduction along the thin filament. One can model the stress–strain relation of Tm using the worm-like chain model with the equation by Bustamante et al.57 The estimated persistence length of Tm is 55–170 nm.29 If we assume an extended molecular length of 40 nm, we can calculate the force required to extend (stretch) Tm. The force required to change the length of Tm from 39.8 to 39.9 nm is 0.6–2.5 nN. These calculations show that if Tm is near fully extended when on the actin filament, it requires significant force to “stretch” it and thus it can be in-elastic. Modulation of this elasticity, as may occur via different Tm isoforms, affords another avenue for striated muscle to tune its calcium response. Additional modulation of about one-third of the length of Tm could also occur via the TnT domain that binds to Tm, a region of TnT showing vast isoform differences.58
The current work strongly supports a long-range influence of the strong cross-bridge on Tn. Our images are underestimates of differences in rates in the different regions when one considers the Gaussian distribution of the detection of light from a point source by the camera and the nature of the imaging system. The current imaging and analysis is on the edge of being able to minimize the light emitted from one region influencing another. A case in point is the pCa 9 TnCIT myofibrils. The ratio is about 0.3 for the first 100 min of the reaction (Figure 5(b)). It should be much lower and confounds the estimates for B state exchange. We can improve our imaging fidelity and use sarcomeres with longer thin filaments to improve the quality of the data. Additionally, we can model the convoluted image data by fitting single sarcomere intensity profiles to the sum of Gaussian distributions of point light sources, especially since it is a very uniform structure.
As a final technical note, Tn binding to actin-Tm is strong, much like chromatin binding proteins in the nucleus59 or proteins in focal adhesion complexes.60 There are subtle changes in Tn subunit interactions with actin-Tm that regulate contraction. There may be similar subtle changes in protein interactions within other complex protein assemblies of the cell. Thus, the methods we developed have potential applications for studying protein interactions in other protein assemblies and how they are influenced by signal transduction. It is a very subtle technique that can be done within the near in situ structure.
Materials and Methods
Proteins
Myofibrils were prepared from rabbit psoas muscle as described by Swartz et al.36 with minor modifications of increasing the extent of Polytron homogenization (2×longer), and decreasing (twofold) the extent of Dounce homogenization and volume of wash buffer to muscle mass. Myofibril protein concentration was determined by the Biuret assay with bovine serum albumin as the standard. Troponin and its subunits were purified from rabbit white muscle as described by Greaser & Gergely.9 Mutant TnCs (chicken skeletal) were prepared and isolated as described61 and their protein concentration determined by the BCA assay using rabbit skeletal TnC as the standard (E280nm ml/mg=0.154, Mr=18,000). Troponin was labeled with Alexa 488 or Alexa 594 maleimide (Molecular Probes, Eugene OR). The protein was reduced by incubation in 100 mM KCl, 10 mM Hepes (pH 7.5), 2 mM MgCl2, 1 mM EGTA, 0.1%(v/v) 2-mercaptoethanol, 10 mM DTT for 30 min at 22 °C, desalted on a G-25 column equilibrated with the same buffer without reducing agent, mixed with 1.5–2 mol dye/mol protein and incubated overnight at 4 °C. The reaction was quenched with excess DTT, free dye removed by desalting on a G-25 column, and protein concentration determined by the BCA assay using unlabeled protein as standard assuming an E280nm ml/mg=0.45 and Mr=75,000. Labeling was >95% in TnI with the remainder in TnC and TnT, and the labeling ratio was 0.8–1.2 mol dye/mol Tn. Alexa 488 and 594 were used interchangeably as there was no influence of the dye on the pattern of exchange.
