

Abstract
Polycyclic aromatic hydrocarbons (PAH) are widespread environmental contaminants, and some are potent carcinogens in rodents. Carcinogenic PAH are activated in cells to metabolites that react with DNA to form stable covalent DNA adducts. It has been proposed [Cavalieri, E. L. & Roger, E. G. (1995) Xenobiotica 25, 677–688] that unstable DNA adducts are also formed and that apurinic sites in the DNA resulting from unstable PAH adducts play a key role in the initiation of cancer. The potent carcinogen dibenzo[a,l]pyrene (DB[a,l]P) is activated in cells to (+)-syn- and (−)-anti-DB[a,l]P-11,12-diol-13,14-epoxide (DB[a,l]PDE), which have been shown to form stable adducts with DNA. To evaluate the importance of unstable PAH adducts, we compared stable adduct formation to apurinic site formation. Stable DB[a,l]PDE adducts were determined by 33P-postlabeling and HPLC. To measure apurinic sites they were converted to strand breaks, and these were monitored by examining the integrity of a particular restriction fragment of the dihydrofolate reductase gene. The method easily detected apurinic sites resulting from methylation by treatment of cells or DNA with dimethyl sulfate or from reaction of DNA with DB[a,l]P in the presence of horseradish peroxidase. We estimate the method could detect 0.1 apurinic site in the 14-kb fragment examined. However, apurinic sites were below our limit of detection in DNA treated directly with (+)-syn- or (−)-anti-DB[a,l]PDE or in DNA from Chinese hamster ovary B11 cells so treated, although in these samples the frequency of stable adducts ranged from 3 to 10 per 14 kb. We also treated the human mammary carcinoma cell line MCF-7 with DB[a,l]P and again could not detect significant amounts of unstable adducts. These results indicate that the proportion of stable adducts formed by DB[a,l]P activated in cells and its diol epoxides is greater than 99% and suggest a predominant role for stable DNA adducts in the carcinogenic activity of DB[a,l]P.
Covalent binding of chemical carcinogens, including the environmental polycyclic aromatic hydrocarbons (PAH), to target cell DNA is a critical first step in the process of tumor initiation (1, 2). Before they can interact with DNA, PAH require metabolic activation to chemically reactive metabolites (3). The most well characterized pathway of activation involves two oxidations of the PAH catalyzed by cytochrome P450s and an intermediate hydrolysis by epoxide hydrolase to produce a reactive vicinal diol epoxide (refs. 3–5, and references cited therein). This pathway of metabolic activation can lead to sterically hindered bay- or fjord-region diol epoxides and has been observed for many carcinogenic PAH, including benzo[a]pyrene (B[a]P) (4), benzo[c]phenanthrene (6), and 7,12-dimethylbenz[a]anthracene (7). Another potential pathway of activation involves one-electron oxidation of the PAH to form a reactive radical cation (8, 9).
The most highly carcinogenic environmental PAH identified to date is dibenzo[a,l]pyrene (DB[a,l]P) (10, 11). It has been detected in several environmental soil and sediment samples (12), in a biologically active fraction of cigarette smoke condensate (13), and in the particulates formed by combustion of smoky coal (14). DB[a,l]P is highly mutagenic in MCL-5 cells, a cell line derived from human B-lymphoblastoid cells that expresses several transfected cytochrome P450 genes and thus is capable of metabolizing PAH (15). Low doses of DB[a,l]P are mutagenic in the V79 Chinese hamster lung cell line cocultured with MCF-7 human mammary carcinoma cells to provide metabolic activation capacity (16). In cells that are able to metabolize PAH, DB[a,l]P is activated to electrophilic metabolites, which react with nucleophiles such as DNA (16). Although DB[a,l]P (Fig. 1) could in theory be metabolized to three regioisomeric diol epoxides—a bay-region 1,2-diol-3,4-epoxide and two fjord-region epoxides, the 3,4-diol-1,2-epoxide and the 11,12-diol-13,14-epoxide—studies of the covalent DNA adducts formed from DB[a,l]P in human and rodent cells in culture demonstrated that only one regioisomeric diol epoxide is formed, the 11,12-diol-13,14-epoxide (17). In addition, activation of DB[a,l]P takes place with high stereoselectivity. Only the (−)-anti-(11R,12S,13S,14R)- and the (+)-syn-(11S,12R,13S,14R)-DB[a,l]PDE (see Fig. 1) were detected (18, 19). Over 73% of the DNA adducts formed by these diol epoxides arose from reaction with deoxyadenosine; the anti-diol epoxide formed 17% deoxyguanosine adducts, but the syn-diol epoxide formed less than 9% (18).
Figure 1.
Structures of the parent hydrocarbon DB[a,l]P and its two metabolically formed fjord-region (−)-anti- and (+)-syn-DB[a,l]PDE.
Cavalieri and Rogan (20) have reported that PAH can also be activated to radical cations that react with DNA to form labile adducts destabilizing the glycosidic bond and leading to loss of the modified base by depurination. For DB[a,l]P activated by 3-methylcholanthrene-induced rat liver microsomes, they reported that 84% of the DNA adducts were depurinating adducts and only 16% were stable adducts (21). Although many of the unstable adducts were reported to be those potentially formed from radical cation reactions, unstable syn-DB[a,l]PDE adducts represented 31% and unstable anti-DB[a,l]PDE adducts represented 3% of the total adducts. Because the stable DB[a,l]PDE adducts represented only 16% of the total adducts, this finding would imply that at least two-thirds of the DB[a,l]PDE adducts are unstable and that the proportion is much greater than this for syn-DB[a,l]PDE adducts (21). They hypothesized that the apurinic (AP) sites formed by depurination of these unstable adducts as well as those formed through reactions of DB[a,l]P radical cation could be involved in tumor initiation by DB[a,l]P (21, 22).
