

Abstract
Background
The anthracycline doxorubicin (DOX) is an effective chemotherapeutic agent used to treat pediatric cancers, but is associated with cardiotoxicity which can manifest many years after the initial exposure. To date, very little is known about the mechanism of this late onset cardiotoxicity.
Methods and Results
To understand this problem, we developed a pediatric model of late onset DOX-induced cardiotoxicity, where juvenile mice were exposed to DOX, using a cumulative dose that did not induce acute cardiotoxicity. These mice developed normally and had no obvious cardiac abnormalities as adults. However, evaluation of the vasculature revealed that juvenile DOX exposure impaired vascular development resulting in abnormal vascular architecture in the hearts with less branching and decreased capillary density. Both physiological and pathological stress induced late onset cardiotoxicity in the adult DOX mice. Moreover, adult mice subjected to myocardial infarction (MI) developed rapid heart failure which correlated with a failure to increase capillary density in the injured area. Progenitor cells participate in regeneration and blood vessel formation after an MI, but DOX mice had fewer progenitor cells in the infarct border zone. Interestingly, DOX treatment reduced proliferation and differentiation of the progenitor cells into cells of cardiac lineages.
Conclusions
Our data suggest that anthracycline treatment impairs vascular development as well as progenitor cell function in the young heart, resulting in an adult heart that is more susceptible to stress.
Keywords: anthracyclines, heart failure, vascularization, cardiac progenitor cells
Introduction
Anthracyclines are very effective anti-neoplastic agents with a broad anti-tumor spectrum, including many solid tumors and leukemias. Unfortunately, their clinical use is limited by progressive and dose-related cardiotoxicity that may not manifest itself until many years after treatment 1. Three distinct types of anthracycline-induced cardiotoxicity have been described: acute, early, and late onset cardiotoxicity 2. Acute anthracycline-induced cardiotoxicity occurs typically within the first week of treatment and is usually reversible on discontinuation of treatment. Early onset cardiotoxicity usually presents within a year after completion of treatment and most patients who develop significant cardiotoxicity have a chronic dilated cardiomyopathy 3. Late onset cardiotoxicity is characterized by a latent period during which cardiac function appears normal and the patient is asymptomatic. This type of cardiotoxicity can manifest as much as 15 years after treatment, and is characterized by progressive left ventricular dysfunction leading to irreversible congestive heart failure. Heart failure is often precipitated by events such as exercise, pregnancy, and acute viral infection 4, 5. This latent cardiac toxicity presents a particularly challenging dilemma when treating pediatric cancers. The only known risk factors for late onset cardiotoxicity are cumulative anthracycline dose and younger age at the time of treatment 6. The frequency of cardiotoxic effects has been reported to be as high as 57% among survivors of childhood cancer 7, and a survey by the Pediatric Cardiomyopathy Registry shows that more than 15% of all adult patients with cardiomyopathy were previously treated for cancer during childhood or adolescence 8.
Since anthracyclines are such effective anti-cancer drugs, their mechanisms of action have been under intense investigation for many years. However, the mechanisms underlying the cardiotoxic effects are still not fully understood and multiple mechanisms have been proposed to explain the actions of anthracyclines. These include reactive oxygen species (ROS) production, apoptosis, disruption in DNA replication and transcription 9, 10. In contrast, very little is known about the underlying mechanisms of the late onset cardiotoxicity, and no animal model has been developed that recapitulates this clinical scenario.
In this study, we developed a juvenile mouse model of anthracycline-mediated late onset cardiotoxicity. Juvenile mice exposed to modest doses of DOX develop normally and have no obvious cardiac abnormalities as adults. However, these hearts have abnormal vasculature and a reduced number of progenitor cells which correlated with an increased sensitivity to physiological and pathological stimulus.
Methods
Animals and Experimental Protocol
FVB/N mice were injected at a postnatal age of 5, 10, 15, and 20 days with an i.p. injection of either doxorubicin (Sigma) or saline. Doxorubicin was injected in a volume of 30-40 μl. The first two doses at 5 and 10 days of age were at a concentration of 1 mg/kg, and the last two injections at 15 and 20 days after birth were 0.5 mg/kg. Control animals were injected with an equivalent volume of saline. Animals were observed daily and weighed weekly. At 8-12 weeks of age, the animals were used for experiments.
Swimming Protocol
Eight to 12-week old saline (n=10) and doxorubicin (n=10) mice swam together in a glass tank in 34°C water with a light current to prevent floating. To get accustomed to swimming, the mice initially swam for 15 min/day. Sessions were gradually increased to 90 min twice a day. After one week mice were swimming for 90 min twice a day for 14 days separated by a 2 h rest period 11. The total swimming time was 21 days. One group of saline (n=10) and doxorubicin (n=10) injected mice were used as sedentary controls. Twenty-four hours after completion of the 21-day swimming regimen, mice were evaluated for cardiac function and then sacrificed for tissue harvest.
In vivo coronary artery ligation and Infarct Size Measurement
Coronary occlusion was performed as previously described 12. Briefly, 8 to 12-week old saline (n=29) and doxorubicin (n=37) treated mice were anesthetized with isoflurane, intubated and ventilated. Pressure controlled ventilation (Harvard Apparatus) was maintained at 9 cm H2O throughout the procedure. An 8-0 silk suture was placed around the proximal left coronary artery and then ligated. The suture was left in place and the animal immediately closed up. Sham-operated mice had a suture placed around the LAD but not tightened. Only mice that survived the procedure were used for experiments. To measure infarct size, mice were sacrificed after 24 h and the hearts harvested. The ventricles were cut into 1 mm sections and stained in 1% triphenyl tetrazolium chloride 13.