Troponin IT was prepared by mixing the subunits 1:1 (mol:mol) in 6 M urea, 400 mM KCl, 20 mM Mes (pH 6.5), 2 mM MgCl2, 0.5 mM EGTA, 1 mM NaN3, 1 mM DTT then serially dialyzed to give 0 M urea in the same buffer. Troponin T, for exchange, was prepared in the same manner. Protein concentrations were determined by spectroscopy using an E280nm ml/mg = 0.46 for TnIT and 0.50 for TnT. Troponin T or TnIT were exchanged for intrinsic Tn with methods modified from Shiratshi.62 Myofibrils were incubated with TnIT or TnT (0.3 mg/1 mg myofibril protein) twice at 25 °C for 2.5 h in 250 mM KCl, 20 mM Mes (pH 6.5), 2 mM MgCl2, 1 mM DTT, 0.6 mM CaCl2, 0.5 mM EGTA, 1 mM NaN3 then washed with pCa 9 buffer.
Troponin exchange
Labeled troponin was mixed with an equal volume of myofibrils giving a final of 1 μM labeled Tn and 0.1 mg/ml myofibrils (100 μl final) and incubated at room temperature. Labeled Tn was 16-fold that of the intrinsic Tn level. To stop the exchange reaction, myofibrils were pelleted, washed once with pCa 9 buffer, plated on coverslips, fixed with 3% (v/v) formaldehyde in pCa 9 buffer for greater than 5 min, washed with rigor buffer (75 mM KCl, 10 mM imidazole (pH 7.2), 2 mM MgCl2, 2 mM EGTA, 1 mM NaN3), drained and mounted on slides on mounting medium (rigor buffer made in 75% (v/v) glycerol with 1 mg/ml of p-phenylenediamine), excess medium wicked off, and coverslips sealed with clear nail polish. Slides were stored at −20 °C until imaged. We did not follow the same myofibril over time but sampled a myofibril population at different times. Labeled Tn exchange was done using native myofibrils or myofibrils that had prior exchange (>90%) with unlabeled TnT or TnIT as described above. For myofibrils containing mutant TnCs, TnIT was first exchanged for intrinsic TnCIT giving TnIT myofibrils followed by addition of 2 μM TnC (saturation confirmed by ATPase assays) to give TnCIT resulting in Tn being reconstituted on the thin filament of the myofibrils. Labeled Tn was added and the exchange done for 16 min. In all cases rabbit skeletal TnCIT was used as the labeled protein whereas different Tns (T, IT, or CIT with C being mutants) were pre-exchanged into the myofibrils.
Solutions and assays
pCa (−log [Ca2+]) solutions were made as described by Swartz et al.37 and contained 1 mM DTT and 1 mg/ml bovine serum albumin (BSA). Myofibrillar ATPase assays were done as described by Swartz et al.63 in a final volume of 100 μl with 10 μg of myofibrillar protein.
Imaging
Myofibrils were imaged using a Nikon Diaphot equipped with epifluorescence illumination, multi-band-pass filters, digital control of white light and excitation shutters and a 100×, NA 1.4, phase-contrast lens. Image light was collected with a 12 bit CoolSnap HQ camera controlled via IPLab (ver 3.2.2) run with a PowerMac 9600. The combination of lens and camera gave a virtual pixel size of 64 nm. Image collection was done using scripts that employed either autoexpose (pattern only) or exposure for fixed time (pattern and intensity, time set below saturation of pixels for longest incubation time). The script would acquire the phase-contrast image, then the fluorescence image, then save the images to disk. The script was set to loop 20 times to give 20 image pairs per slide.
Image analysis and presentation
Raw images were rotated to the horizontal using bilinear interpolation and rough cropped (100 pixels tall ×200 pixels wide) using the phase image to set rotation angle and pixel positions for cropping. The resulting image had the myofibril along the horizontal midline of the image and this was further cropped to give three contiguous sarcomeres (2 full+2 halves) with the image having the Z-line of the central sarcomere in the center of image. For quantitative analysis, the fluorescence image (acquired using fixed exposure time) was corrected for background by subtraction of the mean intensity of a ROI 5 pixels tall ×80 pixels wide, positioned in the upper left of the image. The net image was analyzed for sarcomere intensity by setting the minimum threshold at ten intensity units and obtaining the sum of pixels×intensity of the above threshold pixels. Thresholding at ten was not absolutely necessary but it allowed for quick visualization of the segment (myofibril intensity footprint) and minimized the contribution of low intensity pixels to the sum of low signal myofibrils.