To determine which potential mechanism of activation of the potent carcinogen DB[a,l]P is involved in damaging DNA and ultimately in tumor formation, we developed a procedure to measure the proportion of stable and unstable adducts formed by reaction of DB[a,l]P and DB[a,l]PDE with DNA. Either Chinese hamster ovary B11 cells or DNA isolated from such cells was treated with (+)-syn- or (−)-anti-DB[a,l]PDE, and human mammary carcinoma cells (MCF-7) known to metabolically activate PAH were treated with DB[a,l]P; both stable and unstable adducts were measured. Stable adducts were measured by postlabeling with 33P and separation and quantitation with HPLC. The unstable adducts are reported (21) to result in the formation of AP sites in the DNA; a sensitive assay for such sites was devised, which uses Southern blotting analysis of the integrity of a restriction fragment of the dihydrofolate reductase (DHFR) gene after alkaline hydrolysis. Studies with DNA methylated by dimethyl sulfate (DMS) or incubated with horseradish peroxidase (HRP) plus DB[a,l]P, a system known to activate DB[a,l]P to radical cations (21), demonstrated that the formation of AP sites can be detected by this technique. This analysis allowed a quantitative comparison of the proportion of stable to unstable depurinating adducts formed both in DNA in solution and in DNA in cells.
MATERIALS AND METHODS
Cell Culture.
The Chinese hamster ovary cell line (CHO-B11) with a 50-fold amplified DHFR gene was grown in 150-mm2 tissue culture dishes with minimal essential medium (MEM) supplemented with 10% dialyzed fetal calf serum, penicillin and streptomycin, l-glutamine, nonessential amino acids, and 460 nM methotrexate in 5% CO2/95% air at 37°C (23, 24). The human mammary carcinoma MCF-7 cell line was grown in 175-cm2 flasks with MEM supplemented with 10% fetal calf serum, sodium pyruvate, and nonessential amino acids in 5% CO2/95% air at 37°C.
Preparation and Reaction of DNA.
Cells were lysed and digested with proteinase K (0.1 mg/ml) in TE (10 mM Tris/1 mM EDTA, pH 8.0)/0.5% SDS, pH 8.0, for 10–12 hr at 37°C. DNA was extracted with phenol and chloroform/isoamyl alcohol (24:1), precipitated with 2 vol of 100% ethanol, dissolved in TE, and digested with the restriction endonuclease KpnI (10 units/μg of DNA) at 37°C for 2 hr (24).
KpnI-restricted DNA was reacted with a freshly prepared solution of 0.1 M DMS (Aldrich) in 50 mM sodium cacodylate, pH 8.0, and TE for 4 min at 22°C. After addition of 0.2 vol of 1.0 M 2-mercaptoethanol, 1.5 M sodium acetate, pH 7.0, the DNA was precipitated with ethanol and dissolved in TE. The DNA was stored at 4°C and then heated at 50°C for 6 hr to depurinate at sites of the N-methylpurines (25).
(+)-syn- and (−)-anti-DB[a,l]PDE were synthesized as previously described (26, 27) and dissolved in dry dimethyl sulfoxide (DMSO). KpnI-restricted B11 DNA (1.25 mg) was allowed to react with 76 μM (+)-syn- and 13 μM (−)-anti-DB[a,l]PDE at 37°C for 24 hr and then heated as above.
For reaction of DNA with DB[a,l]P in the presence of HRP/H2O2, KpnI-restricted DNA (500 μg) isolated from CHO-B11 cells was exposed to 20, 40, or 80 μM DB[a,l]P (Chemsyn Science Laboratories, Lenexa, KS) in the presence of 0.5 mM H2O2 and 0.1 mg/ml HRP (Sigma) in 0.067 M potassium phosphate, pH 7. Following a reaction period of 1 hr at 37°C, the DNA was precipitated by addition of 2 vol of ethanol and dissolved in TE (28).
Cell Treatment.
Medium was changed when cells had covered >90% of the surface area of the culture dish. Twenty-four hours later CHO-B11 cells were treated with 150 mM DMS or 1.4, 4.3, or 8.5 μM (+)-syn- or (−)-anti-DB[a,l]PDE or solvent (DMSO) alone. The cells were harvested 1 hr after treatment, and the DNA was isolated and treated as above. The MCF-7 cells were treated with DMSO alone or with 1 or 2 μM DB[a,l]P for 24 hr. The MCF-7 cells were harvested and the isolated DNA was digested as described.
Detection of AP Sites by DNA Cleavage.
After the CHO-B11 DNA samples had been incubated at 50°C, one portion of the DNA reacted with DMS was incubated in the presence of 40 mM methoxyamine (Sigma) at pH 7.2 and 37°C for 30 min to reduce chemically the AP sites and protect them from the subsequent alkaline hydrolysis (25, 29, 30). In a similar way, equal portions of the DNA reacted with DB[a,l]P in the presence of HRP and in cells were incubated with methoxyamine. Samples were then treated with 0.1 M NaOH at 37°C for 30 min to cleave the DNA at all AP sites (24). The DNA was subjected to electrophoresis on a 0.5% alkaline-agarose gel overnight and transferred from the gel to a support membrane (Hybond N+ nylon) in 20× SSPE (3.6 M NaCl/0.2 M NaH2PO4/22 mM EDTA/NaOH to pH 7.7–8.0). The DNA was fixed to the membrane with 0.4 M NaOH and probed with a radiolabeled riboprobe prepared with plasmid pZH5 or pGEM0.69, which recognizes a 14-kb fragment of the hamster DHFR gene or a 20-kb fragment of the human DHFR gene, respectively (Promega Riboprobe System kit). After high stringency washing (0.1× SSPE/0.1% SDS, 65°C), radioactivity associated with the full-length fragment was measured by autoradiography or with a model GS-250, Molecular Imager Phosphor Imaging System (Bio-Rad).
33P-Postlabeling Analysis.
