

Abstract
Purpose
Status epilepticus is a neurological emergency associated with neuronal injury, lasting behavioral disturbance, and a high rate of mortality. Intravenous levetiracetam (LEV), an antiepileptic drug approved to treat partial seizures, has recently been introduced. We sought to determine the effect of LEV administered intravenously in a chemoconvulsant model of status epilepticus.
Methods
We examined the effect of intravenous LEV in the rat lithium-pilocarpine model of status epilepticus. Ten or 30 minutes after the onset of behavioral status epilepticus, animals were treated with LEV (200–1200 mg/kg i.v.) administered in a single bolus. Behavioral responses were recorded. Selected animals had continuous EEG recording before, during and after the administration of LEV. Some animals were sacrificed 24 h after the experiment and processed for histochemical assessment of neuronal injury.
Results
When administered 30 minutes after the onset of behavioral epileptic seizures, transient attenuation of ictal behavior was observed in animals treated with 800 mg/kg or more of LEV. The duration of behavioral attenuation increased sharply as the dose rose to 1000 mg/kg or higher, from a mean of 4 minutes to 23.6 minutes. When administered 10 minutes after seizure onset, 400 mg/kg of LEV resulted in transient ictal behavioral attenuation, and higher doses caused relatively longer periods of attenuation. Pretreatment with LEV prior to pilocarpine also delayed the onset of seizures. EEG recordings, however, showed no significant attenuation of ictal discharge. By contrast, TUNEL staining demonstrated less neuronal injury in hippocampii and other limbic structures in animals that responded behaviorally to LEV.
Conclusions
Intravenous administration of LEV in a chemoconvulsant model of status epilepticus results in attenuation of behavioral manifestations of seizure discharge and in reduction of neuronal injury but does not significantly alter ictal discharge recorded by EEG.
Keywords: Parenteral, Treatment, Seizure, Chemoconvulsant, Neuronal Injury
Introduction
Epilepsy is a chronic neurological disorder characterized by recurrent spontaneous seizures. Status epilepticus (SE), defined as continuous seizure activity lasting over 30 minutes, or repeated seizures between which patients do not recover to their baseline level of consciousness, is a medical emergency with high morbidity and mortality if not treated promptly [1, 2]. Recurrent seizures or status epilepticus may cause injury to hippocampus and other limbic structures in humans and in animal seizure models.
Status epilepticus is typically treated in a protocol-driven fashion beginning with intravenous anticonvulsants. Up to 30% of SE patients do not respond to first or second line antiepileptic drug (AED) treatment and require administration of anesthetic agents, but in this group morbidity and mortality increase significantly [3, 4]. Because no new intravenous treatments have been developed and licensed for the treatment of SE, in spite of the fact that existing treatments frequently fail, it is important to discover new drugs with utility in the initial (pre-anesthetic) treatment of status epilepticus. Whereas traditional AEDs target Na+ channels, T-type Ca++ channels, GABA receptors or glutamate receptors [5–7], it is likely that efficacious alternative agents will aim at new molecular targets to achieve specific therapeutic effects.
Levetiracetam (LEV), a novel anti-epileptic drug that has a unique profile in animal models of partial and generalized seizures, was initially identified as an anticonvulsant in a mouse audiogenic seizure model [8]. It does not have anti-convulsant activity in either the maximal electroshock seizure (MES) or the subcutaneous pentylenetetrazole (PTZ) seizure rodent [9, 10], but is a potent anticonvulsant in genetic rodent models [9, 11, 12], the amygdala kindling model [13–16], and spontaneous recurrent seizure models [14]. LEV has been approved for clinical use in the U.S. since 1999 to treat partial epilepsy [17–19], and more recently to treat tonic clonic and myoclonic seizures. LEV inhibits neuronal hypersynchronization in cultured hippocampal slices [20], and inhibits N-type Ca++ channels [21]. It prevents the upregulation of BDNF and neuropeptide Y mRNAs, and downregulation of NPY1- and NPY5-like receptors in the kindling rat model [22]. LEV binds to the synaptic vesicle protein SV2A [23], where it has been hypothesized to interfere with neurotransmitter release. In animal studies it shows few adverse effects [9, 24], suggesting a high safety margin. The unique characteristics of LEV, including its antiepileptic profile in animal models as well as its molecular target suggest that LEV represents a new class of AED.
The effect of LEV on prevention and treatment of status epilepticus has not been widely studied. In a previous report [9], LEV administered intraperitoneally appeared to attenuate status epilepticus induced by systemic pilocarpine or kainate, but intravenous administration of LEV has not been studied.