The intensity ratio was determined by twofold enlarging the net image with bi-linear interpolation, determining the position of the Z-lines from the phase image, then using these positions to place ROIs (20 pixels tall ×4 pixels wide) ×32, ×12, +8 and +28 pixels from the respective Z-line, with the ROI vertically placed in the middle of the width of the half sarcomere. The mean intensity of the ROI was measured and used to calculate the intensity ratio (non-overlap intensity/overlap intensity). The mean of the six ratios was used to calculate the mean for the myofibril. The mean for the slide (time, pCa, Tn, etc.) was based upon the mean of the myofibrils. Both sarcomere intensity and intensity ratio were dependent upon sarcomere length. Thus, data sets for sarcomere intensity and intensity ratio were restricted to myofibrils with sarcomere lengths of 2.7–2.9 μm (n=5–12 per data point). Image presentation shows intensity within a myofibril not between myofibrils. For this, the image was converted from 12 bit to 8 bit resulting in the lowest intensity pixel being 0 and highest intensity pixel being 256, independent of the original low and high pixel. This allows for comparison of the pattern between myofibrils (images) even though there were large differences in the low and high pixel intensity between myofibrils. For Figures 3 and 4, the three sarcomere-cropped images were converted from 12 bit to 8 bit, and cropped along the midline of the myofibril to give the upper half as phase contrast and the lower as the fluorescence. These images were then montaged with this presentation aiding in anatomical location of the fluorescence intensity within the half sarcomere. For Figure 5, only fluorescence images are presented. The fluorescence images were cropped within the footprint of the sarcomere after converting to 8 bit and montaged with the others for the data set. For Figures 7 and 8, the ROI was delineated within the footprint of the myofibril, and this was converted from 12 to 8 bit. This is done to maximize presentation of subtle intensity differences, a major feature of this work. In all cases, primary selection criteria were sarcomere length and those nearest the mean for intensity ratio of the population sample. This minimized bias in what is presented and was truly representative of the sample population.
Single sarcomere intensity profiles from myofibrils presented in Figure 5(a) were obtained from the 8 bit images using a 4×44 (columns×rows) pixel ROI, placed along the midline of a sarcomere. The column average (four pixels) was calculated and normalized to the peak intensity for each sarcomere. The relative intensity profiles were plotted as a function of pixel (row) position.
Control experiments
To confirm that we were observing exchange of labeled Tn for TnCIT, TnIT or TnT in myofibrils, numerous experiments were done. To determine if we were getting “extra” Tn binding to myofibrils, labeled exchange was done for 4 h at pCa 4 or 9. The total Tn was compared between control and exchanged myofibrils by SDS-PAGE/silver staining and there was no difference in the density of the TnT and TnI bands between control and exchange myofibrils (data not shown). Comparison of the amount of labeled Tn by spectrofluorimetry of dissolved myofibrils and using labeled Tn as the standard showed that there was about 30% more labeled Tn in pCa 4 samples than pCa 9 (data not shown) because the exchange rate is very slow in the non-overlap region at pCa 9 (see Results). The amount of labeled Tn exchanged into the myofibrils was similar to that of the endogenous Tn at pCa 4. This suggests that there was minimal extra Tn binding and there was exchange of labeled protein for intrinsic. The pattern of labeled Tn exchange was independent of labeled Tn concentration when done at 10, 100 and 1000 nM labeled Tn, suggesting that the exchange rate was independent of labeled protein concentration within the range used for these studies (data not shown). An additional concern was inter-subunit exchange, that is exchange of free monomers (TnC, TnI or TnT) or dimer (TnCI) with those in Tn on the thin filament. We have previously done TnC exchange and it was pCa and cross-bridge dependent and rapid (ca ten times faster) compared to the fastest rates observed for whole Tn exchange (unpublished observations). Thus, we did not use labeled TnC in Tn as a probe for the rate of Tn dissociation. Rather, we used Tn that was labeled on TnI. To determine if labeled TnCI would exchange with TnCI in Tn on the thin filament, myofibrils were incubated with labeled TnCI (label in I) and there was very little labeled protein in the myofibrils after 24–48 h incubation (extremely slow). Thus, it was unlikely that TnCI was exchanging as a dimer with TnCI in Tn on the thin filament. Additional experiments to confirm that we were measuring whole Tn exchange involved doing labeled Tn exchange in rabbit psoas myofibrils containing either native or cardiac (c) Tn (done by pre-exchanging bovine cTn for native Tn), then doing labeled Tn exchange using either labeled skeletal (s) Tn or cTn. The labeled cTn had most of the label on cTnT whereas the labeled sTn had most on the sTnI. The exchange patterns were different and were dependent upon the Tn isoform in the myofibril not the labeled Tn used for the exchange. If there were exchange of just TnI and not TnCIT, this would not be the result. Thus, these numerous control experiments strongly suggest that we were measuring the rate of Tn dissociation and that we were not getting significant exchange of TnCI or TnI.
