

Abstract
The multifunctional enzyme apurinic endonuclease 1/redox enhancing factor 1 (Ape1/Ref-1) maintains genetic fidelity through the repair of apurinic sites and regulates transcription through redox-dependent activation of transcription factors. Ape1 can therefore serve as a therapeutic target in either a DNA repair or transcriptional context. Inhibitors of the redox function can be used as either therapeutics or novel tools for separating the two functions for in vitro study. Presently there exist only a few compounds that have been reported to inhibit Ape1 redox activity; here we describe a series of quinones that exhibit micromolar inhibition of the redox function of Ape1. Benzoquinone and naphthoquinone analogs of the Ape1-inhibitor E3330 were designed and synthesized to explore structural effects on redox function and inhibition of cell growth. Most of the naphthoquinones were low micromolar inhibitors of Ape1 redox activity, and the most potent analogs inhibited tumor cell growth with IC50 values in the 10–20 micromolar range.
Introduction
Two fundamental concerns present in managing cellular homeostasis are maintaining DNA fidelity and regulating the expression of the genetic information contained therein. With regard to DNA fidelity, a major recurring event cells must overcome is the formation of apurinic or apyrimidinic (APa) sites, formation of which takes place on the order of 104 times per cell per day by spontaneous glycosidic hydrolysis.1–3 AP sites in DNA have numerous deleterious ramifications, including prohibiting DNA replication, cytotoxicity, and mutagenicity. Spontaneous glycosidic hydrolysis is not the only route to forming AP sites; glycosylases as well as DNA damaging agents can induce AP site formation in DNA.1 The ubiquitous enzyme apurinic/apyrimidic endonuclease 1 (Ape1) is a major component of the base excision repair (BER) pathway and has the responsibility of repairing AP sites throughout the genome. Ape1 possesses multiple enzymatic functions; the most relevant to BER is the 5′ AP-endonuclease activity that initiates the removal of AP sites.
Gene expression is also controlled in part by Ape1. At the same time the BER function of Ape1 was being explored, another enzyme was identified that performed redox-dependent regulation of numerous transcription factors. This enzyme was named redox enhancing factor 1 (Ref-1) and was linked to the regulation of transcription factors such as activator protein 1 (AP-1), hypoxia inducing factor 1 alpha (HIF-1α), and nuclear factor kappa B (NFκB). It was subsequently determined that these were two distinct functions of the same protein, initially thought to reside in two non-overlapping domains but later determined to have a minor degree of overlap.1–5 However, redox or repair can be silenced independently using specific point mutations for each activity, indicating that each function can act independently.
Through both the redox and DNA repair functions Ape1 supports cancer cell proliferation, and elevated expression levels have been shown to correlate to poor patient prognosis.1–3 Ape1 is overexpressed in a number of cancers, where increased levels of DNA repair leads to resistance against DNA damaging agents, and increased redox activity is expected to enhance replication through redox cycling of transcription factors. Therefore Ape1 represents an interesting therapeutic target in different mechanistic contexts. Inhibitors of the BER function of Ape1 can be utilized as a complementary treatment option for those encountering resistance to DNA-damaging agents. Alternatively, inhibition of the redox function of Ape1 might interfere with regulation of transcription and alter a number of stress-induced responses of cancer cells. Recent data indicates that blocking the repair function of Ape1 leads to cell death, while redox activity inhibition leads to decreased cell growth and cytostatic effects.6 Additionally, recent data indicates that blocking Ape1 redox function blocks angiogenesis.6–8 Small molecule inhibitors of the redox function can also serve as tools to separate the two functions of Ape1 without the lethality of knocking out Ape1 completely.9
The design of inhibitors targeting the redox function of Ape1 is hindered by a lack of information regarding the redox active site. Mutation analysis has shown that cysteine 65 is necessary for redox activity; however, in every crystal structure C65 is buried, suggesting that a conformational change might be required to present the relevant redox-active structure.10 Furthermore, there is only one known compound in the literature that has been shown to inhibit the redox function of Ape1.1 To provide structural insight into potential inhibitor specificity for the redox active site, a series of benzoquinones and naphthoquinones has been synthesized based on the structure of (E)-3-(5,6-dimethoxy-3-methyl-1,4-dioxocyclohexa-2,5-dienyl)-2-nonylpropenoic acid (E3330, 1), a known inhibitor of the redox function of Ape1.1 (Figure 1) Analogs with improved physicochemical and binding profiles also have the potential to provide crystallographic data when complexed with the protein to elucidate the structure of the redox active site. The structure of 1 provides five regions for potential exploration in preliminary SAR studies, namely 1) the substituent on the 3-position of the quinone ring, 2) the benzoquinone structure, 3) the substituent α to the carboxyl group, 4) the carboxylate moiety, and 5) the saturation of the substituent at the quinone 2-position.
Figure 1.
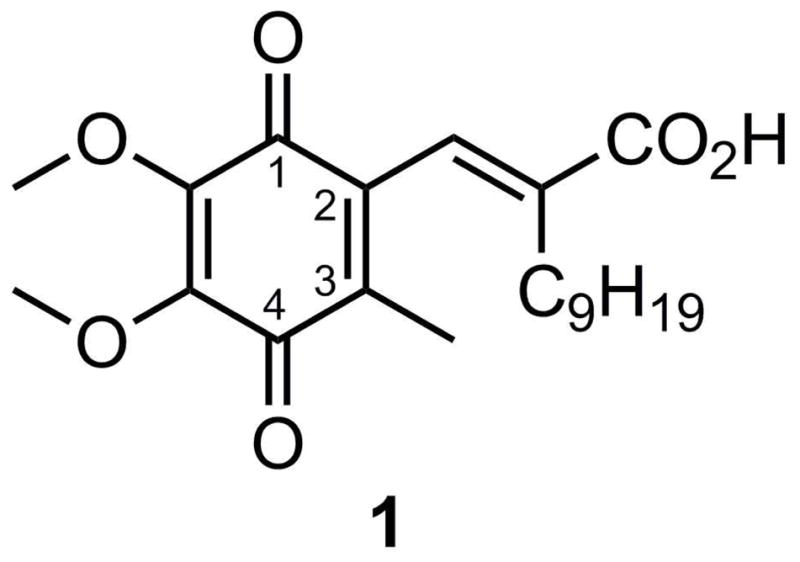
E3330, a reported inhibitor of APE1 redox activity.1
Results and Discussion
Chemistry
The synthesis of benzoquinone 1 (E3330) has been reported in a patent, and key transformations have appeared separately in print.11–12 The patent synthesis begins with the tribromination of p-cresol followed by a Cu(I)-mediated Ullmann reaction to generate 4-methyl-2,3,6-trimethoxyphenol that is alkylated in situ to provide 2,3,4,5-tetramethoxytoluene.12 In our hands the methanolysis of 4-methyl-2,3,6-tribromophenol resulted in a complex mixture that included products derived from reduction of the bromo substituents; these side products were very difficult to remove, and the resulting tetramethoxytoluene could not be obtained pure. To overcome these difficulties we developed an alternative synthesis of 1 (Scheme 1) that was also used for the synthesis of several analogs.10 Bayer-Villiger oxidation of 2,3,4-trimethoxybenzaldehyde 2 and subsequent hydrolysis in situ gives 2,3,4-trimethoxyphenol which is then alkylated to provide 1,2,3,4-tetramethoxybenzene 3 in excellent yield.13 Ring methylation is then performed to provide 2,3,4,5-tetramethoxytoluene in high yield and purity,14 and the aldehyde moiety is introduced using α,α-dichloromethyl methyl ether and titanium (IV) chloride15 to give 4 in 95% yield. The chloroaldehyde intermediate 6 was prepared by formylation of 3 using N-methyl formanilide and POCl3, followed by chlorination with sulfuryl chloride.16 The unsaturated ester moiety was introduced by Emmons condensation of aldehyde 4 or 6 and the appropriate phosphonate. All of the Emmons products derived from aldehyde 4 are formed exclusively as the E isomer. When using the chloro-substituted aldehyde 6, however, the E:Z selectivity is 5:1 when the reaction is run at room temperature; performing the reaction in refluxing toluene provides 100% E isomer. The esters were hydrolyzed and the intermediate acids were oxidized to the benzoquinones 1, 5a–e and 7. The 3-methyl analogs were easily oxidized with either nitric acid or ceric ammonium nitrate (CAN); however, successful oxidation of the 3-chloro analog could only be accomplished using CAN. Finally, the methoxyethyl substituent was introduced by condensation of 4 with the Emmons reagent prepared from α-bromo-γ-butyrolactone to give lactone 8. Treatment of 8 with trimethylorthoformate in acidic methanol generated the ring-opened methoxyethyl analog;17 hydrolysis of the methyl ester and oxidation afforded the quinone 9.
Scheme 1a.

aReagents and Conditions: a) H2O2, H2SO4, MeOH, reflux 3 h; b) K2CO3, MeI, acetone, reflux 2 d; c) i. nBuLi, THF, 0 °C, 1 h; ii. MeI, 0 °C, 2 h; d) CHCl2OCH3, TiCl4, CH2Cl2, 0 °C to rt 4 h; e) NaH, (EtO)2P(O)CHRCO2Et THF, rt 12 h; f) KOH, EtOH, reflux 30 min; g) HNO3, AcOH, EtOAc, rt 4 h; h) NMFA, POCl3, CH2Cl2, rt 2 d; i) SO2Cl2, CH2Cl2, rt, 1 h; j) CAN, MeCN, H2O, 1 h; k) α-bromo-γ-butyrolactone, (EtO)3P, then NaH, toluene, reflux 8 h; l) H2SO4, CH(OCH3)3, MeOH, reflux 12 h; m) KOH, EtOH, reflux 30 min.
Carboxamide analogs of the free acids were also of interest both for SAR and for the potential development of coupled products. However, all efforts to introduce a hydroxyethylamide moiety by reaction with the benzoquinone acids (e.g., 5a) were unsuccessful. Therefore acid 10 was converted to amide 11 and subsequently oxidized to give the desired quinone amide 12 (Scheme 2). The synthesis of 11 was initially attempted using DCC as a reagent; surprisingly, amide 11 was obtained as a 1:1 mixture of E and Z products. In contrast, the E stereochemistry was completely retained when 11 was synthesized using PyBOP in the coupling reaction. Further exploration of coupling conditions using a variety of unsaturated acids and amines showed that activation with either DCC or DIC leads to mixtures of E and Z products, but activation with PyBOP, methyl chloroformate, or oxalyl chloride do not. It is hypothesized that a reversible intramolecular cyclization of the activated carbodiimide intermediate is taking place that leads to loss of the stereochemical integrity of the double bond (Scheme 2). The amidation product E-11 was then readily oxidized using nitric acid to provide hydroxyethylamide 12.
Scheme 2a.

aReagents and conditions: a) DCC, ethanolamine, CH2Cl2, rt; b) PyBOP, Et3N, rt 30 min, then ethanolamine, rt 4 h; c) Ag(II)O, HNO3, AcOH, EtOAc, rt 40 min
The naphthoquinone analogs were constructed via condensation of the appropriate aldehydes and phosphonates followed by ester hydrolysis and oxidation to the quinone. Synthesis of the requisite aldehydes is detailed in Scheme 3. Aldehydes 14a–c were prepared by dichloromethyl methyl ether formylation of the di- or trimethoxynaphthalenes. The methylthio substituent was introduced by lithiation of the hydroxymethylnaphthalene followed by reaction with dimethyl disulfide and subsequent oxidation of the hydroxymethyl group to the aldehyde (14d). The 3-halo substituents in 14e–g were introduced by electrophilic halogenation of 14a. Emmons condensation with the naphthaldehydes showed reduced E selectivity relative to their benzaldehyde counterparts. The greatest E selectivity was observed with 3-methyl aldehyde 14b, which afforded 100% E in refluxing toluene and an E:Z ratio of approximately 5:1 at room temperature. All other analogs produced a significant proportion of Z isomer (20–50%) even in refluxing toluene. The Emmons products 15a–g were converted to the quinones 16a–g by saponification and subsequent oxidation. Nitric acid was sufficient to oxidize the 3-methyl, 3-methoxy, and 3-methylthio analogs, but efficient oxidation of the 3-halo analogs required treatment with argentic oxide.
Scheme 3a.