The DB[a,l]PDE-DNA from CHO-B11 and MCF-7 cells (2.5 μg) was digested with nuclease P1 (0.6 unit) and prostatic acid phosphatase (0.35 unit) in 12.5 mM sodium acetate/3 mM ZnCl2, pH 5.2 at 37°C for 45 min (18). The samples were then evaporated to 5 μl and incubated with [γ-33P]ATP (1.85 MBq; 74 TBq/mmol), T4 polynucleotide kinase (18 units), and kinase buffer (2 μl; 0.5 M Tris/8 mM spermidine/100 mM MgCl2/0.1 M DTT, pH 9.6) at 37°C for 1 hr (30). They were then incubated with snake venom phosphodiesterase (0.013 unit) and apyrase (0.1 unit) for 1 hr at 37°C and stored at −20°C (31).
Detection of Stable Adducts by HPLC Analysis of the 33P-Postlabeled Samples.
The 33P-postlabeled adducts were isolated from unreacted [γ-33P]ATP, [33P]phosphate, and unlabeled nucleosides by chromatography on a Sep-Pak C18 cartridge as previously described (18). Before sample application, the cartridge was conditioned with methanol (10 ml), distilled water (10 ml), and loading buffer (10 ml; 0.5 M potassium phosphate, pH 6.0). The samples were diluted in loading buffer (10 ml), and to ensure maximum retention of the labeled DNA samples, they were applied twice to the cartridge. The cartridge was washed three times with distilled water (10 ml) and once with a diluted basic methanol solution [0.25% ammonium hydroxide (14.5 M)/4.75% methanol/95% distilled water]. The labeled PAH–DNA adducts were eluted from the cartridge with 5% basic methanol solution [2 ml; 5% ammonium hydroxide (14.5 M)/95% methanol].
The DB[a,l]PDE–DNA adducts (106 cpm, as determined by scintillation spectrometry) were analyzed by HPLC on a C18 column (5 μm Ultrasphere, 4.6 × 250 mm; Beckman), with a linear gradient consisting of 0.1 M ammonium phosphate buffer, pH 5.5 (solvent A) and 10% acetonitrile/90% methanol (solvent B) at a flow rate of 1 ml/min. The gradient used was: 44–49% B over 40 min; 49–55% B over 60 min; 55–65% B over 20 min (18). The radioactivity was detected by an on-line radioisotope flow detector (Radiomatic Flow-One Beta; Packard).
RESULTS
Strategy.
To determine the proportions of stable and unstable adducts formed by reaction of DB[a,l]PDE with DNA, we used a standard postlabeling procedure to measure the frequency of stable adducts and developed a sensitive assay for the frequency of AP sites present in the DNA, as a result of the formation of unstable adducts. Cavalieri and Rogan (20) report that these unstable adducts depurinate very rapidly after formation and cannot be attached to the DNA. For stable adducts the DNA was digested to DB[a,l]PDE-adducted dinucleotides with nuclease P1 and prostatic acid phosphatase, the 5′-hydroxyl of the adducted nucleotide was labeled with 33P by using [γ-33P]ATP and T4 kinase, and finally the 3′ nucleotide was removed with snake venom phosphodiesterase to yield a 5′ [33P]DB[a,l]PDE–deoxyribonucleotide. The labeled species were then analyzed by HPLC with radiochemical detection to identify and quantitate the stable DB[a,l]PDE–DNA adducts.
The assay for AP sites in the DNA is based on the specificity of nucleic acid hybridization and the resolving power of alkaline-agarose gels and is adapted from the technique developed to measure repair of alkylation damage in specific sequences of the genome (24). Purified cellular DNA was digested with the restriction endonuclease KpnI and after various treatments, underwent electrophoresis on alkaline-agarose gels and was transferred to a hybridization support membrane. The membrane was probed with a 32P-labeled riboprobe specific for a sequence in the transcription template strand of a 14-kb KpnI fragment at the 5′ end of the DHFR gene. (In the B11 cells, this gene is present in approximately 50 copies, which facilitates the analysis, but this is not an essential requirement. In MCF-7 cells, which have a normal copy number, a probe was used for the 20-kb fragment of the DHFR gene.) A single AP site in one of the DNA fragments, when converted to a strand break by the incubation in strong alkali, removes that fragment from the population that migrates in electrophoresis to the position of 14-kb (or 20-kb for the human DNA) fragments. AP sites randomly introduced to an average of one per fragment result in a reduction in hybridization at the position of full-length fragments to 37% of the control value. A reduction in hybridization to 80%, easily measured, would result from an average of 0.2 site per fragment. Thus the sensitivity of the assay is, in theory, at least 1 site per 60,000 bases and would be capable of detecting AP sites if they occurred even at the level of 10% or so of the expected frequency of stable adducts.
In practice, the hybridization signal in the test DNA must be compared with some control, representing the total amount of fragment in the assay, as only the quantity of full-length fragments is measured. If the control is DNA simply not treated with the damaging agent, care must be taken to ensure that identical amounts of DNA are analyzed, and the possible effects of the necessarily different histories of the samples must be considered. To circumvent these issues Scicchitano and Hanawalt (25) devised a method in which the DNA sample is divided in half just before analysis, one of the two resulting samples is allowed to react with methoxyamine to render AP sites resistant to alkaline-induced strand cleavage, and both samples are incubated with alkali and then analyzed. In addition, a small amount of a control DNA fragment, which also contains the hybridization sequence, is added to the sample just before the analysis to allow further correction for sample loading errors.
Formation of Adducts in Purified DNA.
KpnI-restricted B11 cellular DNA was allowed to react with (+)-syn- or (−)-anti-DB[a,l]PDE for 24 hr to ensure complete reaction and adequate time for depurination at the sites of any unstable adducts. One portion of the reaction mixture was used for 33P postlabeling, and the remainder was used for DNA fragment analysis by gel electrophoresis. To eliminate any possibility that AP sites escaped detection because of failure to precipitate short DNA fragments, the DB[a,l]PDE-reacted DNA was not precipitated or extracted with organic solvents to remove tetraols derived from DB[a,l]PDE before electrophoresis. Initial experiments comparing organic solvent-extracted DNA to the DNA in the complete reaction mixture demonstrated that the presence of tetraols had no effect on the mobility of the 14-kb DHFR gene fragment in alkaline-agarose gels.