Acknowledgements
We thank Drs M. Greaser, S. Lehrer, and M. Geeves for valuable discussions on the work and on earlier versions of the manuscript. This work was supported by the AHA, Midwest Affiliate (to D.R.S. and Z.Y.) and Ohio Valley Affiliate (to J.P.D.) and NIH (AR020792 to J.P.D. and S.B.T.; HL073828 to D.R.S.).
Abbreviations used
- TnI
troponin I
- TnT
troponin T
- TnIT
troponin I and T complex
- TnCIT
troponin C, I, and T complex
- S1
myosin subfragment 1
- FRET
fluorescence resonance engery transfer
References
- 1.Gordon AM, Homsher E, Regnier M. Regulation of contraction in striated muscle. Physiol. Rev. 2000;80:853–924. doi: 10.1152/physrev.2000.80.2.853. [DOI] [PubMed] [Google Scholar]
- 2.Squire JM, Morris EP. A new look at thin filament regulation in vertebrate skeletal muscle. FASEB J. 1998;12:761–771. doi: 10.1096/fasebj.12.10.761. [DOI] [PubMed] [Google Scholar]
- 3.Bremel RD, Weber A. Cooperation within actin filament in vertebrate skeletal muscle. Nature New Biol. 1972;238:97–101. doi: 10.1038/newbio238097a0. [DOI] [PubMed] [Google Scholar]
- 4.Piroddi N, Tesi C, Pellegrino MA, Tobacman LS, Homsher E, Poggesi C. Contractile effects of the exchange of cardiac troponin for fast skeletal troponin in rabbit psoas single myofibrils. J. Physiol. 2003;552:917–931. doi: 10.1113/jphysiol.2003.051615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moss RL, Lauer MR, Giulian GG, Greaser ML. Altered Ca2+ dependence of tension development in skinned skeletal muscle fibers following modification of troponin by partial substitution with cardiac troponin C. J. Biol. Chem. 1986;261:6096–6099. [PubMed] [Google Scholar]
- 6.Spudich JA, Huxley HE, Finch JT. Regulation of skeletal muscle contraction. II. Structural studies of the interaction of the tropomyosin-troponin complex with actin. J. Mol. Biol. 1972;72:619–632. doi: 10.1016/0022-2836(72)90180-5. [DOI] [PubMed] [Google Scholar]
- 7.Hitchcock SE, Huxley HE, Szent-Gyorgyi AG. Calcium sensitive binding of troponin to actin-tropomyosin: a two-site model for troponin action. J. Mol. Biol. 1973;80:825–836. doi: 10.1016/0022-2836(73)90212-x. [DOI] [PubMed] [Google Scholar]
- 8.Potter JD, Gergely J. Troponin, tropomyosin, and actin interactions in the Ca2+ regulation of muscle contraction. Biochemistry. 1974;13:2697–2703. doi: 10.1021/bi00710a007. [DOI] [PubMed] [Google Scholar]
- 9.Greaser ML, Gergely J. Purification and properties of the components from tropinin. J. Biol. Chem. 1973;248:2125–2133. [PubMed] [Google Scholar]
- 10.Takeda S, Yamashita A, Maeda K, Maeda Y. Structure of the core domain of human cardiac troponin in the Ca(2+)-saturated form. Nature. 2003;424:35–41. doi: 10.1038/nature01780. [DOI] [PubMed] [Google Scholar]
- 11.Vinogradova MV, Stone DB, Malanina GG, Karatzaferi C, Cooke R, Mendelson RA, Fletterick RJ. Ca(2+)-regulated structural changes in troponin. Proc. Natl Acad. Sci. USA. 2005;102:5038–5043. doi: 10.1073/pnas.0408882102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heeley DH, Golosinska K, Smillie LB. The effects of troponin T fragments T1 and T2 on the binding of non-polymerizable tropomyosin to F-actin in the presence and absence of troponin I and troponin C. J. Biol. Chem. 1987;262:9971–9978. [PubMed] [Google Scholar]