aReagents and conditions: a) i. H2, 10% Pd/C, THF, rt, 4 h; ii. NaH, Me2SO4,2 h, rt; b) R = H, CH3: TiCl4, CHCl2OCH3, CH2Cl2, 0 °C, 4 h; R = OCH3: i. nBuLi, THF, −78 °C, 3 h; ii. DMF, −78 °C; c) Selectfluor, CH3CN, reflux; d) SO2Cl2, CH2Cl2, rt, 4 h; e) Br2, CH2Cl2, rt, 1 h; f) R = H: NaH, (EtO)2P(O)CHR’CO2Et, THF, rt, 12 h; R = CH3, OCH3, SCH3, F, Cl, Br: NaH, (EtO)2P(O)CHR’CO2Et, PhMe, reflux, 12 h; g) EtOH, KOH, reflux, 1 h; h) R = H, CH3, OCH3> SCH3: HNO3, EtOAc, AcOH, rt, 3 h; R = F, Cl, Br: HNO3, EtOAc, AcOH, Ag(II)O, rt, 2 h; i) LiAlH4, THF, rt, 12 h; j) i. nBuLi, THF, −78 °C; ii. (SCH3)2, −78 °C; k) PCC, CH2Cl2, rt, 8h
An interesting result was observed when a mixture of 3-chloro E- and Z-acids (resulting from hydrolysis of the crude Emmons product 15f) was oxidized using nitric acid and argentic oxide; although the E-quinone product 16f was produced from the E-acid as expected, the Z-quinone product following oxidation was recovered as a methyl ester. The oxidation was carried out on pure Z-acid 18, and the Z-methyl ester 19 was the major product (Scheme 4). In order to rationalize the selective esterification of the Z-carboxylate group, an intramolecular esterification is hypothesized (Scheme 4) in which the Z- oxidation was recovered as a methyl ester. The oxidation was carried out on pure Z-acid 18, and the Z-methyl ester 19 was the major product (Scheme 4). In order to rationalize the selective esterification of the Z-carboxylate group, an intramolecular esterification is hypothesized (Scheme 4) in which the Z-carboxylate attacks the methyl group of the oxonium ion intermediate generated in the oxidation reaction. The selective esterification of the Z-acids is fortuitous in that it provides a convenient strategy to purify otherwise difficult-to-separate E and Z products by generating easily separable E-acid and Z-methyl ester.
Scheme 4.

Finally, the naphthoquinone series was completed with the preparation of methoxyethyl side chain analogs 21a–c, the amide 23, and the bromoacid 26 in which the double bond at the 2-position was fully saturated (Scheme 5). Syntheses of 21 and 23 were accomplished via routes analogous to those previously described for the benzoquinone products. Alcohol 24 (the reduction product of 14a) provided the starting material for the synthesis of 26; conversion to the α,3-dibromide followed by displacement of the benzylic bromide afforded the saturated ester 25. Oxidation and ester hydrolysis gave the desired product.
Scheme 5a.

aReagents and conditions: a) α-bromobutyrolactone, (EtO)3P, then NaH, toluene, reflux 8 h; b) H2SO4, CH(OCH3)3, MeOH, reflux 12 h; c) KOH, EtOH, reflux 30 min; d) R = CH3: HNO3, EtOAc, AcOH, rt 3 h; R = Cl or Br: HNO3, EtOAc, AcOH, Ag(II)O, rt 2 h; e) PyBOP, Et3N, DMF:CH2Cl2 rt 30 min, then ethanolamine, rt 4 h; f) Ag(II)O, HNO3, AcOH, EtOAc, rt 30 min; g) HBr, CH2Cl2; rt 30 min; h) Br2, CH2Cl2, rt 4 h; i) Li(SiMe3)2N, CH3CO2Et, THF, −78 C to rt 4 h; j) HCl, THF, reflux.
Redox, Endonuclease and Cell Growth Inhibition
Compounds were tested for the ability to inhibit the redox function of Ape1 in a gel shift assay (EMSA), and IC50 values were generated for each compound. Inhibition of Hey-C2 ovarian cancer cell growth was also assessed. Compounds were also evaluated as inhibitors of Ape1 endonuclease activity; all compounds were inactive at concentrations up to 100 μM. The redox and cell growth inhibition results are summarized in Tables 1 and 2; a representative EMSA assay is shown in Figure 2. Modification of the alkyl substituent on the double bond of the benzoquinones (Table 1) had only modest effects on the redox activity; replacement of the n-nonyl group (1, IC50 = 10 μM) with a methyl group (5b, IC50 = 3 μM) enhanced redox activity, whereas removal of the alkyl group (5a, IC50 = 40 μM) reduced potency in the redox assay. In contrast, most of the structural modifications in the benzoquinone series diminished cell growth inhibitory activity compared to the lead compound 1. The loss of cell-based activity relative to redox activity is especially dramatic for compounds 5a, 5b and 9. However, these compounds are significantly more hydrophilic than 1, so they may be too polar to permeate the cell effectively. By comparison, most of the naphthoquinones (Table 2) were more potent than 1 as inhibitors of Ape1 redox activity, with IC50 values in the 1–5 micromolar range. Compounds with electronegative substituents in the 3-position of the naphthoquinone ring generally had the highest redox inhibitory activity; in contrast, the 3-methyl compounds were the least active in this assay. The best of the naphthoquinones were 2–4 fold more potent than 1 in growth inhibitory activity and, although the most potent compounds were generally 3-chloro or 3-bromo analogs, the structural modifications investigated here had only modest effects on growth inhibition. It is interesting to note that functionalization of the carboxylic acid as the hydroxyethyl amide did not affect either redox or cell growth inhibitory activity significantly (compounds E-16f and 23).
Table 1.
Redox and growth inhibition of benzoquinone analogs
| Compound | R1 | R2 | R3 | Redox Inhibition IC50, μMa | Growth Inhibition GI50, μMb |
|---|---|---|---|---|---|
| 1 | CH3 | C9H19 | OH | 10 | 35 |
| 5a | CH3 | H | OH | 40 | 250 |
| 5b | CH3 | CH3 | OH | 3 | 130 |
| 5e | CH3 | C4H9 | OH | 15 | 85 |
| 7 | Cl | CH3 | OH | 3 | 45 |
| 9 | CH3 | C2H4OCH3 | OH | 10 | 200 |
| 12 | CH3 | C9H19 | NH(CH2)2OH | 10 | 35 |
Determined in a gel shift EMSA assay; see experimental section for details.
Hey-C2 cells treated with drug for 72 h. Values represent the average of three experiments.
Table 2.
Redox and growth inhibition of naphthoquinone analogs
| Compound | R1 | R2 | R3 | Redox Inhibition IC50, μMa | Growth Inhibition GI50, μMb |
|---|---|---|---|---|---|
| 16a | H | CH3 | OH | 3 | 20 |
| 16b | CH3 | CH3 | OH | 15 | 45 |
| 16c | OCH3 | CH3 | OH | 1 | 40 |
| 16d | SCH3 | CH3 | OH | 1 | 30 |
| 16e | F | CH3 | OH | 4 | 50 |
| 16f (E) | Cl | CH3 | OH | 1 | 15 |
| 16f (Z) | Cl | CH3 | OH | 2 | 30 |
| 16g | Br | CH3 | OH | 2 | 13 |
| 21a | CH3 | C2H4OCH3 | OH | 10 | 30 |
| 21b | Cl | C2H4OCH3 | OH | 3 | 30 |
| 21c | Br | C2H4OCH3 | OH | 1 | 25 |
| 23 | Cl | CH3 | NH(CH2)2OH | 1 | 8 |
| 26 | Br | H (saturated) | OH | 5 | 15 |
Determined in a gel shift EMSA assay; see experimental section for details.
Hey-C2 cells treated with drug for 72 h. Values represent the average of three experiments.
Figure 2.

Inhibition of Ape1 redox activity measured by electrophoretic mobility shift assay. See experimental section for details.
Conclusion
The goal of this work was to exploit 1 as a lead to develop more potent and druggable inhibitors of Ape1 redox activity. Analogs based on the benzoquinone core showed little improvement in enzymatic activity, and most were less effective cell growth inhibitors, presumably because of significant changes in physicochemical properties. Most of the naphthoquinone analogs were highly potent inhibitors of Ape1 redox activity. All of these compounds had favorable physicochemical properties, with cLogP values in the 1–3 range. The most potent compounds have an electron-withdrawing substituent at the 3-position of the naphthoquinone ring. Several of these compounds exhibited IC50 values of ~ 1 μM in the Ape1 redox assay and < 20 μM in the cell growth inhibition assay.
Experimental Procedures
Materials and Methods
All NMR spectra were recorded using a 300 MHz Bruker spectrometer equipped with a 5 mm multinuclear probe. 1H chemical shifts are reported in parts per million using tetramethylsilane as internal standard. 31P NMR spectra were obtained using broadband 1H decoupling, and chemical shifts are reported in parts per million using 1% triphenylphosphine oxide/benzene-d6 as the coaxial reference (triphenylphosphine oxide/benzene-d6 has a chemical shift of +24.7 ppm relative to 85% phosphoric acid). Mass spectral data were obtained from the Purdue University Mass Spectrometry Service, and elemental analyses were performed by the Purdue University Microanalysis Lab. Purity of all tested compounds was assessed by either combustion analysis or HPLC (C18 reverse phase) and found to be 95% or greater. Silica gel grade 60 (230–400 mesh) was used to carry out all flash chromatographic separations. Thin layer chromatography was performed using Analtech glass plates precoated with silica gel (250 microns). Visualization of the plates was accomplished using UV and/or the following stains: 1% 4-(p-nitrobenzyl)pyridine in acetone followed by heating and subsequent treatment with 3% KOH in methanol; or p-anisaldehyde dip (1.85% p-anisaldehyde, 20.5% sulfuric acid, 0.75% acetic acid in 95% ethanol) followed by heating. All anhydrous reactions were carried out under an atmosphere of argon. Tetrahydrofuran was distilled prior to use from sodium using benzophenone ketyl as an indicator. Dichloromethane, triethylamine, pyridine, and acetonitrile were distilled from calcium hydride prior to use. Unless otherwise noted, all other solvents were purchased from Fisher or VWR, and used as received. All chemical reagents were purchased from Aldrich, unless otherwise noted.
Enzymatic Redox Assay
Inhibition of Ape1 redox activity was measured using an electrophoretic mobility shift assay (EMSA).18 Briefly, purified Ape1 protein (10 mg/mL) was reduced with DTT (1.0 mM) at 37 °C for 10 min and then diluted with PBS buffer to final concentrations of Ape1 and DTT of 2 mg / mL and 0.2 mM respectively. A final volume of 18 mL was prepared from EMSA buffer (10 mM Tris [pH 7.5], 50 mM NaCl, 1 mM MgCl2, 1 mM EDTA, 5% [vol/vol] glycerol) to which was added two mL of reduced Ape1 protein and 6 mg of oxidized nuclear extracts (Hey-C2 cells, treated with 0.01 mM diamide for 10 min); and the reaction was incubated at room temperature for 30 min. One mL of poly(dI-dC) · poly(dI-dC) (1 mg/L, Amersham Biosciences, Piscataway, NJ) was added for 5 min followed by one mL of the 5′ hexachlorofluorescein phosphoramidite (HEX)-labeled double-stranded oligonucleotide DNA (0.1 pmol, The Midland Certified Reagent Company, Midland, TX) containing the AP-1 consensus sequence (5′CGCTTGATGACTCAGCCGGAA-3′), and the mixture was further incubated for 30 min. at room temperature. The final concentration of DTT in the redox reactions was 0.02 mM. Samples were loaded on a 5% nondenaturing polyacrylamide gel and subjected to electrophoresis in 0.5X TBE buffer (200 V for 1 h at 4°C) and detected using the Hitachi FMBio II Fluorescence Imaging System (Hitachi Genetic Systems, South San Francisco, CA). The HEX fluorophore is excited by a solid-state laser at 532 nm (Perkin-Elmer) and emits a fluorescent light signal at 560 nm, which is then measured using a 585 nm filter.
Growth Inhibition Assay
Growth inhibition was tested in vitro using the MTS assay following the procedure of Fishel et al.19 Hey-C2 ovarian cancer cells were cultured in RPMI 1640 medium containing 10% fetal calf serum. Cells (2–4,000) were aliquoted into each well of a 96-well plate in triplicate and allowed to adhere overnight. Cells were incubated with drug for 72 h at 37 °C. After 72 h, 0.05 mg/mL MTS reagent was added to each well and incubated at 37°C for 4 h followed by absorbance measurement at 490 nm. The values were standardized to wells containing media alone.