To serve as a positive control for the assay, a portion of this B11 DNA was treated with DMS to produce heat-labile methylpurines (24) and heated at 50°C to produce AP sites. These samples and those from the DNA treated with (+)-syn- or (−)-anti-DB[a,l]PDE were then assayed as described under Materials and Methods (Fig. 2). Incubation of completely untreated DNA with NaOH had no effect on the amount of intact 14-kb fragment present. For the DNA treated with DMS and methyoxyamine, treatment with NaOH had little or no effect on the amount of full-length fragment, judged by comparison with the untreated control DNA, but it greatly reduced the amount of intact fragment in the unprotected DNA, showing that the DMS-treated DNA did indeed contain AP sites.
Figure 2.
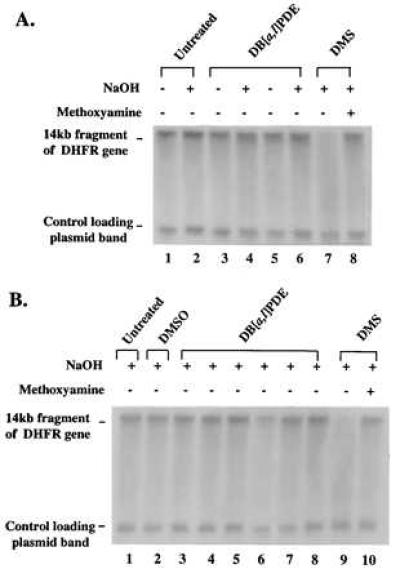
Alkaline-agarose gel electrophoresis of DB[a,l]PDE-reacted B11 cell DNA and DNA from DB[a,l]PDE-treated B11 cells. CHO-B11 DNA and B11 cells in culture were treated with DB[a,l]PDE. The DNA was incubated in the presence or absence of sodium hydroxide (indicated above the lanes) for 30 min at 37°C (see Materials and Methods). DMS-methylated DNA was included as a positive control for the detection of AP sites. (A) KpnI-restricted DNA: untreated (lanes 1 and 2); reacted with 76 μM (+)-syn-DB[a,l]PDE (lanes 3 and 4); reacted with 13 μM (−)-anti-DB[a,l]PDE (lanes 5 and 6); reacted with 0.1 M DMS (lane 7 without methoxyamine treatment and lane 8 with methoxyamine treatment). (B) KpnI-restricted DNA from CHO-B11 cell cultures treated with: no treatment (lane 1); DMSO (lane 2); 1.4, 4.3, or 8.5 μM (+)-syn-DB[a,l]PDE (lanes 3–5); 1.4, 4.3, or 8.5 μM (−)-anti-DB[a,l]PDE (lanes 6–8); or DMS [without methoxyamine (lane 9) or with methoxyamine (lane 10)]. All samples underwent electrophoresis on a 0.5% alkaline-agarose gel and were probed with a 32P riboprobe for the transcribed strand of the 14-kb fragment of the DHFR gene.
However, neither (+)-syn- nor (−)-anti-DB[a,l]PDE treatment had any detectable effect on the amount of full-length fragment either with or without alkaline incubation. The relative amounts of the full-length fragments, corrected for loading by using the plasmid band amounts, were calculated (Fig. 3A); it is clear that the amount of full-length fragment in the DB[a,l]PDE-reacted samples was identical to that in the untreated control, indicating that reaction with DB[a,l]PDE under the conditions used created no unstable adducts measurable by this assay (Fig. 3A). The same result was obtained when methoxyamine pretreatment of the DB[a,l]PDE samples was carried out (data not shown) as would be expected because no AP sites were detected in these samples.
Figure 3.
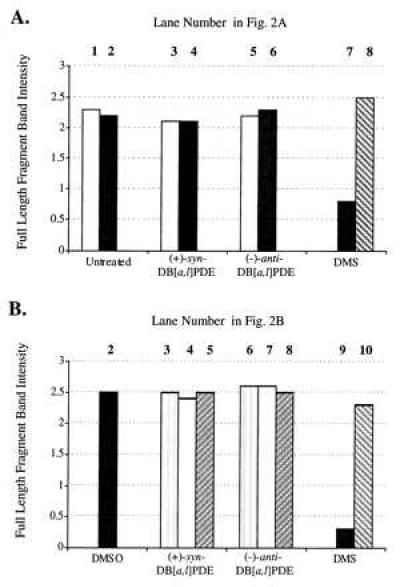
Intensity of the 14-kb band of the DHFR gene of the DNA samples shown in Fig. 2. All band intensities were normalized for the efficiency of sample loading on the gel through the use of a plasmid loading control. (A) DNA that had reacted with DB[a,l]PDE in solution. (B) DNA from cells treated with DB[a,l]PDE. The lane numbers corresponding to the sample in Fig. 2 are indicated above each sample.
The (+)-syn- and (−)-anti-DB[a,l]PDE used in this experiment were chemically reactive with DNA; measurement of stable adducts (Table 1) indicated 3 and 10 adducts per 14 kb, respectively. Thus, for (−)-anti-DB[a,l]BDE, the level of unstable adducts formed must have been well below 2% of that of stable adducts.
Table 1.