- 13.Zhou X, Morris EP, Lehrer SS. Binding of troponin I and the troponin I-troponin C complex to actin-tropomyosin. Dissociation by myosin subfragment 1. Biochemistry. 2000;39:1128–1132. doi: 10.1021/bi992327m. [DOI] [PubMed] [Google Scholar]
- 14.Fuchs F. The binding of calcium to glycerinated muscle fibers in rigor. The effect of filament overlap. Biochim. Biophys. Acta. 1977;491:523–531. doi: 10.1016/0005-2795(77)90297-5. [DOI] [PubMed] [Google Scholar]
- 15.Hofmann PA, Fuchs F. Evidence for a force-dependent component of calcium binding to cardiac troponin C. Am. J. Physiol. 1987;253:541–546. doi: 10.1152/ajpcell.1987.253.4.C541. [DOI] [PubMed] [Google Scholar]
- 16.Pan BS, Solaro RJ. Calcium-binding properties of troponin C in detergent-skinned heart muscle fibers. J. Biol. Chem. 1987;262:7839–7849. [PubMed] [Google Scholar]
- 17.Cantino ME, Eichen JG, Daniels SB. Distributions of calcium in A and I bands of skinned vertebrate muscle fibers stretched to beyond filament overlap. Biophys. J. 1998;75:948–956. doi: 10.1016/S0006-3495(98)77583-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vibert P, Craig R, Lehman W. Steric-model for activation of muscle thin filaments. J. Mol. Biol. 1997;266:8–14. doi: 10.1006/jmbi.1996.0800. [DOI] [PubMed] [Google Scholar]
- 19.Vibert PJ, Haselgrove JC, Lowy J, Poulsen FR. Structural changes in actin-containing filaments of muscle. J. Mol. Biol. 1972;71:757–767. doi: 10.1016/s0022-2836(72)80036-6. [DOI] [PubMed] [Google Scholar]
- 20.Parry DA, Squire JM. Structural role of tropomyosin in muscle regulation: analysis of the X-ray diffraction patterns from relaxed and contracting muscles. J. Mol. Biol. 1973;75:33–55. doi: 10.1016/0022-2836(73)90527-5. [DOI] [PubMed] [Google Scholar]
- 21.Lehman W, Hatch V, Korman V, Rosol M, Thomas L, Maytum R, et al. Tropomyosin and actin isoforms modulate the localization of tropomyosin strands on actin filaments. J. Mol. Biol. 2000;302:593–606. doi: 10.1006/jmbi.2000.4080. [DOI] [PubMed] [Google Scholar]
- 22.Geeves MA, Chai M, Lehrer SS. Inhibition of actin-myosin subfragment 1 ATPase activity by troponin I and IC: relationship to the thin filament states of muscle. Biochemistry. 2000;39:9345–9350. doi: 10.1021/bi0002232. [DOI] [PubMed] [Google Scholar]
- 23.Grabarek Z, Tao T, Gergely J. Molecular mechanism of troponin-C function. J. Muscle Res. Cell. Motil. 1992;13:383–393. doi: 10.1007/BF01738034. [DOI] [PubMed] [Google Scholar]
- 24.Luo Y, Wu JL, Li B, Langsetmo K, Gergely J, Tao T. Photocrosslinking of benzophenone-labeled single cysteine troponin I mutants to other thin filament proteins. J. Mol. Biol. 2000;296:899–910. doi: 10.1006/jmbi.1999.3495. [DOI] [PubMed] [Google Scholar]