Synthetic Procedures
(E)-3-(5,6-dimethoxy-3-methyl-14-dioxocyclohexa-25-dienyl)-2-nonylpropenoic acid (1)
According to the modified procedure of Murphy et al.,20 NaH (0.135 g, 3.38 mmol) was added to a flame-dried 50 mL 3-neck round bottom flask connected to a water-jacketed reflux condenser. The flask was purged with argon, and THF (20 mL) was added to the flask followed by triethyl 2-phosphonoundecanoate (1.08 g, 3.09 mmol) at room temperature. The reaction was stirred at room temperature for 30 min, and then 4 (0.581 g, 2.42 mmol) dissolved in THF (10 mL) was added rapidly at room temperature. The reaction was stirred for another 12 h at room temperature, then acidified with 2 M HCl, diluted with ethyl acetate, and washed with brine. The organic layer was dried, filtered, and condensed. The resulting oil was purified via flash column chromatography (3:17 EtOAc:hexanes) to provide ethyl (E)-3-(6-methyl-2,3,4,5-trimethoxyphenyl)-2-nonylpropenoate (0.603 g, 57%) as a colorless oil. Rf = 0.40 (3:17 EtOAc:hexanes) 1H NMR (CDCl3): δ 0.84 (t, 3H, J = 7.2 Hz); 1.08 – 1.22 (m, 11H); 1.24 – 1.35 (m, 3H); 1.33 (t, 3H); 2.03 (s, 3H); 2.12 (m, 2H); 3.69 (s, 3H); 3.78 (s, 3H); 3.88 (s, 3H); 3.93 (s, 3H); 4.25 (q, 2H); 7.37 (s, 1H).
The ethyl ester (1.57 g, 3.60 mmol) was dissolved in EtOH (12.0 mL) and then KOH (0.41 g, 7.3 mmol) was added and the solution was refluxed for 1 h. The reaction was then acidified with 2 M HCl, extracted with ethyl acetate, washed 2 times with saturated brine, dried, filtered, and condensed. The product was obtained as a light-yellow oil (1.43 g, 97%) following flash chromatography (2:3 Et2O:hexanes 0.5% AcOH). Rf = 0.36 (2:3 Et2O:hexanes 0.5% AcOH) 1H NMR (CDCl3): δ 0.84 (t, 3H); 1.13 (m, 12H); 1.36 (m, 2H); 2.04 (s,3H); 2.14 (m, 2H); 3.70 (s, 3H); 3.79 (s, 3H); 3.89 (s, 3H); 3.93 (s, 3H); 7.537 (s,1H)
Following modifications to the procedures by Shinkawa et al. and Flader et al.,11,21 the unsaturated acid (1.13 g, 2.77 mmol) was dissolved in ethyl acetate (15 mL) at room temperature. HNO3 (0.75 mL) and AcOH (6 drops) were added at room temperature, and the reaction was stirred for 4 h. The reaction was then diluted with EtOAc (20.0 mL) washed with brine, dried over MgSO4, filtered, and condensed. The red oil was then purified by flash chromatography (2:3 Et2O:hexanes 0.5% AcOH) followed by recrystallization from Et2O/hexanes to afford 1 (0.443 g, 42%) as a red solid. Rf = 0.16 (2:3 Et2O:hexanes 0.5% AcOH) mp = 56 – 57 °C (Lit. 68 °C).5 1H NMR (CDCl3): δ 0.84 (t, 3H), 1.18 (bs, 14H); 1.39 (bs, 2H); 1.94 (d, 3H); 2.09 (t, 3H,); 3.99 (s, 3H); 4.02 (s, 3H); 7.26 (d, 1H).
(E)-3-(5,6-dimethoxy-3-methyl-1,4-dioxocyclohexa-2,5-dienyl)-propenoic acid (5a)
Following the method used for the synthesis of 1, the Emmons condensation was performed with 4 (0.323 g, 1.34 mmol) to provide ethyl (E)-3-(6-methyl-2,3,4,5-trimethoxyphenyl)-propenoate (0.343 g, 83%) as a pale yellow oil following chromatography (CH2Cl2 to 1:19 EtOAc:CH2Cl2). Rf = 0.17 (CH2Cl2). 1H NMR (CDCl3): δ 1.32 (t, 3H); 2.26 (s, 3H); 3.76 (s, 3H); 3.78 (s, 3H); 3.88 (s, 3H); 3.94 (s, 3H); 4.24 (q, 2H); 6.52 (d, 1H); 7.77 (d, 1H).
The ethyl ester (0.066 g, 0.21 mmol) was hydrolyzed (KOH, EtOH) to provide the unsaturated acid (0.046 g, 78%) as a light tan solid following flash chromatography (2:3 Et2O:hexanes 0.5% AcOH) or recrystallization from Et2O/hexanes. Rf = 0.16 (2:3 Et2O:hexanes 0.5% AcOH) mp = 96 – 97 °C 1H NMR (CDCl3): δ 3.77 (s, 3H); 3.81 (s, 3H); 3.89 (s, 3H); 3.95 (s, 3H); 6.59 (d, 1H); 7.90 (d, 1H)
The unsaturated acid (0.089 g, 0.31 mmol) was then oxidized to provide 5a (0.024 g, 57%) as a red solid following flash chromatography (1:1 Et2O:hexanes 0.5% AcOH) and subsequent recrystallization from Et2O/hexanes. Rf = 0.06 (2:3 Et2O:hexanes 0.5% AcOH), mp = 116 – 125 °C. 1H NMR (CDCl3): δ 2.19 (s, 3H); 4.00 (s, 6H); 6.76 (d, 1H); 7.60 (d, 1H).
(E)-3-(5,6-dimethoxy-3-methyl-1,4-dioxocyclohexa-2,5-dienyl)-2-methylpropenoic acid (5b)
Following the method used for the synthesis of 1, the Emmons condensation was performed with 4 (0.569g, 2.37 mmol) to provide ethyl (E)-3-(6-methyl-2,3,4,5-trimethoxyphenyl)-2-methylpropenoate (0.674 g, 88%) as a pale yellow oil following chromatography (1:3 EtOAc:hexanes). Rf = 0.48 (1:3 EtOAc:hexanes) 1H NMR (CDCl3): δ 1.31 (t, 3H); 1.71 (d, 3H); 2.01 (s, 3H); 3.67 (s, 3H); 3.77 (s, 3H); 3.87 (s, 3H); 3.91 (s, 3H); 4.24 (q, 2H); 7.47 (d, 1H).
The ethyl ester (0.156 g, 0.481 mmol) was hydrolyzed (KOH, EtOH) to provide the unsaturated acid (0.121 g, 85%) as a light yellow solid following flash chromatography (2:3 Et2O:hexanes 0.5% AcOH) and recrystallization from Et2O/hexanes. Rf = 0.16 (2:3 Et2O:hexanes 0.5% AcOH) mp = 98 – 102 °C 1H NMR (CDCl3): δ 1.76 (d, 3H, J = 1.2 Hz); 2.05 (s, 3H); 3.69 (s, 3H); 3.79 (s, 3H); 3.90 (s, 3H); 3.93 (s, 3H); 7.64 (d, 1H).
The unsaturated acid (0.057 g, 0.19 mmol) was then oxidized to provide 5b (0.016 g, 31%) as a red solid following flash chromatography (1:1 Et2O:hexanes 0.5% AcOH) and recrystallization from Et2O/hexanes. Rf = 0.06 (2:3 Et2O:hexanes 0.5% AcOH) mp = 134 – 136 °C 1H NMR (CDCl3): δ 1.77 (d, 3H, J = 1.2 Hz); 1.95 (d, 3H, J = 1.2 Hz); 4.01 (s, 3H); 4.03 (s, 3H); 7.39 (s, 1H).
(E)-3-(5,6-dimethoxy-3-methyl-1,4-dioxocyclohexa-2,5-dienyl)-2-ethylpropenoic acid (5c)
Following the method used for the synthesis of 1, the Emmons condensation was performed with 4 (0.437 g, 1.82 mmol) to provide ethyl (E)-3-(6-methyl-2,3,4,5-trimethoxyphenyl)-2-ethylpropenoate (0.258 g, 42%) as a colorless oil following chromatography (1:3 EtOAc:hexanes). Rf = 0.50 (1:3 EtOAc:hexanes) 1H NMR (CDCl3): δ 0.94 (t, 3H); 1.33 (t, 3H); 2.02 (s, 3H); 2.15 (q, 2H); 3.69 (s, 3H); 3.78 (s, 3H); 3.89 (s, 3H); 3.92 (s, 3H); 4.26 (q, 2H); 7.36 (s, 1H).
The ethyl ester (0.041 g, 0.12 mmol) was then hydrolyzed to provide the unsaturated acid (0.010 g, 27%) as a gold oil following flash chromatography (2:3 Et2O:hexanes 0.5% AcOH. Rf = 0.16 (2:3 Et2O:hexanes 0.5% AcOH) 1H NMR (CDCl3): δ 0.98 (t, 3H); 2.04 (s, 3H); 2.18 (q, 2H); 3.70 (s, 3H); 3.79 (s, 3H); 3.90 (s, 3H); 3.93 (s, 3H); 7.53 (s, 1H).
The unsaturated acid (0.010 g, 0.033 mmol) was then oxidized to provide 5c (0.003 g, 32%) as a red solid following flash chromatography (1:1 Et2O:hexanes 0.5% AcOH) and recrystallization from Et2O/hexanes. Rf = 0.10 (2:3 Et2O:hexanes 0.5% AcOH) mp = 124 – 131 °C. 1H NMR (CDCl3): δ 1.01 (t, 3H); 1.94 (d, 3H); 2.12 (q, 2H); 3.99 (s, 3H); 4.01 (s, 3H); 7.23 (d, 1H)
(E)-3-(4,5-dimethoxy-2-methyl-3,6-dioxocyclohexa-1,4-dienyl)-2-propylpropenoic acid (5d)
Following the method used for the synthesis of 1, the Emmons condensation was performed with 4 (0.437 g, 1.82 mmol) to provide ethyl (E)-3-(6-methyl-2,3,4,5-trimethoxyphenyl)-2-propylpropenoate (0.275 g, 43%) as a colorless oil following chromatography (1:3 EtOAc:hexanes). Rf = 0.52 (1:3 EtOAc:hexanes) 1H NMR (CDCl3): δ 0.75 (t, 3H, J = 7.2 Hz); 1.33 (t, 3H, J = 7.2 Hz); 1.35 (m, 2H); 2.03 (s, 3H); 2.11 (m, 2H); 3.69 (s, 3H); 3.78 (s, 3H); 3.88 (s, 3H); 3.93 (s, 3H); 4.25 (q, 2H, J = 7.2 Hz); 7.38 (s, 1H).
The ethyl ester (0.034 g, 0.098 mmol) was then hydrolyzed (KOH, EtOH) to provide the unsaturated acid (0.029 g, 92%) as a gold oil following flash chromatography (2:3 Et2O:hexanes 0.5% AcOH). Rf = 0.18 (2:3 Et2O:hexanes 0.5% AcOH) 1H NMR (CDCl3): δ 0.77 (t, 3H); 1.41 (m, 2H); 2.04 (s, 3H); 2.13 (m, 2H); 3.70 (s, 3H); 3.79 (s, 3H); 3.89 (s, 3H); 3.93 (s, 3H); 7.55 (s, 1H).
The unsaturated acid (0.029 g, 0.090 mmol) was then oxidized to provide 5d (0.016 g, 59%) as a red solid following flash chromatography (1:1 Et2O:hexanes 0.5% AcOH) and recrystallization from Et2O/hexanes. Rf = 0.12 (2:3 Et2O:hexanes 0.5% AcOH) mp = 95 – 99 °C 1H NMR (CDCl3): δ 0.76 (t, 3H); 1.41 (m, 2H); 1.91 (d, 3H); 2.04 (m, 2H); 3.95 (s, 3H); 3.98 (s, 3H); 7.24 (s, 1H).