Stable adducts per 14 kb of DB[a,l]PDE-reacted DNA from CHO-B11 cells and per 20 kb of DNA from DB[a,l]P-treated MCF-7 cells
| Type of DB[a,l]PDE | Conc., μM | Stable adducts per 14- and 20-kb fragment |
|---|---|---|
| B11 DNA | ||
| (+)-syn-DB[a,l]PDE | 76 | 3 |
| (−)-anti-DB[a,l]PDE | 13 | 10 |
| B11 cells | ||
| (+)-syn-DB[a,l]PDE | 1.4 | 3 |
| 4.3 | 8 | |
| 8.5 | 7 | |
| (−)-anti-DB[a,l]PDE | 1.4 | 3 |
| 4.3 | 7 | |
| 8.5 | 14 | |
| MCF-7 cells | ||
| DB[a,l]P | 1 | 8 |
| 2 | 10 |
Formation of Adducts in Cells.
We next measured the formation of stable adducts and AP sites in DNA when cells in culture were treated with either (+)-syn- or (−)-anti-DB[a,l]PDE. Cells were treated for 1 hr with a concentration ranging from 1.4 to 8.5 μM and harvested, and their DNA was isolated and analyzed (Fig. 2B). Control DNA from untreated cultures, DNA from DMSO (solvent)-treated cultures, and DNA from DB[a,l]PDE-treated cultures contained 14-kb DHFR bands of nearly equal intensity. DNA from DMS-treated cultures that had been incubated with methoxyamine also contained such a band, but its counterpart, not treated with methoxyamine, was very faint, indicating efficient production of heat-labile lesions. The normalized intensity ratios of the full-length fragment bands (Fig. 3B) show once again no detectable formation of unstable adducts by the hydrocarbon diol epoxides. Stable adducts were detected at all concentrations (Table 1), and their frequency was dose-dependent.
Experiments with DB[a,l]P.
The foregoing provides strong evidence that the reactive diol epoxides at the concentrations we used form few if any unstable adducts by a direct addition mechanism, either in pure DNA or in cells. Unstable adduct formation has been reported to result from another route involving peroxidative activation of the parent compound DB[a,l]P to a radical cation (20). To investigate this, we first incubated purified B11 DNA in various concentrations of DB[a,l]P in the presence of HRP and hydrogen peroxide under conditions reported to activate DB[a,l]P to radical cations (28) and analyzed the DNA as before (Fig. 4). In this experiment, we did observe clear evidence for formation of AP sites. A decrease in the amount of full-length fragments appeared with treatment, in a dose-dependent manner, and was prevented by incubation of the DNA with methoxyamine before incubation in NaOH.
Figure 4.
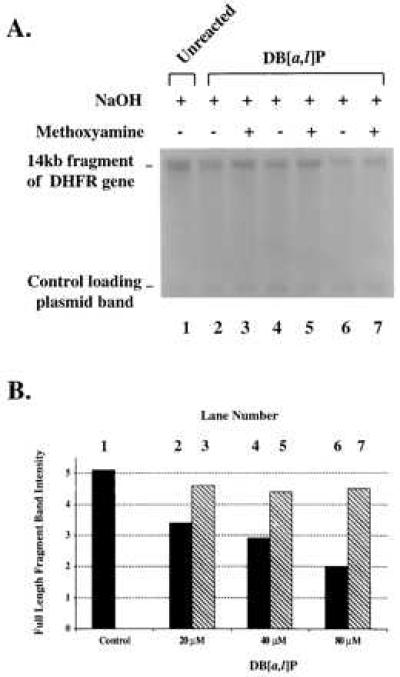
Alkaline-agarose gel electrophoresis of CHO-B11 cell DNA incubated with DB[a,l]P in the presence of HRP/H2O2. The reaction mixture was incubated for 1 hr at 37°C (see Materials and Methods). (A) KpnI-restricted DNA: no DB[a,l]P (lane 1); reacted with 20, 40, or 80 μM DB[a,l]P (lanes 3–7; without and with methoxyamine treatment). (B) Intensity of the 14-kb band of the DHFR gene of the DNA samples. All band intensities were normalized for the efficiency of sample loading on the gel through the use of a plasmid loading control.
We next turned to a human cell line that is capable of metabolically activating the parent hydrocarbon to determine whether unstable adducts could be observed under those conditions. MCF-7 cells were treated with two different concentrations of DB[a,l]P for 24 hr, and the DNA was isolated and analyzed. Again, no evidence was obtained for the formation of unstable adducts (Fig. 5), although stable adducts were easily measured (Table 1). Analysis for unstable adducts in the DNA of cells exposed to DB[a,l]P for only 4 hr gave similar results (data not shown).
Figure 5.

Alkaline-agarose gel electrophoresis of DNA from DB[a,l]P-treated MCF-7 cells. MCF-7 cells in culture were treated with DB[a,l]P for 24 hr (see Materials and Methods). (A) KpnI-restricted DNA from MCF-7 cell cultures treated with: DMSO (lanes 1 and 2); 1 or 2 μM DB[a,l]P (lanes 3–6; without or with methoxyamine). All samples underwent electrophoresis on a 0.5% alkaline-agarose gel and were probed with a 32P riboprobe for the transcribed strand of the 20-kb fragment of the human DHFR gene. (B) Intensity of the 20-kb band of the DHFR gene of the DNA samples. All band intensities were normalized for the efficiency of sample loading on the gel through the use of a plasmid loading control.
Stable Adducts.
The stable adducts in each DNA sample were analyzed by 33P postlabeling and HPLC. The HPLC elution profiles of the (+)-syn- and (−)-anti-DB[a,l]PDE-DNA adducts are shown in Fig. 6. The (+)-syn-DB[a,l]PDE adduct profile (Fig. 6A) from cells treated with 1.4 μM contains two major DNA adducts, which have previously been identified as being trans (65 min) and cis (92 min) addition products of dA with (+)-syn-DB[a,l]PDE (17). The adduct profile of the 1.4 μM (−)-anti-DB[a,l]PDE-treated cell DNA sample (Fig. 6B) contains one major peak, which has been identified as a dA adduct, and three smaller earlier eluting peaks, which contain mainly dG adducts (18). Similar adduct profiles were obtained in cell cultures treated with higher doses of (+)-syn- or (−)-anti-DB[a,l]PDE and in DNA that had reacted with these diol epoxides in solution (data not shown). The HPLC profile of the DNA that had reacted with DB[a,l]P in the presence of HRP (Fig. 6C) demonstrated a very low level of stable adducts (less than 0.05 per 14 kb). In contrast, the adduct profile of the 2 μM DB[a,l]P-treated cell DNA sample (Fig. 6D) demonstrated that DB[a,l]P is metabolized in MCF-7 cells mainly to the (−)-anti-diol epoxide and contained more dA than dG DNA adducts.