- 25.Smith DA, Geeves MA. Cooperative regulation of myosin-actin interactions by a continuous flexible chain II: actin-tropomyosin-troponin and regulation by calcium. Biophys. J. 2003;84:3168–3180. doi: 10.1016/S0006-3495(03)70041-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pirani A, Vinogradova MV, Curmi PM, King WA, Fletterick RJ, Craig R, et al. An atomic model of the thin filament in the relaxed and Ca2+-activated states. J. Mol. Biol. 2006;357:707–717. doi: 10.1016/j.jmb.2005.12.050. [DOI] [PubMed] [Google Scholar]
- 27.Murakami K, Yumoto F, Ohki SY, Yasunaga T, Tanokura M, Wakabayashi T. Structural basis for Ca2+-regulated muscle relaxation at interaction sites of troponin with actin and tropomyosin. J. Mol. Biol. 2005;352:178–201. doi: 10.1016/j.jmb.2005.06.067. [DOI] [PubMed] [Google Scholar]
- 28.Narita A, Yasunaga T, Ishikawa T, Mayanagi K, Wakabayashi T. Ca(2+)-induced switching of troponin and tropomyosin on actin filaments as revealed by electron cryo-microscopy. J. Mol. Biol. 2001;308:241–261. doi: 10.1006/jmbi.2001.4598. [DOI] [PubMed] [Google Scholar]
- 29.Phillips GN, Jr, Chacko S. Mechanical properties of tropomyosin and implications for muscle regulation. Biopolymers. 1996;38:89–95. doi: 10.1002/(SICI)1097-0282(199601)38:1%3C89::AID-BIP7%3E3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 30.Smith DA, Maytum R, Geeves MA. Cooperative regulation of myosin-actin interactions by a continuous flexible chain I: actin-tropomyosin systems. Biophys. J. 2003;84:3155–3167. doi: 10.1016/S0006-3495(03)70040-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brown JH, Zhou Z, Reshetnikova L, Robinson H, Yammani RD, Tobacman LS, Cohen C. Structure of the mid-region of tropomyosin: bending and binding sites for actin. Proc. Natl Acad. Sci. USA. 2005;102:18878–18883. doi: 10.1073/pnas.0509269102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Singh A, Hitchcock-DeGregori SE. Local destabilization of the tropomyosin coiled coil gives the molecular flexibility required for actin binding. Biochemistry. 2003;42:14114–14121. doi: 10.1021/bi0348462. [DOI] [PubMed] [Google Scholar]
- 33.Isambert H, Venier P, Maggs AC, Fattoum A, Kassab R, Pantaloni D, Carlier MF. Flexibility of actin filaments derived from thermal fluctuations. Effect of bound nucleotide, phalloidin, and muscle regulatory proteins. J. Biol. Chem. 1995;270:11437–11444. doi: 10.1074/jbc.270.19.11437. [DOI] [PubMed] [Google Scholar]
- 34.Geeves MA, Lehrer SS. Dynamics of the muscle thin filament regulatory switch: the size of the cooperative unit. Biophys. J. 1994;67:273–282. doi: 10.1016/S0006-3495(94)80478-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lehrer SS, Golitsina NL, Geeves MA. Actin-tropomyosin activation of myosin subfragment 1 ATPase and thin filament cooperativity. The role of tropomyosin flexibility and end-to-end interactions. Biochemistry. 1997;36:13449–13454. doi: 10.1021/bi971568w. [DOI] [PubMed] [Google Scholar]