(E)-3-(5,6-dimethoxy-3-methyl-1,4-dioxocyclohexa-2,5-dienyl)-2-butylpropenoic acid (5e)
Following the method used for the synthesis of 1, the Emmons condensation was performed with 4 (0.650 g, 2.71 mmol) to provide ethyl (E)-3-(6-methyl-2,3,4,5-trimethoxyphenyl)-2-butylpropenoate (0.484 g, 49%) as a yellow oil following chromatography (1:9 EtOAc:hexanes). Rf = 0.54 (1:3 EtOAc:hexanes) 1H NMR (CDCl3): δ 0.73 (t, 3H); 1.12 (m, 2H); 1.29 (m, 2H); 1.32 (t, 3H); 2.03 (s, 3H); 2.13 (m, 2H); 3.69 (s, 3H); 3.78 (s, 3H); 3.88 (s, 3H); 3.92 (s, 3H); 4.25 (q, 2H); 7.37 (s, 1H).
The ethyl ester (0.166 g, 0.453 mmol) was then hydrolyzed (KOH, EtOH) to provide the unsaturated acid (0.144 g, 94%) as a yellow amorphous solid following flash chromatography (2:3 Et2O:hexanes 0.5% AcOH) and recrystallization from Et2O/hexanes. Rf = 0.26 (2:3 Et2O:hexanes 0.5% AcOH) mp = 70 – 85 °C 1H NMR (CDCl3): δ 0.74 (t, 3H); 1.19 (m, 4H); 2.04 (s, 3H); 2.15 (m, 2H); 3.70 (s, 3H); 3.79 (s, 3H); 3.89 (s, 3H); 3.93 (s, 3H); 7.53 (s, 1H).
The unsaturated acid (0.015 g, 0.044 mmol) was then oxidized to provide 5e (0.005 g, 40%) as a red solid following flash chromatography (1:1 Et2O:hexanes 0.5% AcOH) and recrystallization from Et2O/hexanes. Rf = 0.12 (2:3 Et2O:hexanes 0.5% AcOH) mp = 54 – 55 °C 1H NMR (CDCl3): δ 0.82 (t, 3H); 1.22 (m, 2H); 1.39 (m, 2H); 1.95 (d, 3H); 2.10 (m, 2H); 3.99 (s, 3H); 4.02 (s, 3H); 7.24 (s, 1H).
2-Chloro-3,4,5,6-tetramethoxybenzaldehyde (6)
According to a general aryl chlorination procedure by Lopez-Alvarado,22 2,3,4,5-tetramethoxybenzaldehyde (0.767 g, 3.39 mmol) was dissolved in CH2Cl2 (10 mL) at room temperature and then SO2Cl2 (neat, 0.31 mL, 3.7 mmol) was added at room temperature. The reaction was stirred for 1 h and monitored by 1H NMR for completion. The reaction was then diluted with CH2Cl2, washed with brine, dried over MgSO4, filtered, and condensed. The resulting oil was then purified by flash column chromatography (1:4 Et2O:hexanes) to provide 6 (0.861 g, 97%) as a colorless oil. Rf = 0.27 (1:4 Et2O:hexanes) 1H NMR (CDCl3): δ 3.84 (s, 3H); 3.89 (s, 3H); 3.90 (s, 3H); 4.02 (s, 3H); 10.34 (s, 1H).
(E)-3-(3-chloro-5,6-dimethoxy-1,4-dioxocyclohexa-2,5-dienyl)-2-methylpropenoic acid (7)
Following the method used for the synthesis of 1, the Emmons condensation was performed in refluxing toluene with 6 (0.861g, 3.30 mmol) to provide ethyl (E)-3-(2-chloro-3,4,5,6-trimethoxyphenyl)-2-methylpropenoate (0.559 g, 49%) as a colorless oil following chromatography (1:9 iPr2O:hexanes). Rf= 0.29 (1:4 Et2O:hexanes) E:Z = 1:0 in refluxing toluene 1H NMR (CDCl3): δ 1.34 (t, 3H); 1.78 (d, 3H); 3.69 (s, 3H); 3.86 (s, 3H); 3.90 (s, 3H); 3.94 (s, 3H); 4.26 (q, 2H); 7.42 (q, 1H).
The ethyl ester (0.255 g, 0.740 mmol) was then hydrolyzed (KOH, EtOH) to provide the unsaturated acid (0.222 g, 95%) as a red amorphous solid following flash chromatography (2:3 Et2O:hexanes 0.5% AcOH) and recrystallization from Et2O/hexanes. Rf = 0.20 (2:3 Et2O:hexanes 0.5% AcOH) 1H NMR (CDCl3): δ 1.81 (s, 3H); 3.71 (s, 3H); 3.86 (s, 3H); 3.91 (s, 3H); 7.57 (d, 1H)
The unsaturated acid (0.124 g, 0.391 mmol) was dissolved in acetonitrile (10 mL) at room temperature, then ceric ammonium nitrate (0.970 g, 1.77 mmol) dissolved in water (8 ml) was added at room temperature. The reaction was stirred for 30 min and then extracted with CH2Cl2. The organic layer was washed with brine, dried over MgSO4, filtered, and condensed. The red oil was then purified by either flash column chromatography (1:1 Et2O:hexanes 0.5% AcOH) or recrystallization from Et2O/hexanes to afford 7 (0.026 g, 22%) as a red solid. Rf = 0.32 (1:1 Et2O:hexanes 0.5% AcOH) mp = 183 – 185 °C 1H NMR (CDCl3): δ 1.83 (d, 3H); 4.03 (s, 3H); 4.05 (s, 3H); 7.28 (d, 1H).
(E)-4′-(6-methyl-2,3,4,5-tetramethoxyphenyl)-3-ethylidenetetrahydrofuran-2-one (8)
NaH (0.210 g, 5.25 mmol) was added to a flame-dried 100 mL 3-neck round bottom flask connected to a water-jacketed reflux condenser.20 The flask was purged with argon and a drying tube was attached to the top. Toluene (30mL) was added to the flask followed by 2-diethylphosphono-γ-butryrolactone (1.83 g, 8.2 mmol) dissolved in toluene (10 mL) at room temperature. The reaction was heated under reflux for 30 min, and the aldehyde (4, 0.501 g, 2.09 mmol) was dissolved in toluene (5 mL) and added slowly at reflux. The reaction was heated for another 8 h under reflux before being cooled to room temperature, diluted with ethyl acetate, and washed with brine. The organic layer was dried, filtered, and condensed. The resulting oil was purified via flash column chromatography (1:3 EtOAc:hexanes) to provide 8 (0.429 g, 66%) as a white solid. Rf = 0.18 (1:3 EtOAc:hexanes) E:Z = 20:1 in refluxing toluene. Mp = 78 – 79 °C 1H NMR (CDCl3): δ 2.12 (s, 3H); 2.85 (dt, 2H); 3.65 (s, 3H); 3.78 (s, 3H); 3.90 (s, 3H); 3.93 (s, 3H); 4.35 (t, 2H); 7.48 (t, 1H).
(E)-3-(5,6-dimethoxy-3-methyl-1,4-dioxocyclohexa-2,5-dienyl)-2-methoxyethylpropenoic acid (9)
According to a modified procedure of King,17 8 (0.084 g, 0.27 mmol) was added to a flame-dried 10 mL round bottom flask under argon, and a water-jacketed reflux condenser was attached. Anhydrous MeOH (2 mL) was then added, followed by H2SO4 (4 drops) and trimethyl orthoformate (0.31 mL, 4.0 mmol) at room temperature. The reaction was then heated under reflux for 12 h, cooled to room temperature, and the solvent was removed under reduced pressure. The residue was dissolved in EtOAc, washed with saturated brine, dried over MgSO4, filtered, and condensed. The resulting crude product was purified via flash column chromatography (1:3 EtOAc:hexanes) to provide the methyl ester (0.073 g, 76%) as a pale yellow solid. Rf = 0.25 (1:3 EtOAc:hexanes) mp = 35 – 37 °C 1H NMR (CDCl3): δ 2.02 (s, 3H); 2.47 (t, 2H); 3.17 (s, 3H); 3.37 (t, 2H); 3.69 (s, 3H); 3.78 (s, 3H); 3.81 (s, 3H); 3.88 (s, 3H); 3.92 (s, 3H); 7.49 (s, 1H).
The methyl ester (0.074 g, 0.21 mmol) was hydrolyzed following the method for 1 to provide the unsaturated acid (0.070 g, 100%) as a tan solid following flash chromatography (2:3 Et2O:hexanes 0.5% AcOH) and recrystallization from Et2O/hexanes. Rf = 0.16 (2:3 Et2O:hexanes 0.5% AcOH) mp = 97 – 99 °C 1H NMR (CDCl3): δ 2.07 (s, 3H); 2.45 (t, 2H); 3.20 (s, 3H); 3.68 (s, 3H); 3.69 (q, 2H); 3.76 (s, 3H); 3.87 (s, 3H); 3.90 (s, 3H); 7.59 (s, 1H).
The resulting unsaturated acid (0.070 g, 0.21 mmol) was oxidized following the procedure for 1 to provide 9 (0.012 g, 19%) as a red solid following flash chromatography (2:3 Et2O:hexanes 0.5% AcOH) and recrystallization from Et2O/hexanes. Rf = 0.06 (2:3 Et2O:hexanes 0.5% AcOH) mp = 104 – 106 °C; 1H NMR (CDCl3): δ 1.95 (d, 3H); 2.40 (t, 2H); 3.24 (s, 3H); 3.44 (t, 2H); 3.99 (s, 3H); 4.01 (s, 3H); 7.35 (d, 1H).
(E)-N-(2-hydroxyethyl)-3-(6-methyl-2,3,4,5-tetramethoxyphenyl)-2-nonylpropenamide (11)
Dicyclohexylcarbodiimide (0.106 g, 0.514 mmol) was added to a flame dried 10 mL round bottom flask under argon, and then CH2Cl2 (2 mL) was added at room temperature. In a second flame-dried flask under argon a solution of acid (E)-10 (0.167 g, 0.409 mmol) and 1-hydroxybenztriazole (0.008 g, 0.06 mmol) was prepared using DMF (3 mL) and CH2Cl2 (1 mL). The acid/HOBt solution was then added and stirred for 2 h. Ethanolamine (0.040 mL, 0.66 mmol) was then added rapidly at room temperature. The reaction was stirred for an additional 12 h, diluted with EtOAc, washed with brine, dried over MgSO4, filtered, and condensed. The resulting white solid, a mixture of dicyclohexylurea, DCC, and product, was first suspended in approximately 5 mL CH2Cl2. The suspension was then filtered through a pipette containing a cotton plug, and the pipette was washed 2 times with 2 mL of CH2Cl2. The combined fractions were then purified via flash chromatography (1:4 EtOAc:hexanes) to provide two compounds that appeared to be isomers: (E)-11 (0.088 g, 48%, 2:3 EtOAc:hexanes Rf = 0.36) and (Z)-11 (0.063 g, 33%, 2:3 EtOAc:hexanes Rf = 0.71, coeluted with DCC), both as colorless oils. (E)-11: 1H NMR (CDCl3): δ 0.83 (t, 3H); 1.13 (m, 12H); 1.32 (m, 2H); 2.03 (s, 3H); 2.13 (m, 2H); 2.70 (t, 1H); 3.55 (m, 2H); 3.69 (s, 3H); 3.78 (s, 3H); 3.80 (m, 2H); 3.88 (s, 3H); 3.92 (s, 3H); 6.31 (bs, 1H); 6.92 (s, 1H). (Z)-11: 1H NMR (CDCl3): δ 3.19 (s, 3H); 2.14 (m, 2H); 2.42 (m, 2H); 3.68 (m, 1H); 3.75 (s, 3H); 3.78 (s, 3H); 3.88 (s, 3H); 3.92 (s, 3H); 4.07 (m, 1H); 6.64 (s, 1H); 8.53 (d, 1H).