Figure 6.
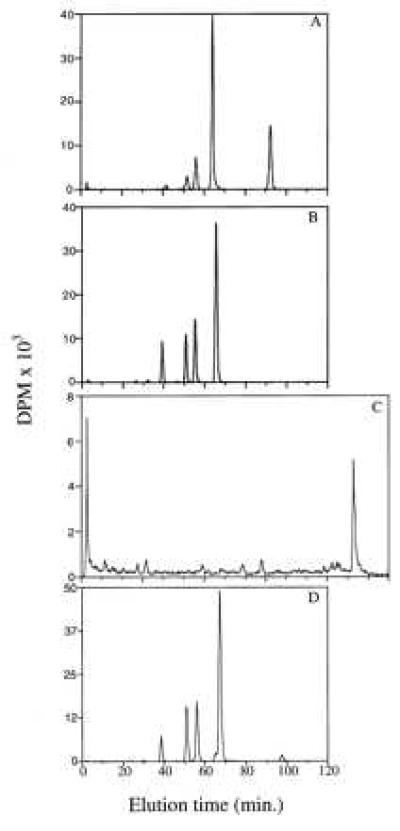
HPLC analysis of the 33P-postlabeled DB[a,l]P-DNA adducts formed in CHO-B11 cells treated for 1 hr with (A): 1.4 μM (+)-syn-DB[a,l]PDE or (B) 1.4 μM (−)-anti-DB[a,l]PDE; (C) the DNA adducts formed by the reaction of 40 μM DB[a,l]P with DNA from CHO-B11 cells in the presence of HRP/H2O2; and (D) the DNA adducts formed in MCF-7 cells treated for 24 hr with 2 μM DB[a,l]P.
The total level of stable DB[a,l]PDE-DNA adducts was calculated based on a B[a]PDE-DNA standard of known modification, which was included with each set of postlabeling samples. The levels of binding are given in Table 1. The number of stable adducts ranged from 3 to 14 per 14 kb in the B11 cells and as high as 10 per 20 kb in the DB[a,l]P-treated MCF-7 cells.
DISCUSSION
Covalent binding of chemical carcinogens to form stable DNA adducts is recognized as one of the important steps in cancer initiation by these compounds. There have been extensive studies of how stable adduct formation can lead to mutation through misincorporation or, in some cases, by causing deletions. Results of most studies with PAH have been consistent with this mechanism of mutation induction. For example, 7,12-dimethylbenz[a]anthracene binds extensively to the deoxyadenosine residues in DNA in cells, and many of the mutations induced by this compound in mice were found to be at dAs within the 61st codon of the ras oncogene (32–36). B[a]P binds extensively to dG residues (5), and many B[a]P-induced mouse tumors contain a mutation at a dG in the 12th codon of the ras protooncogene (37, 38). Extensive studies of mutation induction by the diol epoxides in cells in culture have been consistent with the formation of stable adducts being responsible for the induction of mutations. Recently, Chakravarti et al. (22) proposed that activating mutations induced in the ras protooncogene could result from the formation of unstable adducts that depurinate to leave AP sites. AP sites are known to be mutagenic lesions (39). However, it should be appreciated that Loeb (39) has calculated that over 10,000 AP sites are formed per cell per day, and the repair capacity of cells for such sites appears to be quite efficient (39–42). Cavalieri and coworkers (43) have carefully characterized depurinated adducts formed through one-electron oxidation of PAH. Recently, they reported that PAH diol epoxides may also form unstable adducts, which result in AP sites (21, 22). Because a large number of hydrocarbons is known to be metabolized to diol epoxides it is important to assess the relative frequency of formation of AP sites as compared with stable adducts by reaction of diol epoxides as well as other PAH metabolites with DNA.
To detect and measure the presence of such unstable adducts we examined the formation of alkali-sensitive sites within a 14-kb restriction fragment of the DHFR gene in CHO cells and an analogous 20-kb fragment in cultured human cells (23). In the same DNA, the frequency of stable adducts was measured through a 33P-postlabeling technique. When (+)-syn- or (−)-anti-DB[a,l]PDE was allowed to react with DNA in solution, we were unable to detect the formation of any AP sites. Analysis of the same DNA samples by 33P postlabeling demonstrated that the syn-DB[a,l]PDE-modified DNA contained 3 stable adducts per 14 kb and the anti-DB[a,l]PDE-modified DNA contained 10. Thus, under conditions with very high formation of stable adducts, no unstable adducts were detected. With this assay we were easily able to detect AP sites formed by heating DNA that had reacted with DMS (44).
The possibility that the DB[a,l]PDE could form AP sites in DNA within a cell, even if they were not formed in solution, was investigated by exposing cells in culture to a series of doses of (+)-syn- and (−)-anti-DB[a,l]PDE. Again, although the presence of AP sites resulting from treatment of the cells was readily detected, we were unable to measure any such sites in DNA from cells treated with the diol epoxides. Stable adducts were formed in cellular DNA, at frequencies from 3 to as high as 14 per 14 kb. These results with DB[a,l]PDE are consistent with other studies of PAH diol epoxide–DNA interactions. King et al. (45) reported that anti-B[a]PDE forms only a small proportion of N7 guanine adducts. Osborne and Merrifield (46) examined the ability of syn- and anti-B[a]PDEs to induce single strand breaks in rat liver DNA by determination of changes in the sedimentation coefficient of the DNA in alkaline solution. They found that alkali-labile sites accounted for less than 1% of total binding for (±)-anti-B[a]PDE and (+)-anti-B[a]PDE and between 1 and 2% for (±)-syn-B[a]PDE.