Pilocarpine is a muscarinic cholinergic agonist. The pathophysiology and neuropathology of pilocarpine-induced SE has been thoroughly studied [25]. Intraperitoneal administration of high doses of pilocarpine (300–400 mg/kg), or lower doses of pilocarpine (30–40 mg/kg) after pretreatment with lithium, results in the stereotyped appearance of behavioral and electrographic seizures lasting many hours [26]. Surviving animals typically go on to develop spontaneous recurrent seizures weeks to months after SE. Neuronal injury in rat brain after seizures is highly reminiscent of the injury seen in humans after prolonged SE, affecting mainly hippocampus and limbic structures [27]. The effect of LEV on the onset of pilocarpine-induced SE in mice [28], and on pilocarpine-induced spontaneous recurrent seizures in rats has been studied [14]. Here we sought to determine whether acute treatment with intravenous LEV could stop or attenuate the behavioral and electrographic manifestations of pilocarpine-induced SE and the subsequent development of neuronal injury in rats.
Materials and Methods
Animals
Male Sprague Dawley rats (125–175 g) (Charles River Laboratories; Wilmington, MA.) were used for all experiments. All experiments were conducted under an approved IACUC protocol in accordance with the regulations of the Center for Comparative Medicine and Laboratory Animal Services of Massachusetts General Hospital.
Induction of seizures by lithium-pilocarpine and administration of LEV
18–24 hours prior to pilocarpine, animals were treated with lithium hydrochloride (127 mg/kg (3 meq/kg) i.p.). Thirty minutes prior to pilocarpine administration, animals were treated with scopolamine methyl bromide (1 mg/kg i.p.) to reduce peripheral cholinergic agonist-induced side effects. Animals were then treated with pilocarpine hydrochloride (30 mg/kg i.p.). Using this treatment paradigm, behavioral seizures typically begin within 20–40 minutes. The tail vein was cannulated and LEV was administered as a single bolus through the intravenous catheter at designated timepoints after onset of seizure activity.
Seizure behavior quantification
After pilocarpine injection, animals were closely observed and behavioral changes were recorded. A seizure severity grade was assigned according to a modified Racine scale previously used in our laboratory [29, 30] with regard to the animals' maximal behavioral response (Grade 0, no response; Grade 1, wet dog shake (WDS) and/or behavioral arrest; Grade 2, WDS, staring, pawing, and clonic jerks; Grade 3, WDS, staring, pawing, clonic jerks, rearing and falling; Grade 4, continuous grade 3 seizures for more than 30 minutes (status epilepticus)). Injection of LEV or normal saline (PBS) was undertaken at specific time-points after rats initially achieved Grade 3 seizure severity (rearing and falling). Approximately 90% of all experimental animals achieved Grade 4 seizure severity, and only these animals were utilized for subsequent analyses. Ongoing behavior was observed and recorded. After 2 hours of continuous or intermittent Grade 4 seizure activity, all animals were treated with diazepam (15 mg/kg i.p.) to minimize stress and discomfort and increase survival. Animals were given supplemental dextrose (3% solution) and allowed to survive 24 h prior to sacrifice.
Rat EEG Recording
For EEG recordings adult male Sprague-Dawley rats weighing 200–250 g at the time of surgery were utilized. Rats were anaesthetized with ketamine (70 mg/kg i.p.) (McBedford Laboratories; Bedford, OH) and pentobarbital (25 mg/kg i.p.)(Abbott Laboratories; N. Chicago, IL). Supplemental doses of 15–20mg/kg ketamine were given as needed. Rats were placed in a stereotaxic frame (David Kopf Instruments; Tujunga, CA) and surface recording electrodes (Plastics One; Roanoke, VA) (1/8” stainless steel jewelers screws) were placed in right frontal, left frontal and left parietal locations. A bipolar twist electrode (Plastics One) was placed in the right hippocampus (AP −3.5 mm from bregma, +ML 2.0 mm from bregma, DV −3.7 mm from dura, incisor bar −3.1 mm) using coordinates determined from the atlas of Paxinos and Watson [31]. Electrode pins were inserted into a plastic pedestal (Plastics One), which was then stabilized on the skull with dental acrylic (Benco Dental; Wilkes Barre, PA).
Postoperatively, the animals were housed singly in clear plastic cages and were allowed to recover for 6–8 days during which time they were handled daily before recording was performed. Seizures were induced by pretreatment with lithium and then administration of scopolamine and pilocarpine as described above.
Electroencephalographic activity was monitored in freely moving rats using a dedicated digital EEG machine (Cadwell Laboratories, Inc.; Kennewick, WA). Two control animals treated with intravenous saline were utilized to assess the effects of handling and injection on behavioral and EEG activity during SE. At the end of recording, animals were euthanized with 200 mg/kg i.p. pentobarbital sodium (Abbott). Brains were immediately fixed in 4% paraformaldehyde. Depth electrode placement in the hippocampus was confirmed two days later by gross inspection of coronal brain sections.