- 36.Swartz DR, Greaser ML, Marsh BB. Regulation of binding of subfragment 1 in isolated rigor myofibrils. J. Cell Biol. 1990;111:2989–3001. doi: 10.1083/jcb.111.6.2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Swartz DR, Moss RL, Greaser ML. Calcium alone does not fully activate the thin filament for S1 binding to rigor myofibrils. Biophys. J. 1996;71:1891–1904. doi: 10.1016/S0006-3495(96)79388-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang D, Yancey KW, Swartz DR. Influence of ADP on cross-bridge-dependent activation of myofibrillar thin filaments. Biophys. J. 2000;78:3103–3111. doi: 10.1016/S0006-3495(00)76847-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McKillop DF, Geeves MA. Regulation of the interaction between actin and myosin subfragment 1: evidence for three states of the thin filament. Biophys. J. 1993;65:693–701. doi: 10.1016/S0006-3495(93)81110-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Head JG, Ritchie MD, Geeves MA. Characterization of the equilibrium between blocked and closed states of muscle thin filaments. Eur. J. Biochem. 1995;227:694–699. doi: 10.1111/j.1432-1033.1995.tb20190.x. [DOI] [PubMed] [Google Scholar]
- 41.Brenner B, Kraft T, Yu LC, Chalovich JM. Thin filament activation probed by fluorescence of N-((2-(Iodoacetoxy)ethyl)-N-methyl)amino-7-nitrobenz-2-oxa-1, 3-diazole-labeled troponin I incorporated into skinned fibers of rabbit psoas muscle. Biophys. J. 1999;77:2677–2691. doi: 10.1016/S0006-3495(99)77102-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Davis JP, Rall JA, Alionte C, Tikunova SB. Mutations of hydrophobic residues in the N-terminal domain of troponin C affect calcium binding and exchange with the troponin C-troponin I96-148 complex and muscle force production. J. Biol. Chem. 2004;279:17348–17360. doi: 10.1074/jbc.M314095200. [DOI] [PubMed] [Google Scholar]
- 43.Dahiya R, Butters CA, Tobacman LS. Equilibrium linkage analysis of cardiac thin filament assembly. Implications for the regulation of muscle contraction. J. Biol. Chem. 1994;269:29457–29461. [PubMed] [Google Scholar]
- 44.Wegner A, Walsh TP. Interaction of tropomyosin-troponin with actin filaments. Biochemistry. 1981;20:5633–5642. doi: 10.1021/bi00522a043. [DOI] [PubMed] [Google Scholar]
- 45.Trybus KM, Taylor EW. Kinetic studies of the cooperative binding of subfragment 1 to regulated actin. Proc. Natl Acad. Sci. USA. 1980;77:7209–7213. doi: 10.1073/pnas.77.12.7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tao T, Gong BJ, Leavis PC. Calcium-induced movement of troponin-I relative to actin in skeletal muscle thin filaments. Science. 1990;247:1339–1341. doi: 10.1126/science.2138356. [DOI] [PubMed] [Google Scholar]
- 47.Shitaka Y, Kimura C, Iio T, Miki M. Kinetics of the structural transition of muscle thin filaments observed by fluorescence resonance energy transfer. Biochemistry. 2004;43:10739–10747. doi: 10.1021/bi0492713. [DOI] [PubMed] [Google Scholar]
- 48.Regnier M, Rivera AJ, Wang CK, Bates MA, Chase PB, Gordon AM. Thin filament near-neighbour regulatory unit interactions affect rabbit skeletal muscle steady-state force-Ca(2+) relations. J. Physiol. 2002;540:485–497. doi: 10.1113/jphysiol.2001.013179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ramos CH. Mapping subdomains in the C-terminal region of troponin I involved in its binding to troponin C and to thin filament. J. Biol. Chem. 1999;274:18189–18195. doi: 10.1074/jbc.274.26.18189. [DOI] [PubMed] [Google Scholar]