Isolation of Cardiac Progenitor Cells
Isolation of c-kit positive progenitor cells was done using a protocol adapted from Beltrami et al. 14. Briefly, mouse hearts from 7 day old mouse pups were minced and digested with Collagenase II in J-MEM (supplemented with Hepes, glutamine, taurine, and insulin). The digested heart pieces were filtered through a 30 μm filter, and incubated with anti-CD117-conjugated Miltenyi Biotec Microbeads. C-kit positive cells were isolated by passing cells over a Miltenyi Biotec MiniMACS sorting column. The cells were cultured in Dulbecco’s MEM and Ham’s F12 (ratio 1:1), bFGF (10 ng/ml), EGF (20ng/ml), LIF (10 ng/ml), Hepes (5 mM), and insulin-transferrin-selenite for up to 4 passages.
Statistical Analysis
All values are expressed as means ± Standard Deviation (S.D). Time, treatments and procedures were approximately normally distributed, and were analyzed using ANOVA followed by t-tests. Two factor repeated-measures ANOVA was utilized for mouse weights and Masson’s Trichrome staining. All other variables were analyzed with parametric testing (Student’s t-test). Survival rate was analyzed using the Kaplan-Meier method with the log-rank test. P<0.05 was considered significant. “n” in the figure legends is equal to the sample size per group. All tests were performed using GraphPad Prism 5.
Detailed experimental protocols are described in the Data Supplement.
Results
To study late onset cardiotoxicity, mice were injected with either saline or DOX at 5, 10, 15, and 20 days after birth (Fig. 1A). No acute cardiotoxicity was observed and myocytes appeared normal (Fig 1B) or increases in apoptosis (Fig. 1C) immediately after completion of the injections. Adult hearts from saline and DOX-treated mice appeared morphologically normal (Fig. 1D). DOX-mediated cardiotoxicity is characterized by disorganized myofibrils, increased vacuolization and swelling of organelles 15. However, electron microscopy revealed no degenerative changes suggestive of DOX-mediated toxicity in cardiac myocytes (Fig. 1E). DOX treatment resulted in slower weight gain in pups compared to saline treatment, but this difference diminished as the mice matured (Supplemental Fig. 1). There were no cardiac hypertrophy (Supplemental Fig. 2) and no difference in cardiac function in DOX treated mice in adulthood when compared to saline (Table I).
Figure 1.
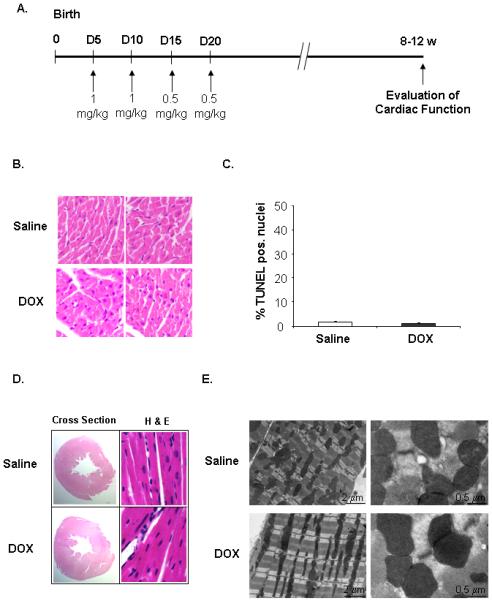
Establishment of a juvenile mouse model of late onset anthracycline-mediated cardiotoxicity. A. Mouse pups were injected with saline or doxorubicin (DOX) at 5, 10, 15, and 20 days after birth. B. H & E staining of heart sections prepared from 21 days old mice. C. TUNEL staining of heart sections prepared from 21 days old mice (n=4). D. H & E staining of adult heart sections. E. Electron micrographs of adult hearts from saline and doxorubicin mice.
TABLE I.
Measurement of cardiac function in vivo
| (n=15) | Saline | DOX | P-value |
|---|---|---|---|
| Heart Rate | 303±96 | 283±87 | 0.58 |
| DP | 93.1±14 | 87.9±13 | 0.32 |
| dP/dTmax | 4731±1109 | 4513±998 | 0.57 |
| dP/dTmin | −3813±858 | −3528±690 | 0.33 |
Since DOX has potent anti-angiogenic effects 16, we investigated if DOX treatment affected vascular development in the juvenile heart. Measurement of blood flow and visualization of the coronary branching pattern revealed that juvenile DOX exposure resulted in reduced blood flow (Fig. 2A) and reduced coronary branching (Fig. 2B) in the adult heart. Although major vessels in DOX hearts appeared to have narrower lumen than saline hearts, examination of the vessels revealed no differences in thickness and composition (Supplemental Fig. 3A-B). In a time-course study, capillary density was unaffected in the young animal during (at Day 11) and immediately after completion (at Day 21) of the DOX injections. However, at day 40 and 60 capillary densities were significantly reduced in the DOX treated animals (Fig. 2C). The vascular endothelial factor (VEGF) is involved in development of vessels and stimulates recruitment and proliferation of endothelial cells 17. Interestingly, myocardial VEGF expression was significantly reduced in the heart at day 21 and remained reduced into adulthood (Fig. 2D and E), suggesting that juvenile DOX exposure might affect vascular development by interfering with VEGF signaling. VEGF levels in the plasma were not different between the saline and DOX treated animals at any time-point (Supplemental Fig. 4).
Figure 2.