For each animal, 2 hours of continuous EEG was visually inspected by an experienced electroencephalographer (AJC) to determine the presence of electrographic seizure activity. Epileptiform activity was identified based on commonly used principles of EEG analysis, including the occurrence of epileptiform spikes. Spikes were recognized on the basis of morphology, electrical field and amplitude. Seizures were identified by a change in the baseline activity with an evolving pattern of higher amplitude rhythmic activity.
Power analysis was used to improve the sensitivity of detecting subtle EEG changes occurring after LEV administration. 300 second blocks of EEG were selected beginning 400 seconds before the first electrographic manifestation of ictal activity, 100 seconds after the first ictal electrographic change, 100 seconds after LEV administration (700 seconds after onset of Stage III ictal behavior), and 1000 seconds after LEV administration. Each block was analyzed to determine power in 8 Hz frequency bands from 0–40 Hz. Power measurements were normalized to the baseline block. Power was then compared between the samples obtained immediately prior to treatment, immediately after treatment, and 15 minutes after treatment.
TUNEL staining of neuronal injury loss
For examination of neuronal injury the in situ nick translation method was used to detect DNA fragmentation. Following sacrifice by transcardiac perfusion with PBS and 4% paraformaldehyde, brains were harvested and post-fixed overnight. 50 μm coronal sections through the hippocampus were cut using a vibratome and collected into wells, each of which contained every sixth section. Sections were stained using the in situ nick-translation method (TUNEL) modified from Wijsman [32]. Briefly, floating sections were incubated in 2x SSC buffer (300 mM NaCl, 30 mM Na citrate) for 20 min at 80 °C to denature the DNA, and then treated with pronase (1μg/ml) for 10 minutes. The reaction is stopped with 2% glycine and incubated in nucleotide complex (10 unit/ml DNA polymerase I, 10 μM each of dCTP, dATP, dGTP, biotin-21dUTP (Clonetech) dissolved in buffer A (50 mM Tris-HCl, pH 7.5, 5 mM MgCl2, 0.005% BSA) for 1 hour at room temperature. The sections were washed, treated with 0.1 % H2O2 for 30 minutes, and then developed with avidin-biotin-peroxidase complex Vectastain ABC detection kit (Vector Lab) for one hour. The colors were developed in buffer containing 2.5% nickel sulfate, 0.04% diaminobenzidine, and 0.0024% H2O2. Sections were then mounted onto slides, air-dried, and counter-stained with cresyl violet, cover slipped and sealed with Permount (Fisher Scientific).
TUNEL data was analyzed by microscopic examination of stained brain sections by two independent observers, both blinded to treatment. Inter-rater agreement was high. In cases where rater's scores differed, an average of the two scores was used. There was never a difference of more than one grade between raters examining specific sections. Different brain regions were examined for the presence or absence of peroxidase reaction product. Only dense nuclear staining was interpreted as positive. The abundance of positively stained neurons in specific structures was graded on a four-plus scale: “0”: no neurons positive, “+”: 0–25% positive, “++”: 25–50% positive, “+++”: 50–75% positive, “++++”: 75–100% positive. Grading of injury in specific structures was performed on sections from a total of 8 saline- and 18 LEV-treated animals and then averaged. Quantification of TUNEL staining was compared using Student's t test. Significance was defined as P < 0.05.
Results
LEV attenuates seizure activity in established status epilepticus
In initial experiments we examined the effect of intravenous LEV administered 30 minutes after animals achieved Grade 3 seizure activity. We chose this behavioral stage as it represents the onset of rearing and falling behavior and is straightforward to ascertain. LEV doses from 100–1200 mg/kg were examined. Following administration of LEV, we observed a dose-dependent attenuation of seizure activity marked by disappearance of rearing and falling behavior. A behavioral effect of LEV was initially observed at a dose of 800 mg/kg where attenuation of Grade 3 seizure activity lasted an average of 6.8 (± 4.1) minutes. At higher doses, duration of attenuation increased to a plateau of approximately 27 minutes at 1000–1200 mg/kg (Figure 1). At these doses, animals appeared sedated within 1 minute of injection and then appeared to recover over 2–5 minutes to walk around their cages with slight head bobbing and exploratory behavior. Gradually rats then developed stronger and stronger body and head jerking until the reappearance of rearing and falling behavior. At doses of 800 and 900 mg/kg, all animals survived. Higher doses were associated with an increasing mortality rate. With doses of 1000 mg/kg or greater, the survival rate dropped to 50–60% (6 out of 10 at 1000 mg/kg, 6 out of 11 at 1100 mg/kg, and 4 out of 8 at 1200 mg/kg).
Figure 1. Dose response relationship between intravenous levetriracetam and duration of attenuation of behavioral seizure activity.