Our method was able to detect AP sites formed in DNA treated with DB[a,l]P under conditions expected to produce radical cations. The relative importance of the radical cations and diol epoxide formation in the activation of DB[a,l]P in cells was investigated by treating MCF-7 cells with DB[a,l]P. DB[a,l]P-treated MCF-7 cells contained up to 10 stable adducts per 20 kb, but no AP site formation was detected.
We interpret these results to indicate that (+)-syn- and (−)-anti-DB[a,l]PDE and DB[a,l]P form mainly stable adducts when they react with DNA either in solution or in cells in culture. Because we were unable to detect any unstable adducts, it is impossible to calculate an accurate ratio of stable to unstable adducts. It is reasonable to assume we could have reliably detected a loss of 10–20% of the full-length fragments, which would result from introduction of 0.1–0.2 site per fragment. Under conditions producing the highest frequencies of stable adducts, this amounts to less than 1–2% of their level and strongly suggests that DB[a,l]P exerts its biological effects through the formation of stable DNA adducts rather than by the induction of AP sites. Although it is possible that AP sites formed by unstable adducts are rapidly repaired in cells and therefore not detectable above background levels, this would suggest that AP sites caused by unstable DB[a,l]P-DNA adducts are unlikely to contribute significantly to the high mutagenic potency of DB[a,l]P. In conclusion, the results of the present study suggest that DB[a,l]P exerts its biological effects through the formation of stable DNA adducts rather than by the induction of unstable adducts that lead to AP sites.
Acknowledgments
This work was supported by National Cancer Institute Grants CA40228 (to W.M.B.) and CA44349 (to C.A.S. at Stanford), Office of Naval Research Grant N0014-86-K-0018 (to C.A.S. at Stanford), and Deutsche Forschungsgemeinschaft Grant SFB302 (to A.S.).
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: PAH, polycyclic aromatic hydrocarbons; DB[a,l]P, dibenzo[a,l]pyrene; (+)-syn-DB[a,l]PDE, DB[a,l]P-11S,12R-diol-13S,14R-epoxide; (−)-anti-DB[a,l]PDE, DB[a,l]P-11R,12S-diol-13S,14R-epoxide; AP, apurinic; DHFR, dihydrofolate reductase; CHO, Chinese hamster ovary; DMS, dimethyl sulfate; DMSO, dimethyl sulfoxide; HRP, horseradish peroxidase; B[a]P, benzo[a]pyrene; B[a]PDE, B[a]P-7,8-diol-9,10-epoxide.
References
- 1.Miller J A. Cancer Res. 1970;30:559–576. [PubMed] [Google Scholar]
- 2.Harvey R G. Polycyclic Aromatic Hydrocarbons: Chemistry and Carcinogenicity. Cambridge, U.K.: Cambridge Univ. Press; 1991. [Google Scholar]
- 3.Conney A H. Cancer Res. 1982;42:4875–4917. [PubMed] [Google Scholar]
- 4.Sims P, Grover P L, Swaisland A, Pal K, Hewer A. Nature (London) 1974;252:326–327. doi: 10.1038/252326a0. [DOI] [PubMed] [Google Scholar]
- 5.Baird W M, Ralston S L. In: Comprehensive Toxicology. Bowden G T, Fisher S M, editors. Oxford, U.K.: Elsevier; 1997. pp. 171–200. [Google Scholar]
- 6.Pruess-Schwartz D, Baird W M, Yagi M, Jerina D M, Pigott M A, Dipple A. Cancer Res. 1987;47:4032–4037. [PubMed] [Google Scholar]
- 7.Moschel R C, Baird W M, Dipple A. Biochem Biophys Res Commun. 1997;76:1092–1098. doi: 10.1016/0006-291x(77)90968-8. [DOI] [PubMed] [Google Scholar]
- 8.Cavalieri E L, Rogan E G. Environ Health Perspect. 1985;64:69–84. doi: 10.1289/ehp.856469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cavalieri E L, Rogan E G. Pharmacol Ther. 1992;55:183–199. doi: 10.1016/0163-7258(92)90015-r. [DOI] [PubMed] [Google Scholar]
- 10.Cavalieri E L, Higginbotham S, RamaKrishna N V S, Devanesan P D, Todorovic R, Rogan E G, Salmasi S. Carcinogenesis. 1991;12:1939–1944. doi: 10.1093/carcin/12.10.1939. [DOI] [PubMed] [Google Scholar]
- 11.Higginbotham S, RamaKrishna N V S, Johansson S L, Rogan E G, Cavalier E L. Carcinogenesis. 1993;14:875–878. doi: 10.1093/carcin/14.5.875. [DOI] [PubMed] [Google Scholar]
- 12.Kozin I S, Gooijer C, Velthorst N H. Anal Chem. 1995;67:1623–1626. [Google Scholar]
- 13.Snook M E, Severson R F, Arrendale R F, Higman H C, Chortyk O T. Beitr Tabakforsch. 1977;9:79–101. [Google Scholar]
- 14.Mumford J L, Harris D B, Williams K. Environ Sci Technol. 1987;21:308–311. doi: 10.1021/es00157a014. [DOI] [PubMed] [Google Scholar]
- 15.Busby W F, Smith H, Crespi C L, Penman B W. Mutat Res. 1995;342:9–16. doi: 10.1016/0165-1218(95)90085-3. [DOI] [PubMed] [Google Scholar]