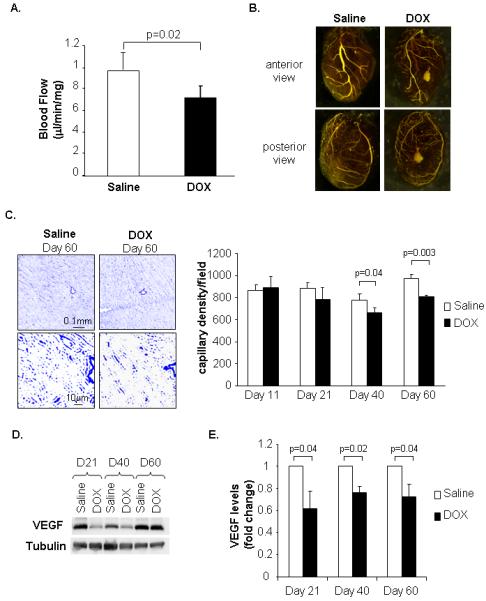
Assessment of blood flow and vascular structure in adult mouse hearts exposed to juvenile saline or DOX treatments. A. Measurement of blood flow in adult hearts using fluorescent microspheres (n=6). B. Microfil polymer injection to visualize coronary branching in hearts. C. Representative CD31 staining of adult heart sections at Day 60 and quantitation of capillary density at 11, 21, 40 and 60 days after birth (n=4). D. Western analysis of VEGF expression in heart. E. Quantitation of VEGF in heart lysates (n=4).
Sufficient vasculature is important for the heart to adapt to increased workload such as exercise. To investigate whether the DOX exposed mice were more sensitive to exercise we subjected saline and DOX adult mice to endurance swimming. After 21 days of swimming, hearts of DOX injected mice displayed cardiac hypertrophy (Fig. 3A) and dilation of the left ventricle (Fig. 3B). In contrast, saline treated mice did not display any significant hypertrophy or dilatation of the ventricle after the swimming, confirming that cardiac hypertrophy was attributable to the combination of DOX treatment and exercise. H & E staining revealed many areas of myofibril disarray in DOX exposed hearts after swimming (Fig. 3C). Also, hearts from DOX treated mice had increased fibrosis after swimming (Fig. 3D). Hemodynamic analysis of mice showed that the ventricular end-diastolic pressure was reduced in swimming saline mice, whereas it was increased after swimming in DOX treated mice (Fig. 3F). These data demonstrate that modest doses of DOX in young mice result in decreased capability of the hearts to adapt to increased workload in adulthood.
Figure 3.
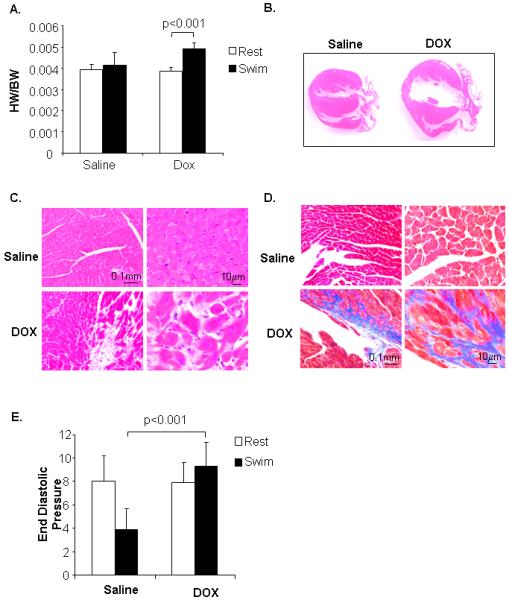
Juvenile doxorubicin treatment increases susceptibility to exercise in adulthood. A. Swimming for 21 days induces cardiac hypertrophy in doxorubicin-treated mice as measured by the ratio of heart weight (HW) to body weight (BW) (n=9). B. H & E staining of heart sections after swimming. C. H & E staining of DOX heart sections after swimming. D. Masson’s trichrome staining of heart sections after swimming. E. Measurement of ventricular end diastolic pressure (mmHg) in sedentary or swimming mice (n=5-8).
Moreover, neovascularization in the border zone after myocardial infarction (MI) is an important beneficial response that limits the development of left ventricular remodeling and deterioration to heart failure 18. To investigate if juvenile DOX exposure impaired the neovascularization response, adult saline and DOX mice were subjected to MI by permanent ligation of the LAD. Interestingly, DOX treated mice were highly sensitive to MI and the survival rate of DOX treated mice was only ~25% compared to ~80% for the saline mice (Fig. 4A). DOX exposed mice had larger (but not significant) infarct size (80.3%) than saline mice (71.3%) (Fig. 4B & C). More importantly, DOX exposed mice had significantly larger area at risk, suggesting differences in the vasculature (Fig. 4D). DOX mice also had extensive fibrosis compared to saline treated mice after MI (Fig. 4E). Even though there was no increase in fibrosis under non-stressed condition between the saline and DOX hearts (data not shown), the amount of fibrosis was significantly increased in the remote (non-risk) area in the DOX heart. In addition, measurement of capillary density in the infarct border zone 4 days after MI showed that DOX-exposed mice had reduced neovascularization after MI compared to saline mice (Fig. 5A). Also, the abundance of staining for α-SMA was attenuated in DOX hearts in the border zone (Fig. 5B). Visualization of the vasculature post-MI confirmed that DOX hearts had reduced vessel formation in the border zone (Fig. 5C). These results suggest that DOX treatment affects vascular development in the juvenile heart and puts the adult heart at greater risk for ischemic injury.