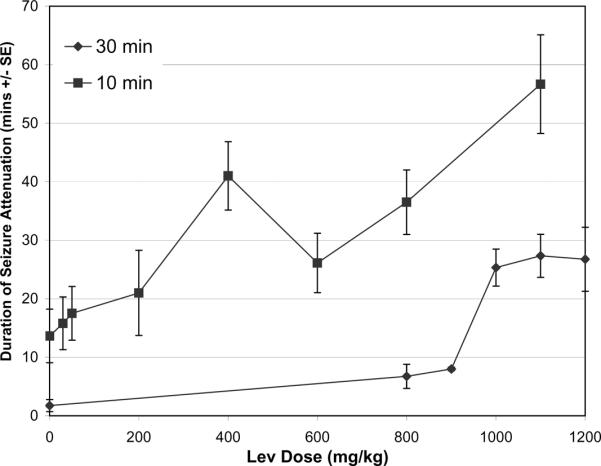
LEV was administered 10 (squares) or 30 (diamonds) minutes after the initial Grade 3 seizure. For 10 minute groups: saline N=11; 30mg/kg N=5, 50mg/kg N=6, 200mg/kg N=5, 400 mg/kg N=5, 600 mg/kg N=9, 800mg/kg N=6, 1100 mg/kg N=6. For LEV 30 minutes after SE: saline N=4, 800 mg/kg N=4, 900 mg/kg N=7, 1000 mg/kg N=6, 1100 mg/kg N= 6, 1200 mg/kg N=4. Error bars represent standard error of the mean of each sample group.
Clinical studies suggest that earlier treatment of status epilepticus may be more effective. We therefore examined the effect of LEV when administered 10 minutes after the onset of pilocarpine-induced seizures. Control animals were treated with vehicle alone (n=11) (Figure 1). After doses as low as 200 mg/kg LEV injected 10 minutes after the onset of seizures, ictal behavior was briefly attenuated (21 minutes compared to 13.6 minutes in controls, At higher doses, longer periods of attenuation were observed, with an average duration of attenuation of 56.6 (±20) minutes after LEV 1100 mg/kg. The behavioral changes were similar to those observed when animals were given LEV 30 minutes after seizure onset, with longer period of slight head bobbing and walking-seeking behavior. Once severe seizure activity recurred, animals again showed strong jerking accompanied by rearing and falling.
We compared seizure latency between animals pretreated with LEV (1100mg/kg) immediately before administration of pilocarpine (n=5) to animals treated with pilocarpine alone (n=13). In untreated animals, the average time to seizure onset for rats after pilocarpine injection was 26 (± 3.96) minutes. Following LEV pretreatment, one animal had no seizures. The average time to seizure onset in the remaining 4 animals was 97 (± 38) minutes (p=0.00003, t-test) confirming a result previously demonstrated by others [9, 28].
Next we examined the effect of intravenous LEV on electrographic ictal activity. We examined both scalp and depth recordings from the hippocampus. One week after the surgical placement of EEG recording electrodes, rats were pretreated with LiCl and then seizures were induced with pilocarpine. We examined several doses of LEV, ranging from 50–1000 mg/kg, injected 10 minutes after the onset of seizures in twelve animals. In saline-treated animals both surface and depth electrodes displayed continuous spikes and spike and wave discharge that correlated with Grade 3 behavioral seizure activity. No dose of LEV resulted in a consistent change in the appearance of either the cortical or hippocampal EEG, although in some animals treated with 800 mg/kg LEV, the EEG became slightly discontinuous with slightly less frequent epileptic discharges of lower amplitude. Power analysis was used to improve the sensitivity of detecting subtle EEG changes occurring after LEV administration. There was no significant change in power in any frequency domain examined. While there was a slight decrease in the overall amplitude of the EEG after LEV administration, this decrease was not statistically significant (Figure 2). In the terminal phase of the experiments, complete cessation of ictal electrographic discharge did not occur after diazepam administration (15 mg/kg) but was observed after pentobarbital administration.
Figure 2. EEG recording from an animal prior (upper tracing) and 11 minutes after (lower tracing) treatment with LEV 800 mg/kg administered 10 minutes after the onset of status epilepticus.

Recording is bi-polar between left and right central skull screws. Vertical bar = 75 μV, horizontal bar = 1 sec).
We used histopathological assessment of DNA fragmentation to examine the effect of LEV treatment on neuronal injury in limbic structures after pilocarpine-induced status epilepticus. For these experiments animals were treated with intravenous diazepam 2 hours after the onset of seizures to reduce convulsive behavior and promote survival. Animals were sacrificed 24 hours later. In control animals (N=8), after seizures neuronal injury was found in the CA1 region of the hippocampal pyramidal cell layer, amygdale, specific thalamic nuclei, and in cingulate and perirhinal cortex (Figure 3). In rats treated with LEV (800–1200 mg/kg, N = 18) either 10 minutes (N = 9) or 30 minutes (N = 9) after pilocarpine-induced seizures, we found a statistically significant reduction in neuronal injury in CA1, thalamus, amygdala and cortex as compared to controls (Figure 4). In our sample, the timing of LEV administration (10 or 30 mins after behavioral onset) did not affect the magnitude of reduction in neuronal injury.