(E)-N-(2-hydroxyethyl)-3-(5,6-dimethoxy-3-methyl-1,4-dioxocyclohexa-2,5-dienyl)-2-nonyl-propenamide (E-12)
Amide (E)-11 (0.088 g, 0.20 mmol) was added to a flame-dried round bottom flask and dissolved in EtOAc (7.0 mL). Then AcOH (5 drops) and HNO3 (0.5 mL) were added and the reaction was stirred for 3 hours at room temperature. The colorless oil easily dissolved in EtOAc and the reaction turned orange upon addition of HNO3. The reaction was then diluted with EtOAc (40.0 mL), washed 3 times with brine, dried over MgSO4, filtered, and condensed to provide an orange oil. The oil was then purified using flash chromatography (1:2 - 1:1 EtOAc:hexanes) and recrystallized from acetone/hexane to provide (E)-12 (0.037 g, 45%) as a red oil. Rf = 0.24 (2:3 EtOAc:hexanes) 1H NMR (CDCl3): δ 0.84 (t, 3H); 1.17 (m, 12 H); 1.33 (m, 2H); 1.93 (d, 3H); 2.09 (t, 2H); 2.51 (t, 1H); 3.52 (q, 2H); 3.79 (dd, 2H); 3.98 (s, 3H); 4.02 (s, 3H); 6.48 (bs, 1H); 6.53 (s, 1H).
1,4-Dimethoxy-2-naphthaldehyde (14a)
Following the procedure of Evans et al.,23 1,4-naphthoquinone (13a, 10.00 g, 63.20 mmol) and Pd/C (10 wt%, 0.994 g) were added to a flame-dried 500 mL round bottom flask at room temperature under Argon. THF (250.0 mL) was then added at room temperature, the reaction vessel was covered in foil, and then purged with hydrogen gas for 10 min. A balloon filled with H2 was attached and the reaction stirred for 4 h at room temperature. The reaction was then purged with Argon before adding NaH (5.66 g, 142 mmol) slowly at 0 °C. After 10 min dimethyl sulfate (13.0 mL, 137 mmol) was added slowly at 0 °C. The reaction mixture then became too thick to stir, so THF (50 mL) was added. The dark green mixture was allowed to stir at room temperature for 4 h. The reaction was then filtered through celite, washed with brine, dried over MgSO4, and condensed. The resulting solid was then purified by flash chromatography (EtOAc:hexanes) or recrystallized from Et2O:hexanes to provide 1,4-dimethoxynaphthalene (11.74 g, 99%) as pink needles. Rf = 0.76 (CH2Cl2) mp = 74 – 78 °C (Lit. 84 – 86 °C)18 1H NMR (CDCl3): δ 3.95 (s, 6H); 6.69 (s, 2H), 7.50 (m, 2H); 8.20 (m, 2H). Following a modified procedure of Ito et al.,24 to a flame-dried 50 mL round bottom flask was added 1,4-dimethoxynaphthalene (1.34 g, 7.12 mmol) dissolved in CH2Cl2 (10.0 mL) and then cooled to 0 °C under Argon. Then TiCl4 (1 M in CH2Cl2, 8.0 mL, 8.0 mmol) was added slowly at 0 °C followed by α,α-dichloromethyl methyl ether (0.71 mL, 8.0 mmol) at 0 °C. The reaction was stirred at 0 °C for 3 h, poured into water and stirred for 10 min at room temperature. The reaction was then extracted with EtOAc, washed with saturated brine, dried over MgSO4, filtered, and condensed. The resulting oil was purified using flash chromatography (CH2Cl2) to provide 27 (1.43 g, 93%) as a white solid that was recrystallized from Et2O:hexanes to provide white needles. Rf = 0.62 (CH2Cl2) mp = 108 – 109 °C (Lit. 120 – 121 °C)22 1H NMR (CDCl3): δ 4.01 (s, 3H); 4.08 (s, 3H); 7.11 (s, 1H); 7.62 (m, 2H); 8.23 (m, 2H); 10.56 (s, 1H).
1,4-dimethoxy-3-methyl-2-naphthaldehyde (14b)
1,4-dimethoxy-2-methylnaphthalenewas prepared from 13b (1.90 g, 9.39 mmol) as described above for 14a to give 1.490 g (69%) of the product as a white solid that was recrystallized from Et2O/hexanes to provide white needles. Rf = 0.57 (3:7 EtOAc:hexanes) mp = 78 – 80 °C (Lit. 83.5 – 83.7 °C)22 1H NMR (CDCl3): δ 2.63 (s, 3H); 3.85 (s, 3H); 4.05 (s, 3H); 7.59 (dt, 2H); 8.14 (dd, 2H).
Compound 14b was prepared from 1,4-dimethoxy-2-methylnaphthalene(3.54 g, 20.6 mmol) as described above for 14a to give 4.02 g (97%) of the product as a low melting white solid following flash chromatography (1:19 EtOAc:hexanes). Rf = 0.60 (3:7 EtOAc:hexanes) mp = 29 – 31 °C (Lit. 35.9 – 36.3)221H NMR (CDCl3): δ 2.43 (s, 3H); 3.85 (s, 3H); 3.95 (s, 3H); 7.45 (dt, 2H); 8.09 (dd, 2H).
1,3,4-trimethoxy-2-naphthaldehyde (14c)
1,2,4-Trimethoxynaphthalene was prepared from 13h (5.23 g, 33.3 mmol) as described above for 14a to give 5.19 g (71%) of the product as a red oil that solidified in the freezer. Rf = 0.17 (1:9 EtOAc:hexanes); 0.50 (CH2Cl2) Mp = oil (Lit. 38 – 40 °C)20 1H NMR (CDCl3): δ 3.91 (s, 3H); 3.98 (s, 3H); 3.99 (s, 3H); 6.63 (s, 1H); 7.40 (m, 2 H); 8.09 (dd, 2H) According to the procedure by Syper et al.,23 1,2,4-trimethoxynaphthalene(1.07 g, 4.90 mmol) was dissolved in THF (20.0 mL) at 0 °C under argon, and then nBuLi (1.6 M in hexanes, 4.0 mL, 10.0 mmol) was added at 0 °C. The reaction was stirred at 0 °C for 5 h before DMF (0.850 mL, 11.0 mmol) was added at 0 °C. The reaction was stirred for another 30 min at room temperature before quenching with water. The reaction was diluted with ethyl ether and washed with saturated brine. The organic layer was dried over MgSO4, filtered, and condensed. Following purification via column chromatography (CH2Cl2 to 1:9 EtOAc: CH2Cl2) pure aldehyde 14c (1.07 g, 89%) was obtained as a yellow solid. Rf = 0.25 (1:9 EtOAc:hexanes); 0.20 (CH2Cl2) mp = 43 – 45 °C (Lit. 53 °C d)231H NMR (CDCl3): δ 3.99 (s, 3H); 4.00 (s, 3H); 4.03 (s, 3H); 7.55 (m, 2H); 8.15 (dd, 2H); 10.57 (s, 1H)
1,4-Dimethoxy-3-methylthio-2-naphthaldehyde (14d)
Pyridinium chlorochromate (PCC, 1.03 g, 4.78 mmol) was added to a flame-dried 250 mL round bottom flask followed by dry CH2Cl2 (90 mL) at room temperature under argon. 1,4-dimethoxy-2-hydroxymethyl-3-methylthionaphthalene, synthesized following the procedure of Flader et al.,21 (0.514 g, 1.94 mmol) was dissolved in CH2Cl2 (10 mL) and added slowly at room temperature. The reaction was stirred for a further 12 h at room temperature before being poured into a slurry of florisil, MgSO4, and CH2Cl2. After stirring, the suspension was filtered through celite and condensed. Purification by flash chromatography (1:9 EtOAc:hexanes)gave 14d (0.308 g, 1.17 mmol, 62%) as an amorphous yellow solid. Rf = 0.26 (1:9 EtOAc:hexanes) 1H NMR (CDCl3): δ 2.47 (s, 3H); 4.01 (s, 3H); 4.04 (s, 3H); 7.62 (m, 2H); 8.13 (d, 1H); 8.21 (d, 1H); 10.70 (s, 1H).
1,4-Dimethoxy-3-fluoro-2-naphthaldehyde (14e)
Aldehyde 14a (0.517 g, 2.39 mmol) and MeCN (20 mL) were added to a flame-dried 50 mL round bottom flask under argon, and Selectfluor (1.25 g, 3.37 mmol) was added. The reaction was then heated to reflux for 8 h, then cooled and extracted with EtOAc. The organic layer was washed 3 times with brine, dried over MgSO4, filtered, and condensed to provide the crude aldehyde as a yellow solid. Following flash chromatography (3:1 CH2Cl2:hexanes) aldehyde 14e (0.254 g, 45%) was obtained as a yellow solid. Rf = 0.36 (3:1 CH2Cl2:hexanes) mp = 69 – 71 °C 1H NMR (CDCl3): δ 4.07 (s, 3H); 4.09 (d, 3H); 7.54 (t, 1H); 7.66 (t, 1H); 8.18 (m, 2H); 10.54 (s, 1H).
3-Chloro-1,4-dimethoxy-2-naphthaldehyde (14f)
Following a general aromatic chlorination procedure reported by Lopez-Alvarado,22 aldehyde 14a (2.12 g, 9.81 mmol) was dissolved in CH2Cl2 (15 mL) at room temperature in a flame-dried 50 mL round bottom flask under argon. Neat SO2Cl2 (0.92 mL, 11 mmol) was added at room temperature and the argon line was replaced with a drying tube. After 20 h the reaction was complete by NMR, and the solution was diluted with CH2Cl2 (50 mL), washed with brine, dried over MgSO4, and filtered. The resulting solid was purified by flash chromatography (1:1 CH2Cl2:hexanes) to provide 14f (1.63 g, 66%) as white needles. Rf = 0.21 (1:1 CH2Cl2:hexanes) mp = 94 – 96 °C 1H NMR (CDCl3): δ 3.99 (s, 3H); 4.05 (s, 3H); 7.64 (m, 2H); 8.17 (dd, 2H); 10.61 (s, 1H).
Ethyl (E)-3-(1,4-dimethoxynaphthalen-2-yl)-2-methylpropenoate (15a)
NaH (0.303 g, 7.59 mmol) was added to a flame-dried 50 mL round bottom flask connected to a water-jacketed reflux condenser. The flask was purged with argon and a drying tube was attached to the top of the condenser. Toluene (20 mL) was added to the flask followed by triethyl 2-phosphonopropionate (1.0 ml, 4.6 mmol) at room temperature. The reaction was heated under reflux for 30 min, and aldehyde 14a (0.542 g, 2.51 mmol) was dissolved in toluene (5 mL) and added slowly at reflux. The reaction was heated for another 8 h under reflux. The reaction was then cooled to room temperature, acidified with 2 M HCl, diluted with ethyl acetate, and washed with brine. The organic layer was dried, filtered, and condensed. The resulting oil was purified via flash column chromatography (1:19 iPr2O:hexanes) to provide 15a (0. 554 g, 73%) as a colorless oil. Rf = 0.30 (1:19 iPr2O:hexanes, 4 developments), E:Z = 20:1. 1H NMR (CDCl3): δ 1.36 (t, 3H); 2.10 (d, 3H); 3.91 (s, 3H); 3.96 (s, 3H); 4.29 (q, 2H); 6.70 (s, 1H); 7.51 (m, 2H); 7.96 (d, 1H); 8.15 (m, 2H).
Ethyl (E)-3-(1,4-dimethoxy-3-methylnaphthalen-2-yl)-2-methylpropenoate (15b)
Compound 15b was prepared from 14b (0.506 g, 2.20 mmol) as described above for 15a to give 0.541 g (1.42 mmol, 65%) of the product as a colorless oil following flash chromatography (1:19 iPr2O:hexanes). Rf (E)-15b = 0.22; (Z)-15b = 0.15 (1:19 iPr2O:hexanes, 4 developments), E:Z = 20:1. (E)-15b 1H NMR (CDCl3): δ 1.36 (t, 3H); 1.79 (s, 3H); 2.28 (s, 3H); 3.73 (s, 3H); 3.87 (s, 3H); 4.30 (q, 2H); 7.49 (m, 2H); 7.70 (s, 1H); 8.07 (m, 2H).
Ethyl (E)-3-(1,3,4-trimethoxynaphthalen-2-yl)-2-methylpropenoate (15c)
Compound 15c was prepared from 14c (0.330 g, 1.33 mmol) as described above for 15a to give 0.119 g (27%) of the product as a yellow oil following flash chromatography (1:9 Et2O:hexanes) Rf= (E)-15c = 0.17; (Z)-15c = 0.11 (1:19 iPr2O:hexanes, 4 developments) E:Z = 11:9 in refluxing toluene 1H NMR (CDCl3): δ 1.36 (t, 3H); 1.87 (d, 3H); 3.75 (s, 3H); 3.87 (s, 3H); 3.98 (s, 3H); 4.29 (q, 2H); 7.47 (m, 2H); 7.73 (d, 1H); 8.09 (m, 2H).