Figure 4.
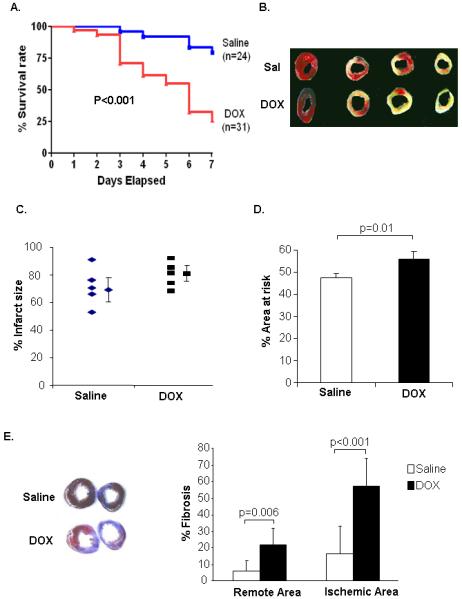
Juvenile doxorubicin treatment increases susceptibility to myocardial infarction in adulthood. A. Kaplan-Meier analysis of survival after myocardial infarction. B. TTC staining of heart sections. C. Infarct size measurement by TTC staining 24 h post MI (n=5). D. Area at risk in saline and DOX hearts. E. Masson’s trichrome staining of heart sections 7 days after myocardial infarction (n=6).
Figure 5.
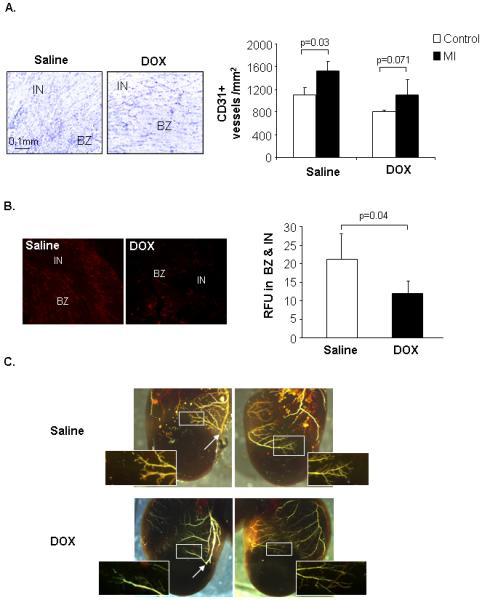
Evaluation of vascular changes in adult hearts after myocardial infarction. A. Representative CD31 staining of heart sections 4 days post-MI and quantitation of capillary density (n=3). B. Representative α-SMA staining of heart sections 4 days post-MI and quantitation of relative fluorescence (RFU, n=3) C. Microfil polymer injection and corrosion casting of hearts 10 days post-MI. Arrow marks the site of ligation. Infarct (IN) and border zone (BZ).
Myocardial infarction is associated with migration of progenitor cells into the damaged area where they participate in regeneration and blood vessel formation 19-21. Immunostaining of adult saline heart sections for the presence of cells positive for the common stem cell marker c-kit 22 revealed small c-kit+ cells with high nucleus/cytoplasm ratio in the border zone of the infarct by 4 days after MI (Fig. 6A, B and C). Interestingly, there were significantly fewer c-kit+ cells in the border zone of animals that had been exposed to DOX as pups (Fig. 6C). We also investigated whether the MI stimulated differentiation of the CPCs into cells of the different lineages as has been previously reported 23-26. At 7 days post-MI, 63% and 44% of c-kit+ cells stained positive for endothelial cell lineage markers Flk-1 and CD31, respectively in saline hearts. Importantly, 29% and 21% of the c-kit+ cells stained positive for Flk-1 and CD31 in DOX hearts (Data Supplement Fig. 5). In saline hearts 53% of the c-kit+ cells were positive for GATA-4 and 62% were positive for MEF2C suggesting that these cells are committed to a myocyte lineage (Supplemental Fig. 6). In contrast, only 32% and 38% of DOX exposed c-kit+ cells expressed GATA-4 and MEF2C, respectively. There was no significant difference in c-kit+ that were positive for smooth muscle cell marker α-SMA (Supplemental Fig. 7). Although the c-kit+ cells may not be directly committed to myogenesis in our model, the data suggest that juvenile DOX exposure affects differentiation of CPCs into myocytes and endothelial cells but not into smooth muscle cells.
Figure 6.
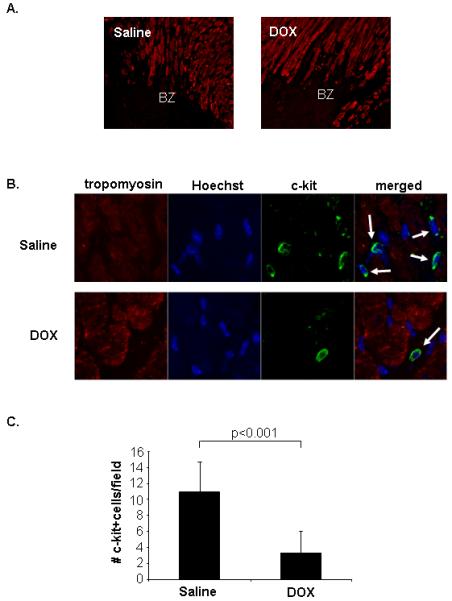
Juvenile doxorubicin exposure results in reduced number of progenitor cells in the border zone (BZ) of an infarct in the adult heart. Hearts sections prepared 4 days after ligation of the LAD were stained with anti-tropomyosin (red) and anti-c-kit (green). Nuclei were counterstained with Hoechst 33342 (blue). A. Dead myocytes in the border zone of an infarct stain negative for tropomyosin. B. Detection of c-kit positive cells in the BZ. C. Quantitation of c-kit+ progenitor cells in the BZ (n=4).