Figure 3. Neuronal injury manifested by DNA fragmentation after Pilocarpine-Induced Status Epilepticus is reduced after treatment with intravenous levetiracetam.

Rats were sacrificed 24 h after induction of SE. Only dense black nuclear staining was interpreted TUNEL staining. The number of positively TUNEL stained neurons in specific limbic structures were counted and compared to the total number of neurons (counter-stained with cresyl violet). Treatment with LEV (A, C) reduced number of neurons displaying DNA fragmentation compared to animals treated with PBS alone (B, D) in CA1 (A, B), entorrhinal cortex (C, D). Note dense black nuclei in positively stained cells. Bar = 500 microns.
Figure 4. Magnitude of histological injury after levetiracetam treatment of pilocarpine-induced status epilepticus.

Control (n=8; solid bars) and levetiracetam (800–1200 mg/kg treatments pooled) pre-treated (n=18; striped bars). * Indicates p=<0.05. Injury score: “0” = no neurons positive; “1”= 0–25% positive; “2”= 25–50% positive; “3”= 50–75% positive; “4”= 75–100% positive. Error bars represent standard error of the mean of each sample group.
Discussion
The main finding of our study is that intravenous levetiracetam treatment transiently attenuates the behavioral response associated with pilocarpine-induced status epilepticus in a dose-dependent fashion, but does not significantly change the electrographic appearance of ictal activity. Earlier treatment has a more prolonged behavioral effect than delayed treatment. Moreover, pretreatment with intravenous LEV significantly delayed the onset of convulsive activity after administration of systemic pilocarpine. In addition, treatment with high doses of LEV attenuated pilocarpine-status-induced neuronal injury in hippocampus as assayed by analysis of DNA fragmentation.
We chose to use a well-characterized chemoconvulsant model of status epilepticus to examine the effect of acute intravenous administration of LEV on ongoing seizure activity, and used clinical observation of ictal behavior as our primary endpoint to assess efficacy. Because pilocarpine induces a highly stereotyped behavioral response that progresses through a series of well defined clinical stages of severity, we were able to detect partial responses to treatment, which we captured as “time in grade”. Based on the hypothesis that intravenous LEV would reduce seizure intensity, we chose to define a reduction in severity grade from Grade 3 to Grade 2 as a response. Moreover, we were able to quantify the duration of reduction to allow statistical analysis of the effect and dose comparison. Using this approach we established a clear dose-response relationship between LEV dose and duration of grade reduction. Our results complement Mazarati's finding in the self-sustaining status epilepticus model that intravenous injection of LEV 10 minutes after perforant path stimulation shortened seizure duration at doses of 200 mg/kg or greater, and in combination with diazepam, LEV suppressed seizures immediately [13].
Importantly, we found that the dose response curve is shifted to the left when LEV is administered 10 minutes, as compared to 30 minutes, after clinical seizure onset. A similar finding in a study of diazepam treatment of pilocarpine-induced seizures [33] supports the clinical observation that earlier treatment is more likely to suppress status epilepticus in patients [34].
We also examined the effect of pretreatment with intravenous LEV on the latency to onset of pilocarpine-induced seizures and confirmed that pretreatment significantly delayed the appearance of initial behavioral changes [9, 28]. We therefore speculate that LEV raises the threshold for occurrence of status epilepticus and may have a role in preventing status in individuals who are predisposed to this serious clinical condition.
Although the behavioral effect of intravenous LEV treatment after the onset of seizures was incomplete, i.e. seizures were not fully terminated, we found clear evidence of a reduction in seizure-induced neuronal injury as assayed by staining for DNA fragmentation. Gibbs found that LEV administration early after seizure onset protected against mitochondrial dysfunction in the self-sustaining status epilepticus model, suggesting one possible mechanism of LEV-mediated neuroprotection in status epilepticus [35, 36].
The mechanism of behavioral attenuation in the absence of change in EEG remains unclear. Whereas many antiepileptic drugs target Na+ channels, T-type Ca++ channels, the GABAergic systems, or glutamate receptors. LEV has an atypical anticonvulsant profile in animal models where it is active against audiogenic seizures and seizures induced by kindling stimulation, but not against maximal electroconvulsive shock or pentylenetetrazole-induced seizures. The mechanism of action of LEV remains incompletely defined. LEV binds to the synaptic vesicle protein SV2A with high affinity [23]. SV2A is a glycoprotein that exists in all synaptic vesicles membranes and plays an important role in synaptic vesicle cycling and neurotransmitters release into the synaptic cleft. An SV2A mouse knockout model manifests abnormal neurotransmission that results in early development failure, severe seizures and death [37]. It is possible that transient LEV-mediated modulation of SV2A function could underlie the transient behavioral response we observed even in the absence of a detectable effect on electrographic discharge.