Ethyl (E)-3-(1,4-dimethoxy-3-methylsulfanylnaphthalen-2-yl)-2-methylpropenoate (15d)
Compound 15d was prepared from 14d (0.308 g, 1.17 mmol) as described above for 15a to give 0.231 g (55%) of the product as an amorphous yellow solid following flash chromatography (1:9 EtOAc:hexanes) and recrystallization from Et2O/hexanes. Rf = 0.26 (1:9 EtOAc:hexanes); E:Z = 7:3. 1H NMR (CDCl3): δ 1.37 (t, 3H); 1.82 (s, 3H); 2.37 (s, 3H); 3.72 (s, 3H); 4.00 (s, 3H); 4.30 (q, 2H); 7.54 (m, 2H); 7.86 (s, 1H); 8.10 (m, 2H).
Ethyl (E)-3-(1,4-dimethoxy-3-fluoronaphthalen-2-yl)-2-methylpropeonoate (15e)
Compound 15e was prepared from 14e (0.216 g, 0.922 mmol) as described above for 15a to give 0.226 g (77%) of the product as a yellow oil following flash chromatography (1:19 Et2O:hexanes). (0.093 g pure E; 0.128 g E/Z mixture) Rf(E)-15e 0.26; (Z)-15e = 0.17 (1:19 Et2O:hexanes, 3 developments); E:Z = 5:1. 1H NMR (CDCl3): δ 1.36 (t, 3H); 1.91 (t, 3H); 3.80 (s, 3H); 4.05 (d, 3H); 4.30 (q, 2H); 7.51 (m, 2H); 7.67 (s, 1H); 8.09 (dd, 1H); 8.14 (dd, 1H).
Ethyl (E)-3-(3-chloro-1,4-dimethoxynaphthalen-2-yl)-2-methylpropenoate (15f)
Compound 15f was prepared from 14f (2.93 g, 11.7 mmol) as described above for 15a to give 2.56 g (65%) of the product as a yellow solid following flash chromatography (1:19 Et2O:hexanes) and recrystallization from Et2O/hexanes. (1.50 g E, yellow solid; 0.960 g E/Z mixture, gold oil; 0.101 g Z, gold solid) Rf(E)-15f= 0.32; (Z)-15f = 0.24 (1:19 iPr2O:hexanes, 4 developments); E:Z = 15:5. mp = 108 – 109 °C 1H NMR (CDCl3): δ 1.37 (t, 3H); 1.83 (s, 3H); 3.75 (s, 3H); 3.98 (s, 3H); 4.30 (q, 2H); 7.55 (m, 2H); 7.68 (s, 1H); 8.11 (m, 2H).
Ethyl (E)-3-(3-bromo-1,4-dimethoxynaphthalen-2-yl)-2-methylpropenoate (15g)
Compound 15g was prepared from 14g (0.515 g, 1.75 mmol) as described above for 15a to give 0.373 g (56%) of the product as a white solid following flash chromatography (1:19 iPr2O:hexanes) and recrystallization from Et2O/hexanes. (0.314 g E; 0.059 g Z) Rf(E)-15g = 0.26; (Z)-15g = 0.19 (1:19 iPr2O:hexanes, 4 developments); E:Z = 17:3. mp = 102 – 103 °C 1H NMR (CDCl3): δ 1.37 (t, 3H); 1.81 (d, 3H); 3.74 (s, 3H); 3.97 (s, 3H); 4.31 (q, 2H); 7.56 (m, 2H); 7.65 (d, 1H); 8.11 (m, 2H).
(E)-3-(1,4-naphthoquinon-2-yl)-2-methylpropenoic acid (16a)
Emmons ester 15a (0.231 g, 0.769 mmol) was dissolved in EtOH (10 mL) and then KOH (0.40 g, 7.13 mmol) was added to the reaction. The reaction was heated to reflux and stirred at this temperature for 30 min. The reaction was then cooled, acidified, and extracted with ethyl acetate. The organic layer was washed with brine, dried over MgSO4, filtered, and condensed. The resulting acid was then used without further purification in the next step. Alternatively, it can be purified via flash chromatography (1:3 EtOAc:hexanes 0.5% AcOH) or by recrystallization from Et2O/hexanes to provide (E)-3-(1,4-dimethoxynaphthalen-2-yl)-2-methylpropenoic acid (0.175 g, 83%) as a white solid. Rf = 0.24 (1:3 EtOAc:hexanes 0.5% AcOH) mp = 149 – 155 °C 1H NMR (CDCl3): δ 2.16 (d, 3H); 3.86 (s, 3H); 3.99 (s, 3H); 6.75 (s, 1H); 7.53 (m, 2H); 8.15 (d, 1H); 8.17 (m, 2H).
The crude acid (0.114 g, 0.419 mmol) was dissolved in ethyl acetate (10 mL) at room temperature, then HNO3 (1 mL) and AcOH (3 drops) were added at room temperature. The reaction was stirred at room temperature for 4 h and then diluted with ethyl acetate and washed with brine. The organic layer was dried over MgSO4, filtered, and condensed. The yellow oil was then purified by either flash column chromatography (2:3 Et2O:hexanes 0.5% AcOH) or recrystallization from Et2O/hexanes to afford 16a (0.059 g, 58%) as a yellow solid. Rf = 0.43 (2:3 Et2O:hexanes 0.5% AcOH) mp = 205 °C d 1H NMR (CDCl3): δ 2.13 (d, 3H,); 6.99 (s, 1H); 7.79 (m, 3H); 8.11 (m, 2H).
(E)-3-(3-methyl-1,4-naphthoquinon-2-yl)-2-methylpropenoic acid (16b)
(E)-3-(1,4-dimethoxy-3-methylnaphthalen-2-yl)-2-methylpropenoic acid was prepared from 15b (0.176 g, 0.560 mmol) as described above for 16a to give 0.134 g (83%) of the product as a light yellow solid following flash chromatography (3:17 acetone:hexanes 0.5% AcOH) and recrystallization from Et2O/hexanes. Rf = 0.20 (1:3 EtOAc:hexanes 0.5% AcOH) mp = 153 – 155 °C 1H NMR (CDCl3): δ 1.80 (s, 3H); 3.75 (s, 3H); 3.89 (s, 3H); 7.51 (m, 2H); 7.72 (s, 1H); 8.09 (m, 2H).
Compound 16b was prepared from (E)-3-(1,4-dimethoxy-3-methylnaphthalen-2-yl)-2-methylpropenoic acid (0.083 g, 0.29 mmol) as described above for 16a to give 0.048 g (0.19 mmol, 65%) of the product as a yellow solid following flash chromatography (3:17 acetone:hexanes 0.5% AcOH) and recrystallization from Et2O/hexanes. Rf = 0.30 (2:3 Et2O:hexanes) mp = 195 – 196 °C. 1H NMR (CDCl3): δ 1.81 (bs, 3H); 2.11 (bs, 3H); 7.58 (m, 1H); 7.73 (m, 2H); 8.09 (m, 2H).
(E)-3-(3-methoxy-1,4-naphthoquinon-2-yl)-2-methylpropenoic acid (16c)
(E)-3-(1,3,4-trimethoxy-naphthalen-2-yl)-2-methylpropenoic acid was prepared from 15c (0.073 g, 0.22 mmol) as described above for 16a to give 0.067 g (100%) of the product as a tan solid following flash chromatography (1:3 EtOAc:hexanes 0.5% AcOH) and recrystallization from Et2O/hexanes. Rf = 0.13 (1:3 EtOAc: hexanes 0.5% AcOH) mp = 119 – 123 °C 1H NMR (CDCl3): δ 1.91 (s, 3H); 3.77 (s, 3H); 3.89 (s, 3H); 3.99 (s, 3H); 7.49 (m, 2H); 7.91 (s, 1H); 8.10 (t, 2H).
Compound 16c was prepared from (E)-3-(1,3,4-trimethoxynaphthalen-2-yl)-2-methylpropenoic acid (0.060 g, 0.20 mmol) as described above for 16a to give 0.015 g (27%) of the product as a yellow solid following flash chromatography (2:3 Et2O:hexanes 0.5% AcOH) and recrystallization from Et2O/hexanes. Rf = 0.43 (2:3 Et2O:hexanes 0.5% AcOH) mp = 205 °C d 1H NMR (CDCl3): δ 1.83 (d, 3H); 4.15 (s, 3H); 7.53 (d, 1H); 7.73 (m (5), 2H); 8.08 (m, 2H).
(E)-3-(3-methylthio-1,4-naphthoquinon-2-yl)-2-methylpropenoic acid (16d)
(E)-3-(1,4-dimethoxy-3-methylthionaphthalen-2-yl)-2-methylpropenoic acid was prepared from 15d (0.165 g, 0.476 mmol) as described above for 16a to give 0.152 g (100%) of the product as an orange solid following flash chromatography (1:3 EtOAc:hexanes 0.5% AcOH) and recrystallization from Et2O/hexanes. Rf = 0.18 (1:3 EtOAc:hexanes 0.5% AcOH) 1H NMR (CDCl3): δ 1.82 (d, 3H); 2.37 (s, 3H); 3.72 (s, 3H); 4.00 (s, 3H); 7.51 (m, 2H); 7.99 (d, 1H); 8.07 (m, 2H).
Compound 16d was prepared from (E)-3-(1,4-dimethoxy-3-methylthionaphthalen-2-yl)-2-methylpropenoic acid (0.180 g, 0.565 mmol) as described above for 16a to give 0.063 g (38%) of the product as a red fluffy solid following flash chromatography (2:3 Et2O:hexanes 0.5% AcOH) and recrystallization from Et2O/hexanes. Rf = 0.45 (2:3 Et2O:hexanes 0.5% AcOH) mp = 195 – 196 °C 1H NMR (CDCl3): δ 1.81 (d, 3H); 2.58 (s, 3H); 7.56 (d, 1H); 7.72 (m, 2H); 8.09 (m, 2H).
(E)-3-(3-fluoro-1,4-naphthoquinon-2-yl)-2-methylpropenoic acid (16e)
(E)-3-(1,4-dimethoxy-3-fluoronaphthalen-2-yl)-2-methylpropenoic acid was prepared from 15e (0.128 g, 0.402 mmol) as described above for 16a to give 0.097 g (83%) of the product as a white solid following flash chromatography (1:4 acetone:hexanes 0.5% AcOH) and recrystallization from Et2O/hexanes. Rf = 0.13 (1:4 acetone:hexanes 0.5% AcOH) mp = 157 – 158.5 °C 1H NMR (CDCl3): δ 1.93 (d, 3H); 3.82 (s, 3H); 4.06 (d, 3H); 7.52 (m, 2H); 7.83 (d, 1H); 8.10 (d,1H); 8.15 (d, 1H) 19F NMR (CDCl3): δ −137.38.
(E)-3-(1,4-dimethoxy-3-fluoronaphthalen-2-yl)-2-methylpropenoic acid (0.097 g, 0.33 mmol) was dissolved in ethyl acetate (10 mL) at room temperature, then HNO3 (1 mL) and AcOH (6 drops) are added at room temperature. Silver (II) oxide (0.309 g, 2.49 mmol) was then added and the reaction was stirred vigorously at room temperature for 1 h. The mixture was filtered through a pasteur pipette and cotton plug, the plug was washed with ethyl acetate, and the combined organic layers were washed with brine. The organic layer was dried over MgSO4, filtered, and condensed. The yellow oil was then purified by either flash column chromatography (1:1 Et2O:hexanes 0.5% AcOH) or recrystallization from Et2O/hexanes to afford 16e (0.020 g, 23%) as a yellow solid. Rf = 0.08 (1:4 EtOAc:hexanes 0.5 % AcOH) mp = 185 – 190 °C d 1H NMR (CDCl3): δ 1.94 (d, 3H); 7.45 (s, 1H); 7.80 (m, 2H); 8.15 (m, 2H) 1H NMR (DMSO): δ 1.83 (d, 3H); 7.15 (s, 1H); 7.91 (m, 2H); 8.06 (m, 2H).