In contrast to the adult heart, the juvenile heart contains a large population of CPCs 27. To investigate if DOX treatment had an effect on CPCs in the juvenile heart, heart sections from mouse pups at 12 days of age were stained for the presence of c-kit. We found that DOX-exposed mice had significantly fewer c-kit+ cells (Fig. 7A and B), suggesting that DOX might be harmful to CPCs. Hearts from saline-injected mice contained many clusters of c-kit+ cells, which are thought to reflect intense expansion of progenitor cells 28. In contrast, hearts of DOX injected pups had fewer clusters of cells positive for c-kit, suggesting that DOX might inhibit proliferation of CPCs. The cell cycle inhibitor p16INK4a was significantly upregulated in c-kit+ cells in DOX exposed hearts at 12 days of age (Fig. 7C and D). CPC isolated from hearts of DOX treated mice also incorporated less BrdU into the DNA in vitro compared to saline (Fig. 7E), consistent with reduced proliferation. This suggests that juvenile doxorubicin treatment might have permanently reduced the number of resident progenitor cells in these hearts.
Figure 7.
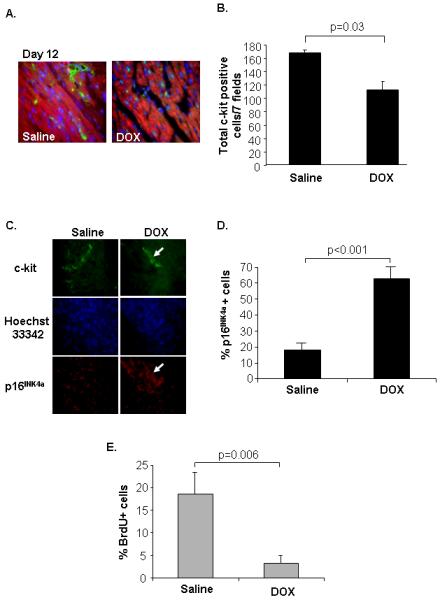
Doxorubicin treatment causes a reduction in c-kit+ progenitor cells in juvenile hearts. A. Mouse pups were injected with saline or 1 mg/kg doxorubicin at 5 and 10 days after birth and heart sections, prepared at 12 days after birth, were stained with anti-c-kit (green), anti-tropomyosin (red), and Hoechst 33342 (blue). B. Quantitation of c-kit+ cells in heart sections (n=3). C. Heart sections from 12 days old mice were stained with anti-c-kit and anti-p16INK4a. Nuclei were counterstained with Hoechst 33342. D. Quantitation of p16INK4a positive progenitor cells in tissue sections (n=3). E. C-kit+ progenitor cells isolated from saline or doxorubicin injected mice at Day 12 had reduced BrdU incorporation in vitro (n=4).
To further investigate the effect of DOX treatment on proliferation of CPCs, we isolated c-kit+ cells from mouse hearts and treated with DOX for 72 h. Treatment of CPCs with 10 nM or 100 nM DOX attenuated proliferation by 40% and 50% respectively (Fig. 8A). In parallel experiments, DOX did not induce cell death at these concentrations, confirming that the reduced number of cells was not due to cell death (Fig. 8B). Inhibition of proliferation was also confirmed by BrdU incorporation studies, where fewer DOX-treated cells incorporated BrdU into the DNA (Fig. 8C). Although an equal number of cells were plated before treatment, wells containing cells treated with saline always had significantly more cells after 72 h compared to DOX-treated (Supplemental Fig. 8). DOX treatment also reduced telomerase activity (Fig. 8D) and induced expression of the senescence marker p16INK4a (Fig. 8E and F), suggesting that DOX induces senescence in CPCs.
Figure 8.
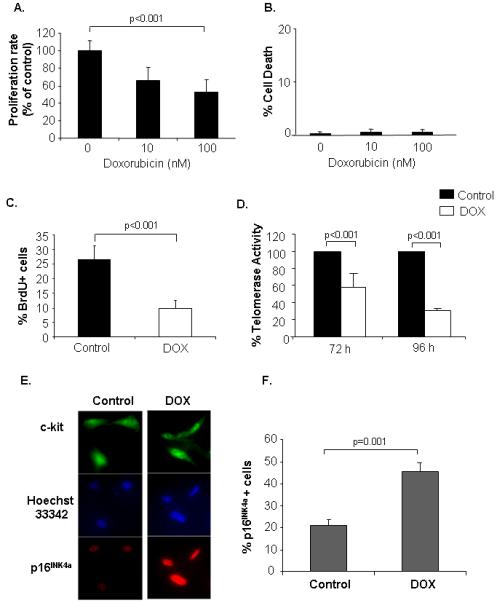
Doxorubicin treatment inhibits proliferation of isolated CPCs. Cells were treated with doxorubicin for 72 h and then assessed for (A) cell proliferation by MTT assay (n=3), and (B) cell death by Trypan Blue exclusion assay (n=3). C. Cells were treated with 100 nM doxorubicin and proliferation was determined by BrdU incorporation (n=4). D. Cells were exposed to vehicle or 100 nM DOX for 72 h or 96 h and then assayed for telomerase activity using the TRAP assay (n=3). E. Isolated CPCs were treated with 100 nM DOX for 72 h and then fixed and stained for the presence of c-kit and p16INK4a. F. Quantitation of p16INK4a positive cells (n=4).