- 16.Ralston S L, Coffing S, Seidel A, Luch A, Platt K L, Baird W M. Chem Res Toxicol. 1997;10:687–693. doi: 10.1021/tx9700275. [DOI] [PubMed] [Google Scholar]
- 17.Ralston S L, Lau H H S, Seidel A, Luch A, Platt K L, Baird W M. Cancer Res. 1994;54:887–890. [PubMed] [Google Scholar]
- 18.Ralston S L, Seidel A, Luch A, Platt K L, Baird W M. Carcinogenesis. 1995;16:2899–2907. doi: 10.1093/carcin/16.12.2899. [DOI] [PubMed] [Google Scholar]
- 19.Ralston S L, Lau H H S, Seidel A, Luch A, Platt K L, Baird W M. Polycycl Arom Compds. 1994;6:199–206. [Google Scholar]
- 20.Cavalieri E L, Rogan E G. Xenobiotica. 1995;25:677–688. doi: 10.3109/00498259509061885. [DOI] [PubMed] [Google Scholar]
- 21.Li L-M, Todorovic R, Rogan E G, Cavalieri E L, Ariese F, Suh M, Jankowiak R, Small G J. Biochemistry. 1995;34:8043–8049. doi: 10.1021/bi00025a010. [DOI] [PubMed] [Google Scholar]
- 22.Chakravarti D, Pelling J C, Cavalieri E L, Rogan E G. Proc Natl Acad Sci USA. 1995;92:10422–10426. doi: 10.1073/pnas.92.22.10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bohr V A, Smith C A, Okumoto D S, Hanawalt P C. Cell. 1985;40:359–369. doi: 10.1016/0092-8674(85)90150-3. [DOI] [PubMed] [Google Scholar]
- 24.Smith C A, Mellon I. In: Advances in Mutagenesis Research I. Obe G, editor. Berlin: Springer; 1990. pp. 153–194. [Google Scholar]
- 25.Scicchitano D A, Hanawalt P C. Proc Natl Acad Sci USA. 1989;86:3050–3054. doi: 10.1073/pnas.86.9.3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luch A, Glatt H, Platt K L, Oesch F, Seidel A. Carcinogenesis. 1994;15:2507–2516. doi: 10.1093/carcin/15.11.2507. [DOI] [PubMed] [Google Scholar]
- 27.Frank H, Luch A, Oesch F, Seidel A. Polycycl Arom Compds. 1996;10:109–116. [Google Scholar]
- 28.Rogan E G, Cavalieri E L, Tibbels S R, Cremosi P, Wamer C D, Nagel D L, Tomer K B, Cerny R L, Gross M L. J Am Chem Soc. 1988;110:4023–4029. [Google Scholar]
- 29.Talpaert-Borle M, Liuzzi M. Biochim Biophys Acta. 1983;740:410–416. doi: 10.1016/0167-4781(83)90089-1. [DOI] [PubMed] [Google Scholar]
- 30.Liuzzi M, Talpaert-Borle M. In: DNA Repair: A Laboratory Manual of Research Procedures. Friedberg E C, Hanawalt P C, editors. Vol. 3. New York: Dekker; 1988. pp. 443–457. [Google Scholar]
- 31.Baird W M, Lau H H, Schmerold I, Coffing S L, Brozich S L, Lee H, Harvey R G. IARC Sci Publ. 1993;124:217–226. [PubMed] [Google Scholar]
- 32.Bigger C A H, Sawicki J T, Blake D M, Raymond L G, Dipple A. Cancer Res. 1983;43:5647–5651. [PubMed] [Google Scholar]
- 33.Dipple A, Pigott M, Moschel R C, Costantino N. Cancer Res. 1983;43:4132–4135. [PubMed] [Google Scholar]
- 34.Balmain A, Pragnell I B. Nature (London) 1983;303:72–74. doi: 10.1038/303072a0. [DOI] [PubMed] [Google Scholar]
- 35.Sukumar S, Notario V, Martin-Zanca D, Barbacid M. Nature (London) 1983;306:658–661. doi: 10.1038/306658a0. [DOI] [PubMed] [Google Scholar]
- 36.Quintanilla M, Brown K, Ramsden M, Balmain A. Nature (London) 1986;322:78–80. doi: 10.1038/322078a0. [DOI] [PubMed] [Google Scholar]
- 37.You M, Candrian U, Maronpot R R, Stoner G D, Anderson M W. Proc Natl Acad Sci USA. 1989;86:3070–3074. doi: 10.1073/pnas.86.9.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bailleul B, Brown K, Ramsden M, Akhurst R J, Fee F, Balmain A. Environ Health Perspect. 1989;81:23–27. doi: 10.1289/ehp.898123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Loeb L A. Cancer Res. 1989;49:5489–5496. [PubMed] [Google Scholar]
- 40.Lindahl T, Nyberg B. Biochemistry. 1972;11:3610–3818. doi: 10.1021/bi00769a018. [DOI] [PubMed] [Google Scholar]
- 41.Schaaper R M, Loeb L A. Proc Natl Acad Sci USA. 1981;78:1773–1777. doi: 10.1073/pnas.78.3.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boiteux S, Laval J. Biochemistry. 1982;21:6746–6751. doi: 10.1021/bi00269a020. [DOI] [PubMed] [Google Scholar]
- 43.Rogan E G, Devanesan P D, RamaKrishna N V S, Higginbotham S, Padmavathi N S, Capman K, Cavalieri E L, Jeong H, Jankowiak R, Small G J. Chem Res Toxicol. 1993;6:356–363. doi: 10.1021/tx00033a017. [DOI] [PubMed] [Google Scholar]
- 44.Hoffmann G R. Mutat Res. 1980;75:63–129. doi: 10.1016/0165-1110(80)90028-7. [DOI] [PubMed] [Google Scholar]
- 45.King H W S, Osborne M R, Brookes P. Chem Biol Interact. 1979;24:345–353. doi: 10.1016/0009-2797(79)90082-6. [DOI] [PubMed] [Google Scholar]
- 46.Osborne M, Merrifield K. Chem Biol Interact. 1985;53:183–195. doi: 10.1016/s0009-2797(85)80095-8. [DOI] [PubMed] [Google Scholar]