Relatively high doses of LEV were required to attenuate ictal behavioral activity in our study in comparison to the doses used to block spontaneous seizures or kindling seizures, and relative to doses used clinically. Mazarati [13] and Gibbs [35] also found a relatively high dose requirement in their studies of LEV activity against self-sustaining status epilepticus. We did not measure LEV serum levels in our study. It is possible that rodents metabolize LEV rapidly, especially in the setting of status, leading to reduced bioavailability. In rats the elimination half-life of LEV in serum is between 1.8–2.8 h [38] whereas in humans the serum half-life is 6–8 h [39], demonstrating a substantial species difference in pharmacokinetics. Additional pharmacokinetic studies will be required to address this issue. Interestingly, while LEV is rapidly absorbed and transported across the blood-brain barrier, there is a significant delay between Tmax (serum) and Tmax (CSF) (0.25–0.50 h vs. 1.33–1.92 h) [38]. Because the efficacy of treatment of SE is highly dependent on the duration of SE prior to treatment, it is possible that a high LEV dose is required to achieve adequate CSF concentrations in a timely manner. It is also possible that the systemic chemoconvulsant model we used dictates the need for unusually high doses of LEV. In particular, the chemoconvulsant remains in circulation throughout the treatment period, presenting an ongoing challenge to homeostatic mechanisms. Moreover, because LEV does not appear to act as a specific neurotransmitter antagonist, its unique mechanism of action may impose a higher dose requirement to achieve efficacy. This notion is supported indirectly by the relatively high dose requirement in clinical use, usually expressed in grams per day rather than milligrams per day as with many anticonvulsant drugs.
We have shown that intravenous LEV, administered at high doses, reduces the intensity of the behavioral response to pilocarpine in the lithium-pilocarpine model of status epilepticus. We have also demonstrated that intravenous LEV, again at high dose, reduces the severity of neuronal injury in hippocampus after Li-Pilo SE. It is therefore surprising that we were unable to demonstrate significant change in the ictal EEG pattern recorded from animals with Li-Pilo SE treated with LEV. This paradox focuses attention on the mechanism of the behavioral and protective effects of LEV. While a specific biochemical effect of LEV might mediate the behavioral and neuroprotective responses we observed, an alternative hypothesis is that LEV acts downstream of the cerebral cortex to modify the expression of epileptic activity. If true, this hypothesis would raise the concern that treatment of status with intravenous LEV may convert convulsive SE into non-convulsive SE by acting to dissociate cortical structures from sub-cortical output pathways. Additional studies will be required to address these issues.
Acknowledgements
This study was supported by an investigator initiated grant from UCB Pharma. Dr. Zheng received support from the American Epilepsy Society.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest Dr. Cole has received consulting fees and honoraria from UCB Pharma, Abbott Laboratories, GlaxoSmithKline, and Pfizer. No other authors have conflicts to disclose.
References
- 1.DeLorenzo RJ, et al. A prospective, population-based epidemiologic study of status epilepticus in Richmond, Virginia. Neurology. 1996;46(4):1029–35. doi: 10.1212/wnl.46.4.1029. [DOI] [PubMed] [Google Scholar]
- 2.DeLorenzo RJ, et al. Epidemiology of status epilepticus [see comments] J Clin Neurophysiol. 1995;12(4):316–25. [PubMed] [Google Scholar]
- 3.Mayer SA, et al. Refractory Status Epilepticus: Frequency, Risk Factors, and Impact on Outcome. Arch Neurol. 2002;59(2):205–210. doi: 10.1001/archneur.59.2.205. [DOI] [PubMed] [Google Scholar]
- 4.Walker MC. The epidemiology and management of status epilepticus. Current Opinions in Neurology. 1998;11(2):149–54. doi: 10.1097/00019052-199804000-00012. [DOI] [PubMed] [Google Scholar]