Discussion
Several laboratories have identified resident progenitor cells in the heart that have the capacity to differentiate into the various cardiac cells both in vitro and in vivo 14, 29-31. These cells are thought to exist in the heart to facilitate growth during adolescence and to provide a mechanism for minor repair. Senescence and death of CPCs correlate with development of heart failure 32. That children are at higher risk for late onset cardiotoxicity than adults could be explained by the fact that the heart is still growing and very likely contains more cardiac progenitor cells than the adult myocardium. C-kit positive cells are present in juvenile human hearts and it was reported that c-kit+ cells were more abundant in children with congenital heart disease and that this increase correlated with an increase in apoptotic myocytes 33. Thus, stress to the young human heart appears to induce mobilization of progenitor cells to the damaged myocardium. Our studies are currently limited to the mouse, but it will be important to verify our findings in human hearts.
Progenitor cells migrate into the border zone following myocardial infarction, where they participate in myocardial regeneration, blood vessel formation, and remodeling 19, 20, 34. We found a reduction in the number of c-kit+ progenitor cells in juvenile mouse hearts after DOX treatment, which correlated with fewer c-kit+ cells in the border zone of an infarct in the adults, compared to saline treated mice. These mice were more susceptible to MI with greater infarcts and increased mortality. Interestingly, mice heterozygous for a defective c-kit receptor had a reduced number of c-kit+ cells in the infarcted heart, which also correlated with increased injury and mortality after MI 34. Clearly, functional c-kit+ progenitor cells are important for repairing a heart in response to stress such as an MI. Our findings suggest that juvenile DOX treatment have permanently reduced the number of CPCs and reduced their differentiation in response to injury. Thus, a minor ischemic event that would cause minimal or no damage in a healthy person with functional progenitor cells might result in more significant damage in a person with fewer progenitor cells migrating to and differentiating into cardiac cells at the site of injury. Accumulation of such stresses over time may be one of the factors that predispose children exposed to anthracyclines to late-onset cardiomyopathy. Interestingly, studies have reported that survivors of Hodgkin’s lymphoma were at increased risk of an infarction and had an increased risk of dying from it 35, 36. These studies also found that the risks were greater in patients treated with anthracyclines when young than in patients that had been treated at older ages.
DOX induce senescence by reducing telomerase activity in many different cell types 37, 38. Telomerase activity is also essential for proliferation of pluripotent stem cells and for certain tissue-specific self-renewing stem cells 32, 39. Neural stem cells isolated from telomerase deficient mice were incapable of expansion in vitro, suggesting that telomere function is important for proliferation of stem cells 40. Interestingly, these mice had a reduced number of resident neuronal stem cells which had impaired proliferation. Heart biopsies from old patients with heart failure showed that c-kit+ progenitor cells had undergone senescence and substantial telomeric shortening, suggesting that these cells can no longer compensate for lost myocytes and therefore may exacerbate the development of heart failure 32. Thus, telomerase in cycling progenitor cells counters progressive shortening of the telomeres and promotes growth and survival of progenitor cells.
In this study DOX exposure had long-lasting effects on vascular development, resulting in a vascular tree with fewer ramifications, thereby limiting the capacity to respond to increased demand. This is not simply a developmental defect but also a reflection of limited progenitor capacity, since neovascularization after MI was also impaired. Similarly, c-kit mutant mice had reduced angiogenesis after MI 34. Neovascularization in the border zone after an infarction is an important process to prevent the development of left ventricular remodeling and deterioration to heart failure. Our results suggest that juvenile DOX treatment particularly affects the microvasculature and implies that childhood DOX exposure puts the adult heart at greater risk for ischemic injury due to a barely adequate blood supply. Moreover, defective c-kit+ cells can affect the myofibroblast repair response after an MI which is important for stabilization of the scar 41. Since we observed a reduction in the formation of α-SMA positive cells in the border zone after an MI in the DOX mice, it is possible that DOX exposure has affected the function of the myofibroblasts.
Occasionally the DOX mice died from cardiac rupture. This is likely attributable to insufficient angiogenesis which is essential for the repair process and function of the fibroblasts. Following an infarction, wound healing ensues, a complex process that involves inflammation, new tissue formation and remodeling. A defect in infarct healing and remodeling can cause cardiac rupture 42. Moreover, endothelial progenitor cells (EPCs) are recruited to ischemic regions, improving neovascularization 43, and it is likely that DOX reduces the pools of other progenitor cells including EPCs. It is also possible that DOX exposure has impaired the heart’s ability to produce and secrete homing factors that will attract progenitor cells to the site of injury or has made the heart a hostile environment that prevents implantation of circulating progenitor cells. This has important implications not only for anthracycline cardiomyopathy, but for other childhood exposures that may affect progenitor cell pools.
Our model provides new insight into the mechanism of late onset cardiotoxicity in mice. However, the similarity and relevance of this model to anthracycline-induced cardiomyopathy in humans are currently unknown and needs to be determined. Also, since the focus of this study was on late-onset cardiotoxicity, we limited this study to the heart and we did not examine other tissues. Our findings suggest that antecedent doxorubicin exposure has lasting effects on the progenitor cells that participate in cardiac repair and neovascularization. Progenitor cells have therapeutic potential, and exogenously administered stem cells have been shown to successfully engraft and improve left ventricular function in animal models of myocardial infarctions 44, 45. Thus, it will be important to explore whether replacing or protecting the CPCs will ameliorate late onset anthracycline-induced cardiotoxicity.