- 50.Davis JP, Rall JA, Reiser PJ, Smillie LB, Tikunova SB. Engineering competitive magnesium binding into the first EF-hand of skeletal troponin C. J. Biol. Chem. 2002;277:49716–49726. doi: 10.1074/jbc.M208488200. [DOI] [PubMed] [Google Scholar]
- 51.Tobacman LS, Sawyer D. Calcium binds cooperatively to the regulatory sites of the cardiac thin filament. J. Biol. Chem. 1990;265:931–939. [PubMed] [Google Scholar]
- 52.Shitaka Y, Kimura C, Miki M. The rates of switching movement of troponin T between three states of skeletal muscle thin filaments determined by fluorescence resonance energy transfer. J. Biol. Chem. 2005;280:2613–2619. doi: 10.1074/jbc.M408553200. [DOI] [PubMed] [Google Scholar]
- 53.Maytum R, Geeves MA, Lehrer SS. A modulatory role for the troponin T tail domain in thin filament regulation. J. Biol. Chem. 2002;277:29774–29780. doi: 10.1074/jbc.M201761200. [DOI] [PubMed] [Google Scholar]
- 54.Tobacman LS, Nihli M, Butters C, Heller M, Hatch V, Craig R, et al. The troponin tail domain promotes a conformational state of the thin filament that suppresses myosin activity. J. Biol. Chem. 2002;277:27636–27642. doi: 10.1074/jbc.M201768200. [DOI] [PubMed] [Google Scholar]
- 55.Zhang Z, Jin JP, Root DD. Binding of calcium ions to an avian flight muscle troponin T. Biochemistry. 2004;43:2645–2655. doi: 10.1021/bi035067o. [DOI] [PubMed] [Google Scholar]
- 56.Ringkob TP, Swartz DR, Greaser ML. Light microscopy and image analysis of thin filament lengths utilizing dual probes on beef, chicken, and rabbit myofibrils. J. Anim. Sci. 2004;82:1445–1453. doi: 10.2527/2004.8251445x. [DOI] [PubMed] [Google Scholar]
- 57.Bustamante C, Marko JF, Siggia ED, Smith S. Entropic elasticity of lambda-phage DNA. Science. 1994;265:1599–1600. doi: 10.1126/science.8079175. [DOI] [PubMed] [Google Scholar]
- 58.Briggs MM, Lin JJ, Schachat FH. The extent of amino-terminal heterogeneity in rabbit fast skeletal muscle troponin T. J. Muscle Res. Cell Motil. 1987;8:1–12. doi: 10.1007/BF01767259. [DOI] [PubMed] [Google Scholar]
- 59.Misteli T. Protein dynamics: implications for nuclear architecture and gene expression. Science. 2001;291:843–847. doi: 10.1126/science.291.5505.843. [DOI] [PubMed] [Google Scholar]
- 60.Petit V, Thiery JP. Focal adhesions: structure and dynamics. Biol. Cell. 2000;92:477–494. doi: 10.1016/s0248-4900(00)01101-1. [DOI] [PubMed] [Google Scholar]
- 61.Tikunova SB, Rall JA, Davis JP. Effect of hydrophobic residue substitutions with glutamine on Ca(2+) binding and exchange with the N-domain of troponin C. Biochemistry. 2002;41:6697–6705. doi: 10.1021/bi011763h. [DOI] [PubMed] [Google Scholar]
- 62.Shiraishi F, Kambara M, Ohtsuki I. Replacement of troponin components in myofibrils. J. Biochem. (Tokyo) 1992;111:61–65. doi: 10.1093/oxfordjournals.jbchem.a123719. [DOI] [PubMed] [Google Scholar]
- 63.Swartz DR, Moss RL, Greaser ML. Characteristics of troponin C binding to the myofibrillar thin filament: extraction of troponin C is not random along the length of the thin filament. Biophys. J. 1997;73:293–305. doi: 10.1016/S0006-3495(97)78070-6. [DOI] [PMC free article] [PubMed] [Google Scholar]