(E)-3-(3-chloro-1,4-naphthoquinon-2-yl)-2-methylpropenoic acid (16f)
Emmons ester 15f (0.959 g, 2.90 mmol) was hydrolyzed as described for 16a to provide (E)-3-(3-chloro-1,4-dimethoxynaphthalen-2-yl)-2-methylpropenoic acid (0.872 g, 98%) as a tan solid following flash chromatography (3:17 acetone:hexanes 0.5% AcOH) and recrystallization from Et2O/hexanes. Rf = 0.08 (3:17 acetone:hexanes 0.5% AcOH) mp = 155 – 157 °C 1H NMR (CDCl3): δ 1.88 (s, 3H); 3.77 (s, 3H); 3.99 (s, 3H); 7.58 (m, 2H); 7.85 (s, 1H); 8.123 (m, 2H).
Following the procedure for 16e, (E)-3-(3-chloro-1,4-dimethoxynaphthalen-2-yl)-2-methylpropenoic acid (0.146 g, 0.476 mmol) was oxidized to provide 16f (0.087 g, 0.32 mmol, 67%) as a yellow solid following flash chromatography (2:3 acetone:hexanes 0.5% AcOH) and recrystallization from Et2O/hexanes. Rf = 0.44 (2:3 acetone:hexanes 0.5% AcOH) mp = 229 – 230 °C. 1H NMR (CDCl3): δ 1.87 (d, 3H); 7.45 (d, 1H); 7.79 (m, 2H); 8.13 (m, 1H); 8.19 (m, 1H).
(E)-3-(3-bromo-1,4-naphthoquinon-2-yl)-2-methylpropenoic acid (16g)
(E)-3-(3-bromo-1,4-dimethoxynaphthalen-2-yl)-2-methylpropenoic acid was prepared from 15g (0.314 g, 0.828 mmol) as described above for 16a to give 0.291 g (100%) of the product as a white solid following recrystallization from Et2O/hexanes. Rf = 0.32 (1:1 Et2O:hexanes 0.5% AcOH) mp = 143 – 145 °C 1H NMR (CDCl3): δ 1.86 (d, 3H); 3.77 (s, 3H); 3.99 (s, 3H); 7.57 (m, 2H); 7.82 (d, 1H); 8.13 (m, 2H).
Following the procedure for 16e, (E)-3-(3-bromo-1,4-dimethoxynaphthalen-2-yl)-2-methyl-propenoic acid (0.291 g, 0.829 mmol) was oxidized to provide 16g (0.122 g, 46%) as a yellow solid following flash chromatography (1:1 Et2O:hexanes 0.5% AcOH) and recrystallization from Et2O/hexanes. Rf = 0.32 (1:1 Et2O:hexanes 0.5% AcOH) mp = 186 – 189 °C. 1H NMR (CDCl3): δ 1.85 (d, 3H); 7.33 (m, 1H); 7.91 (m, 2H); 8.14 (m, 2H).
(E)-4′-(1,4-dimethoxy-2-methylnaphthalen-2-yl)-3-ethylidenetetrahydrofuran-2-one (20a)
Lactone 20a was prepared following the procedure for 8 using aldehyde 14b (0.486 g, 2.11 mmol) to provide 0.629 g (100%) of product as a white solid following flash chromatography (1:4 EtOAc:hexanes) and recrystallization from Et2O/hexanes. Rf = 0.32 (1:3 EtOAc:hexanes); E:Z = 100:0. mp = 105 – 107 °C 1H NMR (CDCl3): δ 2.37 (s, 3H); 2.92 (dt, 2H); 3.70 (s, 3H); 3.87 (s, 3H); 4.40 (t, 2H); 7.52 (m, 2H); 7.69 (t, 1H); 8.09 (m, 2H).
(E)-4′-(3-chloro-1,4-dimethoxynaphthalen-2-yl)-3-ethylidenetetrahydrofuran-2-one (20b)
Lactone 20b was prepared following the procedure for 8 using aldehyde 14f (0.484 g, 1.94 mmol) to provide 0.311 g (51%) of the product as a colorless oil following flash chromatography (1:9 EtOAc:hexanes). Rf = 0.13 (3:37 EtOAc:hexanes, developed 4x); E:Z = 9:1. 1H NMR (CDCl3): δ 2.94 (dt, 2H); 3.73 (s, 3H); 3.99 (s, 3H); 4.41 (t, 2H); 7.58 (m, 2H); 7.77 (t, 1H, 3 Hz); 8.12 (m, 2H).
(E)-4′-(3-bromo-1,4-dimethoxynaphthalen-2-yl)-3-ethylidenetetrahydrofuran-2-one (20c)
Lactone 20c was prepared following the procedure for 8 using aldehyde 14g (0.588 g, 1.99 mmol) to provide 0.307 g (42%) of the product as a colorless oil following flash chromatography (3:17 EtOAc:hexanes). Rf = 0.09 (1:9 EtOAc:hexanes); E:Z = 3:1. 1H NMR (CDCl3): δ 2.92 (dt, 2H, J = 3, 7.2 Hz); 3.73 (s, 3H); 3.98 (s, 3H); 4.41 (t, 2H, J = 7.2 Hz); 7.59 (m, 2H); 7.74 (t, 1H); 8.12 (m, 2H).
(E)-3-(3-methyl-1,4-naphthoquinon-2-yl)-2-methoxyethylpropenoic acid (21a)
The methyl ester was prepared from lactone 20a (0.243 g, 0.815 mmol) following the procedure for 9 to provide 0.249 g (59%) as tan crystals following flash chromatography (1:3 EtOAc:hexanes) and recrystallization from Et2O/hexanes. Rf = 0.43 (1:3 EtOAc:hexanes) mp = 64 – 65 °C 1H NMR (CDCl3): δ 2.28 (s, 3H); 2.55 (t, 2H); 3.07 (s, 3H); 3.37 (t, 2H); 3.75 (s, 3H); 3.85 (s, 3H); 3.86 (s, 3H); 7.48 (m, 2H); 7.71 (s, 1H); 8.07 (m, 2H).
The resulting methyl ester (0.104 g, 0.302 mmol) was hydrolyzed following the procedure for 9 to provide (E)-3-(1,4-dimethoxy-3-methylnaphthalen-2-yl)-2-methoxyethylpropenoic acid (0.076 g, 77%) as a tan solid following flash chromatography (1:3 EtOAc:hexanes 0.5% AcOH) and recrystallization from Et2O/hexanes. Rf = 0.10 (1:3 EtOAc:hexanes 0.5% AcOH) mp = 145 – 148 °C 1H NMR (CDCl3): δ 2.30 (s, 3H); 2.57 (t, 2H); 3.17 (s, 3H); 3.44 (t, 2H); 3.77 (s, 3H); 3.88 (s, 3H); 7.50 (m, 2H); 7.86 (s, 1H); 8.08 (dt, 2H).
Compound 21a was prepared from (E)-3-(1,4-dimethoxy-3-methylnaphthalen-2-yl)-2-methoxyethylpropenoic acid (0.076 g, 0.23 mmol) following the procedure for 9 to provide 0.049 g (70%) of the product as a yellow solid following flash chromatography (2:3 Et2O:hexanes 0.5% AcOH) and recrystallization from Et2O/hexanes. Rf = 0.45 (2:3 Et2O:hexanes 0.5% AcOH) mp = 147 – 148 °C 1H NMR (CDCl3): δ 2.12 (d, 3H); 2.45 (t, 2H); 3.20 (s, 3H); 3.44 (t, 2H); 7.53 (d, 1H); 7.73 (m, 2H); 8.09 (m, 2H).
(E)-3-(3-chloro-1,4-naphthoquinon-2-yl)-2-methoxyethylpropenoic acid (1b)
The methyl ester was prepared from lactone 20b (0.098 g, 0.30 mmol) following the procedure for 9 to provide 0.064 g (58%) as tan crystals following flash chromatography (1:3 EtOAc:hexanes) and recrystallization from Et2O/hexanes. Rf = 0.44 (1:3 EtOAc:hexanes) mp = 61 – 62 °C 1H NMR (CDCl3): δ 2.59 (t, 2H); 3.12 (s, 3H); 3.39 (t, 2H); 3.78 (s, 3H); 3.39 (s, 3H); 3.98 (s, 3H); 7.55 (m, 2H); 7.68 (s, 1H); 8.10 (m, 2H).
The methyl ester (0.064 g, 0.17 mmol) was hydrolyzed following the procedure for 9 to provide (E)-3-(3-chloro-1,4-dimethoxynaphthalen-2-yl)-2-methoxethylpropenoic acid (0.067 g, quantitative) of the product as a tan solid following flash chromatography (1:3 EtOAc:hexanes 0.5% AcOH) and recrystallization from Et2O/hexanes. Rf = 0.12 (1:3 EtOAc:hexanes 0.5% AcOH) mp = 145 – 146 °C 1H NMR (CDCl3): δ 2.60 (t, 2H); 3.21 (s, 3H); 3.45 (t, 2H); 3.80 (s, 3H); 3.98 (s, 3H); 7.56 (m, 2H); 7.80 (s, 1H); 8.11 (m, 2H).
Compound 21b was prepared from (E)-3-(3-chloro-1,4-dimethoxynaphthalen-2-yl)-2-methoxethylpropenoic acid (0.067 g, 0.19 mmol) as described for 16e to provide 0.032 g (53%) of the product as a yellow solid following flash chromatography (2:3 acetone:hexanes 0.5% AcOH) and recrystallization from Et2O/hexanes. Rf = 0.48 (2:3 acetone:hexanes 0.5% AcOH) mp = 160 °C d 1H NMR (CDCl3): δ 2.52 (t, 2H); 3.17 (s, 3H); 3.45 (t, 2H); 7.47 (s, 1H); 7.78 (m, 2H); 8.12 (m, 1H); 8.18 (m, 1H).
(E)-3-(3-bromo-1,4-naphthoquinon-2-yl)-2-methoxyethylpropenoic acid (21c)
The methyl ester was prepared from lactone 20c (0.257 g, 0.708 mmol) following the procedure for 9 to provide 0.290 g (100%) of the product as tan crystals following flash chromatography (1:3 EtOAc:hexanes) and recrystallization from Et2O/hexanes. Rf = 0.42 (1:3 EtOAc:hexanes) mp = 66 – 67 °C 1H NMR (CDCl3): δ 2.57 (t, 2H); 3.12 (s, 3H); 3.39 (t, 2H); 3.77 (s, 3H); 3.86 (s, 3H); 3.97 (s, 3H); 7.42 (m, 2H); 7.65 (s, 1H); 8.10 (m, 2H).
The methyl ester (0.179 g, 0.437 mmol) was hydrolyzed following the procedure for 9 to provide (E)-3-(3-bromo-1,4-dimethoxynaphthalen-2-yl)-2-methoxyethylpropenoic acid (0.060 g, 35%) of the product as a tan solid following flash chromatography (1:3 EtOAc:hexanes 0.5% AcOH) and recrystallization from Et2O/hexanes. Rf = 0.12 (1:3 EtOAc:hexanes 0.5% AcOH) mp = 158 – 160 °C 1H NMR (CDCl3): δ 2.57 (t, 3H); 3.27 (s, 3H); 3.45 (t, 3H); 3.79 (s, 3H); 3.98 (s, 3H); 7.57 (m, 2H); 7.72 (s, 1H); 8.12 (m, 2H).
Compound 21c was prepared from (E)-3-(3-bromo-1,4-dimethoxynaphthalen-2-yl)-2-methoxyethylpropenoic acid (0.060 g, 0.15 mmol) as described for 16e to provide 0.029 g (53%) of the product as a yellow solid following flash chromatography (2:3 acetone:hexanes 0.5% AcOH) and recrystallization from Et2O/hexanes. Rf = 0.45 (2:3 acetone:hexanes 0.5% AcOH) mp = 168 – 172 °C 1H NMR (CDCl3): δ 2.52 (t, 2H); 3.18 (s, 3H); 3.46 (t, 2H); 7.41 (s, 1H); 7.78 (m, 2H); 8.12 (m, 1H); 8.19 (m, 1H).