Clinical Perspective
Anthracyclines such as doxorubicin are effective chemotherapeutic agents used for treatment of many cancers. Unfortunately, their clinical use is limited by the risk of severe cardiotoxicity and heart failure that may not manifest until years later. Anthracyclines are of special concern in pediatric oncology since the youngest children are at the greatest risk of developing late-onset cardiotoxicity. In this study, we present the first evidence that anthracycline exposure in juvenile mice reduces the size of the cardiac progenitor pool, impairs their ability to differentiate into cells of cardiac and vascular lineages, and adversely affects vascular development in the heart. Despite normal development and hemodynamic function as adults, these mice develop rapid heart failure in response to physiological or pathological stress. This animal model recapitulates many of the features of late-onset anthracycline toxicity in humans, and explains why younger children—who have more dividing progenitor cells—are more vulnerable. Our findings suggest that anthracycline-mediated effects on the progenitor cells might underlie the reduced ability to accomplish physiologic hypertrophy as well as cardiac repair and neovascularization. Injection of stem cell mobilizing factors has been shown to enhance migration of bone marrow cells to the heart and to attenuate acute doxorubicin cardiotoxicity. The therapeutic potential of progenitor cells has been demonstrated in animal models where successful engraftment of progenitor cells improved left ventricular function after myocardial infarction. If progenitor cells are required for neovascularization accompanying physiologic hypertrophy or repair, then stem cell mobilization or replacement may ameliorate late-onset heart failure due to anthracyclines.
Supplementary Material
Acknowledgments
FUNDING SOURCES
This work was supported by NIH grants P01HL085577 (MAS), R37HL091102 (MAS), HL087023 (ÅBG), HL060590 (RAG), HL071091 (RAG), and HL092136 (ÅBG & RAG).
Footnotes
Subject Codes: [11] Other heart failure; [129] Angiogenesis; [130] Animal models of human disease
DISCLOSURES
There are no conflicts to disclose.
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Singal PK, Iliskovic N. Doxorubicin-induced cardiomyopathy. N Engl J Med. 1998;339:900–905. doi: 10.1056/NEJM199809243391307. [DOI] [PubMed] [Google Scholar]
- 2.Broder H, Gottlieb RA, Lepor NE. Chemotherapy and cardiotoxicity. Rev Cardiovasc Med. 2008;9:75–83. [PMC free article] [PubMed] [Google Scholar]
- 3.Lefrak EA, Pitha J, Rosenheim S, Gottlieb JA. A clinicopathologic analysis of adriamycin cardiotoxicity. Cancer. 1973;32:302–314. doi: 10.1002/1097-0142(197308)32:2<302::aid-cncr2820320205>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 4.Ali MK, Ewer MS, Gibbs HR, Swafford J, Graff KL. Late doxorubicin-associated cardiotoxicity in children. The possible role of intercurrent viral infection. Cancer. 1994;74:182–188. doi: 10.1002/1097-0142(19940701)74:1<182::aid-cncr2820740129>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 5.Pai VB, Nahata MC. Cardiotoxicity of chemotherapeutic agents: incidence, treatment and prevention. Drug Saf. 2000;22:263–302. doi: 10.2165/00002018-200022040-00002. [DOI] [PubMed] [Google Scholar]
- 6.Kremer LC, van der Pal HJ, Offringa M, van Dalen EC, Voute PA. Frequency and risk factors of subclinical cardiotoxicity after anthracycline therapy in children: a systematic review. Ann Oncol. 2002;13:819–829. doi: 10.1093/annonc/mdf167. [DOI] [PubMed] [Google Scholar]
- 7.Paulides M, Kremers A, Stohr W, Bielack S, Jurgens H, Treuner J, Beck JD, Langer T. Prospective longitudinal evaluation of doxorubicin-induced cardiomyopathy in sarcoma patients: a report of the late effects surveillance system (LESS) Pediatr Blood Cancer. 2006;46:489–495. doi: 10.1002/pbc.20492. [DOI] [PubMed] [Google Scholar]
- 8.Giantris A, Abdurrahman L, Hinkle A, Asselin B, Lipshultz SE. Anthracycline-induced cardiotoxicity in children and young adults. Crit Rev Oncol Hematol. 1998;27:53–68. doi: 10.1016/s1040-8428(97)10007-5. [DOI] [PubMed] [Google Scholar]
- 9.Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev. 2004;56:185–229. doi: 10.1124/pr.56.2.6. [DOI] [PubMed] [Google Scholar]
- 10.Doroshow JH. Effect of anthracycline antibiotics on oxygen radical formation in rat heart. Cancer Res. 1983;43:460–472. [PubMed] [Google Scholar]
- 11.Matsumoto K, Ishihara K, Tanaka K, Inoue K, Fushiki T. An adjustable-current swimming pool for the evaluation of endurance capacity of mice. J Appl Physiol. 1996;81:1843–1849. doi: 10.1152/jappl.1996.81.4.1843. [DOI] [PubMed] [Google Scholar]