- 5.Urbanska EM, et al. NMDA- But Not Kainate-Mediated Events Reduce Efficacy of Some Antiepileptic Drugs Against Generalized Tonic-Clonic Seizures in Mice. Epilepsia. 1999;40(11):1507–1511. doi: 10.1111/j.1528-1157.1999.tb02033.x. [DOI] [PubMed] [Google Scholar]
- 6.Fraser CM, S.G., Butler E, Thompson GG, Lindsay K, Duncan R, Howatson A, Brodie MJ. Effects of valproate, vigabatrin and tiagabine on GABA uptake into human astrocytes cultured from fetal and adult brain tissue. Epileptic Disorders. 1999;1(3):153–7. [PubMed] [Google Scholar]
- 7.Zona C, N.I., Marchetti C, Klitgaard H, Bernardi G, Margineanu DG. Levetiracetam does not modulate neuronal voltage-gated Na+ and T-type Ca2+ currents. Seizure. 2001;10(4):279–286. doi: 10.1053/seiz.2000.0504. [DOI] [PubMed] [Google Scholar]
- 8.Gower AJ, et al. ucb L059, a novel anti-convulsant drug: pharmacological profile in animais. European Journal of Pharmacology. 1992;222(2–3):193–203. doi: 10.1016/0014-2999(92)90855-x. [DOI] [PubMed] [Google Scholar]
- 9.Klitgaard H, et al. Evidence for a unique profile of levetiracetam in rodent models of seizures and epilepsy. European Journal of Pharmacology. 1998;353(2–3):191–206. doi: 10.1016/s0014-2999(98)00410-5. [DOI] [PubMed] [Google Scholar]
- 10.Loscher W, Honack D. Profile of ucb L059, a novel anticonvulsant drug, in models of partial and generalized epilepsy in mice and rats. Eur J Pharmacol. 1993;232(2–3):147–58. doi: 10.1016/0014-2999(93)90768-d. [DOI] [PubMed] [Google Scholar]
- 11.Gower AT, et al. Effects of levetiracetam, a novel antiepileptic drug, on convulsant activity in two genetic rat models of epilepsy. Epilepsy Research. 1995;22(3):207–213. doi: 10.1016/0920-1211(95)00077-1. [DOI] [PubMed] [Google Scholar]
- 12.Yan H, et al. Separation of antiepileptogenic and antiseizure effects of levetiracetam in the spontaneously epileptic rat (SER) Epilepsia. 2005;46(8):1170–7. doi: 10.1111/j.1528-1167.2005.35204.x. [DOI] [PubMed] [Google Scholar]
- 13.Mazarati AM, et al. Anticonvulsant effects of levetiracetam and levetiracetamdiazepam combinations in experimental status epilepticus. Epilepsy Research. 2004;58(2–3):167–174. doi: 10.1016/j.eplepsyres.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 14.Glien M, et al. Effects of the Novel Antiepileptic Drug Levetiracetam on Spontaneous Recurrent Seizures in the Rat Pilocarpine Model of Temporal Lobe Epilepsy. Epilepsia. 2002;43(4):350–357. doi: 10.1046/j.1528-1157.2002.18101.x. [DOI] [PubMed] [Google Scholar]
- 15.De Smedt T, et al. Rapid kindling in preclinical anti-epileptic drug development: The effect of levetiracetam. Epilepsy Research. 2005;67(3):109–116. doi: 10.1016/j.eplepsyres.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 16.Loscher W, Honack D, Rundfeldt C. Antiepileptogenic effects of the novel anticonvulsant levetiracetam (ucb L059) in the kindling model of temporal lobe epilepsy. J Pharmacol Exp Ther. 1998;284(2):474–9. [PubMed] [Google Scholar]
- 17.Hovinga C. Levetiracetam: a novel antiepileptic drug. Pharmacotherapy. 2001;21(11):1375–88. doi: 10.1592/phco.21.17.1375.34432. [DOI] [PubMed] [Google Scholar]
- 18.Shorvon S, et al. Multicenter double-blind, randomized, placebo-controlled trial of levetiracetam as add-on therapy in patients with refractory partial seizures. European Levetiracetam Study Group. Epilepsia. 2000;41(9):1179–86. doi: 10.1111/j.1528-1157.2000.tb00323.x. [DOI] [PubMed] [Google Scholar]
- 19.Shorvon SD, et al. Multicenter Double-Blind, Randomized, Placebo-Controlled Trial of Levetiracetam as Add-On Therapy in Patients with Refractory Partial Seizures. Epilepsia. 2000;41(9):1179–1186. doi: 10.1111/j.1528-1157.2000.tb00323.x. [DOI] [PubMed] [Google Scholar]
- 20.Margineanu DG, Klitgaard H. Inhibition of neuronal hypersynchrony in vitro differentiates levetiracetam from classical antiepileptic drugs. Pharmacol Res. 2000;42(4):281–5. doi: 10.1006/phrs.2000.0689. [DOI] [PubMed] [Google Scholar]
- 21.Niespodziany I, Klitgaard H, Margineanu DG. Levetiracetam inhibits the high-voltage-activated Ca2+ current in pyramidal neurones of rat hippocampal slices. Neuroscience Letters. 2001;306(1–2):5–8. doi: 10.1016/s0304-3940(01)01884-5. [DOI] [PubMed] [Google Scholar]