(E)-N-(2-hydroxyethyl)-3-(3-chloro-1,4-dioxonaphthoquinon-2-yl)-2-methylpropenamide (23)
Compound 22 (0.117 g, 0.381 mmol) was converted to the hydroxyethylamide prepared as described for 11 to provide 0.110 g (83%) of the intermediate as a white solid following flash chromatography (2:3 EtOAc:hexanes). Rf = 0.38 (1:1 acetone:hexanes) mp = 165 – 167 °C 1H NMR (CDCl3): δ 1.86 (d, 3H); 2.68 (bs, 1H, OH); 3.60 (q, 2H); 3.75 (s, 3H); 3.84 (t, 2H); 3.98 (s, 3H); 6.42 (bs, 1H, NH); 7.43 (s, 1H); 7.55 (m, 2H); 8.10 (m, 2H)
The dimethoxynaphthalene intermediate (0.050 g, 0.14 mmol) was then oxidized following the procedure for 16e to provide 23 (0.027 g, 60%) as a yellow solid following flash chromatography (2:3 EtOAc:hexanes) and recrystallized from acetone/hexane. Rf = 0.25 (1:1 acetone:hexanes) mp = 120 °C d 1H NMR (CDCl3): δ 1.87 (s, 3H); 2.51 (bs, 1H, OH); 3.56 (q, 2H); 3.81 (m, 2H); 6.46 (bs, 1H, NH); 7.03 (s, 1H); 7.77 (m, 2H); 8.10 (m, 1H); 8.17 (m, 1H).
Ethyl 3-(3-bromo-1,4-dimethoxynaphthalen-2-yl)-propionate (25)
Following a modified procedure of Clegg et al.,26 lithium hexamethyldisilazide (LiHMDS, 1 M in THF, 1.5 ml, 1.5 mmol) was added to a flame-dried round bottom flask containing THF (20 mL) at −78 °C. Ethyl acetate (0.15 ml, 1.5 mmol) was then added slowly at −78 °C and stirred at −78 °C for 30 min. 2-bromo-3-bromomethyl-1,4-dimethoxynaphthalene (0.415 g, 1.15 mmol) was then dissolved in THF (10 mL) and added at −78 °C, at which point the reaction was allowed to warm to room temperature and then stirred for 4 h. The reaction was monitored by TLC (CH2Cl2) for disappearance of the less polar starting material. Additional LiHMDS (2.0 mL, 2.0 mmol) and EtOAc (0.70 mL, 0.70 mmol) were added and the reaction was monitored for the disappearance of starting material. When no starting material was present (ca. 10 h) the reaction was poured into water, extracted with ethyl acetate, washed with brine, dried over MgSO4, filtered, and condensed. The product was the purified by column chromatography (CH2Cl2 to 1:19 EtOAc:CH2Cl2) to provide 25 (0.361g, 86%) as a yellow oil. Rf = 0.43 (CH2Cl2) 1H NMR (CDCl3): δ 1.26 (t, 3H); 2.62 (m, 2H); 3.29 (m, 2H); 3.91 (s, 3H); 3.95 (s, 3H); 4.17 (q, 2H); 7.51 (m, 2H); 8.02 (m, 1H); 8.07 (m, 1H).
3-(3-Bromo-1,4-naphthoquinon-2-yl)-propionic acid (26)
Compound 25 (0.361g, 0.983 mmol) was oxidized following the procedure for 16e to provide ethyl (3-bromo-1,4-naphthoquinon-2-yl)-propionate (0.135 g, 41%) as a yellow solid following recrystallization from acetone/hexanes. Rf = 0.44 (1:4 acetone:hexanes) mp = 78 – 79 °C 1H NMR (CDCl3): δ 1.23 (t, 3H); 2.59 (t, 2H); 3.16 (t, 2H, J = 7.8 Hz); 4.12 (q, 2H); 7.74 (m, 2H); 8.13 (m, 2H).
The resulting ethyl ester (0.108, 0.320 mmol) was dissolved in THF (5 mL) at room temperature and then 2 M HCl (4 mL) was added at room temperature. The homogeneous solution was then stirred vigorously and heated under reflux for 24 h, or until no starting material was observed by TLC (1:4 EtOAc:hexanes). The reaction was then cooled and diluted with EtOAc (30 mL), then washed 2 times with saturated brine, dried over MgSO4, filtered, and condensed. If any starting ester was still present, the resulting yellow oil was first purified via flash column chromatography (1:4 acetone:hexanes 0 to 0.5 % AcOH) followed by recrystallization from Et2O/hexanes to provide 26 (0.077 g, 78%) as fine yellow needles. Rf = 0.14 (1:3 EtOAc:hexanes 0.5% AcOH) Mp = 156 – 157 °C 1H NMR (CDCl3): δ 2.65 (t, 2H); 3.15 (m, 2H); 7.74 (m, 2H); 8.13 (m, 2H).
Acknowledgments
Financial support for this work was provided by the National Cancer Institute (T32 CA09634 to RLN and CA94025, CA106298 and CA121168 to MRK), the Purdue University – Indiana University Collaborative Biomedical Research Program (to RFB and MRK), and the Riley Children’s Foundation (to MRK).
Footnotes
Abbreviations: AP, apurinic/apyrimidinic; Ape1, apurinic/apyrimidinic endonuclease 1; BER, base excision repair; EMSA, electrophoretic mobility shift assay; Ref-1, redox-enhancing factor 1.
References
- 1.Evans AR, Limp-Foster M, Kelley MR. Going APE over ref-1. Mutat Res. 2000;461:83–108. doi: 10.1016/s0921-8777(00)00046-x. [DOI] [PubMed] [Google Scholar]
- 2.Xanthoudakis S, Miao GG, Curran T. The redox and DNA-repair activities of Ref-1 are encoded by nonoverlapping domains. Proc Natl Acad Sci U S A. 1994;91:23–27. doi: 10.1073/pnas.91.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tell G, Damante G, Caldwell D, Kelley MR. The intracellular localiztion of APE1/Ref-1: more than a passive phenomenon? Antioxid Redox Signal. 2005;7:367–384. doi: 10.1089/ars.2005.7.367. [DOI] [PubMed] [Google Scholar]
- 4.Tell G, Quadrifoglio F, Tiribelli C, Kelley MR. The many functions of APE1/Ref-1; not only a DNA-repair enzyme. Antioxid Redox Signal. 2009;11:601–620. doi: 10.1089/ars.2008.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Izumi T, Brown DB, Naidu CV, Bhakat KK, Macinnes MA, Saito H, Chen DJ, Mitra S. Two essential but distinct functions of the mammalian abasic endonuclease. Proc Natl Acad Sci U S A. 2005;102:5739–5743. doi: 10.1073/pnas.0500986102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luo M, Delaplane S, Jiang A, Reed A, He Y, Fishel M, Nyland RL, Borch RF, Qiao X, Georgiadis MM, Kelley MR. Role of the multifunctional DNA repair and redox signaling protein Ape1/Ref-1 in cancer and endothelial cells: small-molecule inhibition of the redox function of Ape1. Antioxid Redox Signal. 2008;10:1853–1867. doi: 10.1089/ars.2008.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zou GM, Karikari C, Kabe Y, Handa H, Anders RA, Maitra A. The Ape-1/Ref-1 redox antagonist E3330 inhibits the growth of tumor endothelium and endothelial progenitor cells: therapeutic implications in tumor angiogenesis. J Cell Physiol. 2009;219:209–218. doi: 10.1002/jcp.21666. [DOI] [PubMed] [Google Scholar]
- 8.Zou GM, Maitra A. Small-molecule inhibitor of the AP endonuclease 1/REF-1 E3330 inhibits pancreatic cancer cell growth and migration. Mol Cancer Ther. 2008;7:2012–2021. doi: 10.1158/1535-7163.MCT-08-0113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang Y, Guo C, Fishel ML, Wang ZY, Vasko MR, Kelley MR. Role of APE1 in differentiated neuroblastoma SH-SY5Y cells in response to oxidative stress: Use of APE1 small molecule inhibitors to delineate APE1 function. DNA Repair. 2009 doi: 10.1016/j.dnarep.2009.08.003. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Georgiadis MM, Luo M, Gaur RK, Delaplane S, Li X, Kelley MR. Evolution of the redox function in mammalian apurinic/apyrimidinic endonuclease. Mutat Res. 2008;643:54–63. doi: 10.1016/j.mrfmmm.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shinkawa N, Ichino T, Tsujii M, Tsuruki T, Kuroda H. Production of p-dimethoxybenzene derivatives. 05-148175 Japanese Patent.
- 12.Keinan E, Doron E. Total synthesis of linear polyprenoids. 2. Improved preparation of the aromatic nucleus of ubiquinone. J Org Chem. 1987;52:3872–3875. [Google Scholar]
- 13.Tremblay MS, Sames D. A new fluorogenic transformation: development of an optical probe for coenzyme Q. Org Lett. 2005;7:2417–2420. doi: 10.1021/ol0507569. [DOI] [PubMed] [Google Scholar]
- 14.Hansen CA, Dean AB, Draths KM, Frost JW. Synthesis of 1,2,3,4-Tetrahydroxybenzene from D-Glucose: Exploiting myo-Inositol as a Precursor to Aromatic Chemicals. J Amer Chem Soc. 1999;121:3799–3800. [Google Scholar]
- 15.Ohkawa S, Terao S, Terashita Z, Shibouta Y, Nishikawa K. Dual inhibitors of thromboxane A2 synthase and 5-lipoxygenase with scavenging activity of active oxygen species (AOS). Synthesis of a novel series of (3-pyridylmethyl)benzoquinone derivatives. J Med Chem. 1991;34:267–276. doi: 10.1021/jm00105a042. [DOI] [PubMed] [Google Scholar]
- 16.Syper L, Kloc K, Mlochowski J. Synthesis of ubiquinone and menaquinone analogues by oxidative demethylation of alkenylhydroquinone ethers with argentic oxide or ceric ammonium nitrate in the presence of 2,4,6-pyridine-tricarboxylic acid. Tetrahedron. 1980;36:123–129. [Google Scholar]
- 17.King SA. Orthoester-Dependent Alcoholysis of Lactones. Facile Preparation of 4-Alkoxybutanoates and 5-Alkoxypentanoates. J Org Chem. 1994;59:2253–2256. [Google Scholar]
- 18.Gius D, Cao XM, Rauscher FJr, Cohen DR, Curran T, Sukhatme VP. Transcriptional activation and repression by Fos are independent functions: the C terminus represses immediate-early gene expression via CArG elements. Mol Cell Biol. 1990;10:4243–4255. doi: 10.1128/mcb.10.8.4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fishel ML, He Y, Smith ML, Kelley MR. Manipulation of base excision repair to sensitize ovarian cancer cells to alkylating agent temozolomide. Clin Cancer Res. 2007;13:260–267. doi: 10.1158/1078-0432.CCR-06-1920. [DOI] [PubMed] [Google Scholar]
- 20.Murphy JA, Rasheed F, Roome SJ, Scott KA, Lewis N. Intramolecular termination of radical-polar crossover reactions. Journal Chem Soc, Perkin. 1998;1:2331–2339. [Google Scholar]
- 21.Flader C, Liu J, Borch RF. Development of novel quinone phosphorodiamidate prodrugs targeted to DT-diaphorase. J Med Chem. 2000;43:3157–3167. doi: 10.1021/jm000179o. [DOI] [PubMed] [Google Scholar]
- 22.Lopez-Alvarado P, Avendano C, Menendez JC. Efficient, multigram-scale synthesis of three 2,5-dihalobenzoquinones. Syn Comm. 2002;32:3233–3239. [Google Scholar]
- 23.Evans PA, Brandt TA. Hypervalent Iodine Chemistry: Mechanistic Investigation of the Novel Haloacetoxylation, Halogenation, and Acetoxylation Reactions of 1,4-Dimethoxynaphthalenes. J Org Chem. 1997;62:5321–5326. [Google Scholar]
- 24.Ito T, Ikemoto T, Yamano T, Mizuno Y, Tomimatsu K. Practical synthesis of (R)-(+)-6-(1,4-dimethoxy-3-methyl-2-naphthyl)-6-(4-hydroxyphenyl)hexanoic acid: a key intermediate for a therapeutic drug for neurodegenerative diseases. Tetrahedron: Asymmetry. 2003;14:3525–3531. [Google Scholar]
- 25.Syper L, Mlochowski J, Kloc K. Synthesis of oxiranylquinones as new potential bioreductive alkylating agents. Tetrahedron. 1983;39:781–792. [Google Scholar]
- 26.Clegg NJ, Paruthiyil S, Leitman DC, Scanlan TS. Differential Response of Estrogen Receptor Subtypes to 1,3-Diarylindene and 2,3-Diarylindene Ligands. J Med Chem. 2005;48:5989–6003. doi: 10.1021/jm050226i. [DOI] [PubMed] [Google Scholar]