- 22.Husum H, et al. Levetiracetam prevents changes in levels of brain-derived neurotrophic factor and neuropeptide Y mRNA and of Y1- and Y5-like receptors in the hippocampus of rats undergoing amygdala kindling: implications for antiepileptogenic and mood-stabilizing properties. Epilepsy & Behavior. 2004;5(2):204–215. doi: 10.1016/j.yebeh.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 23.Lynch BA, et al. The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proceedings of the National Academy of Sciences. 2004;101(26):9861–9866. doi: 10.1073/pnas.0308208101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lamberty Y, Margineanu DG, Klitgaard H. Absence of Negative Impact of Levetiracetam on Cognitive Function and Memory in Normal and Amygdala-Kindled Rats. Epilepsy & Behavior. 2000;1(5):333–342. doi: 10.1006/ebeh.2000.0098. [DOI] [PubMed] [Google Scholar]
- 25.Turski L, et al. Review: cholinergic mechanisms and epileptogenesis. The seizures induced by pilocarpine: a novel experimental model of intractable epilepsy. Synapse. 1989;3(2):154–71. doi: 10.1002/syn.890030207. [DOI] [PubMed] [Google Scholar]
- 26.Treiman DM, Walton NY, Kendrick C. A progressive sequence of electroencephalographic changes during generalized convulsive status epilepticus. Epilepsy Res. 1990;5(1):49–60. doi: 10.1016/0920-1211(90)90065-4. [DOI] [PubMed] [Google Scholar]
- 27.Cavalheiro EA, et al. Long-Term Effects of Pilocarpine in Rats: Structural Damage of the Brain Triggers Kindling and Spontaneous I Recurrent Seizures. Epilepsia. 1991;32(6):778–782. doi: 10.1111/j.1528-1157.1991.tb05533.x. [DOI] [PubMed] [Google Scholar]
- 28.Oliveira AA, et al. Evaluation of levetiracetam effects on pilocarpine-induced seizures: Cholinergic muscarinic system involvement. Neuroscience Letters. 2005;385(3):184–188. doi: 10.1016/j.neulet.2005.05.048. [DOI] [PubMed] [Google Scholar]
- 29.Weiss S, Cataltepe O, Cole AJ. Anatomical studies of DNA fragmentation in rat brain after systemic kainate administration. Neuroscience. 1996;74(2):541–551. doi: 10.1016/0306-4522(96)00148-0. [DOI] [PubMed] [Google Scholar]
- 30.Hu RQ, et al. Neuronal stress and injury in C57/BL mice after systemic kainic acid administration. Brain Research. 1998;810(1–2):229. doi: 10.1016/s0006-8993(98)00863-4. [DOI] [PubMed] [Google Scholar]
- 31.Paxinos G, Watson C. The rat brain in stereotactic coordinates. Academic Press; San Diego: 1986. [Google Scholar]
- 32.Wijsman JH, et al. A new method to detect apoptosis in paraffin sections: in situ end- labeling of fragmented DNA. J. Histochem. Cytochem. 1993;41(1):7–12. doi: 10.1177/41.1.7678025. [DOI] [PubMed] [Google Scholar]
- 33.Walton NY, Treiman DM. Response of status epilepticus induced by lithium and pilocarpine to treatment with diazepam. Exp Neurol. 1988;101(2):267–75. doi: 10.1016/0014-4886(88)90010-6. [DOI] [PubMed] [Google Scholar]
- 34.Lowenstein D. Treatment options for status epilepticus. Curr Opin Pharmacol. 2005;5(3):334–9. doi: 10.1016/j.coph.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 35.Gibbs JE, Walker MC, Cock HR. Levetiracetam: Antiepileptic Properties and Protective Effects on Mitochondrial Dysfunction in Experimental Status Epilepticus. Epilepsia. 2006;47(3):469–478. doi: 10.1111/j.1528-1167.2006.00454.x. [DOI] [PubMed] [Google Scholar]
- 36.Gibbs JE, Cock HR. Administration of Levetiracetam after prolonged status epilepticus does not protect from mitochondrial dysfunction in a rodent model. Epilepsy Research. 2007;73(2):208–212. doi: 10.1016/j.eplepsyres.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 37.Crowder KM, et al. Abnormal neurotransmission in mice lacking synaptic vesicle protein 2A (SV2A) Proceedings of the National Academy of Sciences. 1999;96(26):15268–15273. doi: 10.1073/pnas.96.26.15268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Doheny HC, et al. Blood and cerebrospinal fluid pharmacokinetics of the novel anticonvulsant levetiracetam (ucb L059) in the rat. Epilepsy Research. 1999;34(2–3):161–168. doi: 10.1016/s0920-1211(98)00104-1. [DOI] [PubMed] [Google Scholar]
- 39.Patsalos PN. Pharmacokinetic profile of levetiracetam: toward ideal characteristics. Pharmacology & Therapeutics. 2000;85(2):77. doi: 10.1016/s0163-7258(99)00052-2. [DOI] [PubMed] [Google Scholar]