- 12.Tarnavski O, McMullen JR, Schinke M, Nie Q, Kong S, Izumo S. Mouse cardiac surgery: comprehensive techniques for the generation of mouse models of human diseases and their application for genomic studies. Physiol Genomics. 2004;16:349–360. doi: 10.1152/physiolgenomics.00041.2003. [DOI] [PubMed] [Google Scholar]
- 13.Gustafsson AB, Sayen MR, Williams SD, Crow MT, Gottlieb RA. TAT protein transduction into isolated perfused hearts: TAT-apoptosis repressor with caspase recruitment domain is cardioprotective. Circulation. 2002;106:735–739. doi: 10.1161/01.cir.0000023943.50821.f7. [DOI] [PubMed] [Google Scholar]
- 14.Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, Kasahara H, Rota M, Musso E, Urbanek K, Leri A, Kajstura J, Nadal-Ginard B, Anversa P. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell. 2003;114:763–776. doi: 10.1016/s0092-8674(03)00687-1. [DOI] [PubMed] [Google Scholar]
- 15.Sun X, Zhou Z, Kang YJ. Attenuation of doxorubicin chronic toxicity in metallothionein-overexpressing transgenic mouse heart. Cancer Res. 2001;61:3382–3387. [PubMed] [Google Scholar]
- 16.Amoh Y, Li L, Katsuoka K, Hoffman RM. Chemotherapy targets the hair-follicle vascular network but not the stem cells. J Invest Dermatol. 2007;127:11–15. doi: 10.1038/sj.jid.5700486. [DOI] [PubMed] [Google Scholar]
- 17.Byrne AM, Bouchier-Hayes DJ, Harmey JH. Angiogenic and cell survival functions of vascular endothelial growth factor (VEGF) J Cell Mol Med. 2005;9:777–794. doi: 10.1111/j.1582-4934.2005.tb00379.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kocher AA, Schuster MD, Szabolcs MJ, Takuma S, Burkhoff D, Wang J, Homma S, Edwards NM, Itescu S. Neovascularization of ischemic myocardium by human bone-marrow-derived angioblasts prevents cardiomyocyte apoptosis, reduces remodeling and improves cardiac function. Nat Med. 2001;7:430–436. doi: 10.1038/86498. [DOI] [PubMed] [Google Scholar]
- 19.Urbanek K, Rota M, Cascapera S, Bearzi C, Nascimbene A, De Angelis A, Hosoda T, Chimenti S, Baker M, Limana F, Nurzynska D, Torella D, Rotatori F, Rastaldo R, Musso E, Quaini F, Leri A, Kajstura J, Anversa P. Cardiac stem cells possess growth factor-receptor systems that after activation regenerate the infarcted myocardium, improving ventricular function and long-term survival. Circ Res. 2005;97:663–673. doi: 10.1161/01.RES.0000183733.53101.11. [DOI] [PubMed] [Google Scholar]
- 20.Limana F, Zacheo A, Mocini D, Mangoni A, Borsellino G, Diamantini A, De Mori R, Battistini L, Vigna E, Santini M, Loiaconi V, Pompilio G, Germani A, Capogrossi MC. Identification of myocardial and vascular precursor cells in human and mouse epicardium. Circ Res. 2007;101:1255–1265. doi: 10.1161/CIRCRESAHA.107.150755. [DOI] [PubMed] [Google Scholar]
- 21.Kuang D, Zhao X, Xiao G, Ni J, Feng Y, Wu R, Wang G. Stem cell factor/c-kit signaling mediated cardiac stem cell migration via activation of p38 MAPK. Basic Res Cardiol. 2007 doi: 10.1007/s00395-007-0690-z. [DOI] [PubMed] [Google Scholar]
- 22.Kondo M, Wagers AJ, Manz MG, Prohaska SS, Scherer DC, Beilhack GF, Shizuru JA, Weissman IL. Biology of hematopoietic stem cells and progenitors: implications for clinical application. Annu Rev Immunol. 2003;21:759–806. doi: 10.1146/annurev.immunol.21.120601.141007. [DOI] [PubMed] [Google Scholar]
- 23.Fransioli J, Bailey B, Gude NA, Cottage CT, Muraski JA, Emmanuel G, Wu W, Alvarez R, Rubio M, Ottolenghi S, Schaefer E, Sussman MA. Evolution of the c-kit-positive cell response to pathological challenge in the myocardium. Stem Cells. 2008;26:1315–1324. doi: 10.1634/stemcells.2007-0751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Altarche-Xifro W, Curato C, Kaschina E, Grzesiak A, Slavic S, Dong J, Kappert K, Steckelings M, Imboden H, Unger T, Li J. Cardiac c-kit+AT2+ Cell Population is Increased in Response to Ischemic Injury and Supports Cardiomyocyte Performance. Stem Cells. 2009 doi: 10.1002/stem.171. [DOI] [PubMed] [Google Scholar]
- 25.Linke A, Muller P, Nurzynska D, Casarsa C, Torella D, Nascimbene A, Castaldo C, Cascapera S, Bohm M, Quaini F, Urbanek K, Leri A, Hintze TH, Kajstura J, Anversa P. Stem cells in the dog heart are self-renewing, clonogenic, and multipotent and regenerate infarcted myocardium, improving cardiac function. Proc Natl Acad Sci U S A. 2005;102:8966–8971. doi: 10.1073/pnas.0502678102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Limana F, Germani A, Zacheo A, Kajstura J, Di Carlo A, Borsellino G, Leoni O, Palumbo R, Battistini L, Rastaldo R, Muller S, Pompilio G, Anversa P, Bianchi ME, Capogrossi MC. Exogenous high-mobility group box 1 protein induces myocardial regeneration after infarction via enhanced cardiac C-kit+ cell proliferation and differentiation. Circ Res. 2005;97:e73–83. doi: 10.1161/01.RES.0000186276.06104.04. [DOI] [PubMed] [Google Scholar]
- 27.Gude N, Muraski J, Rubio M, Kajstura J, Schaefer E, Anversa P, Sussman MA. Akt promotes increased cardiomyocyte cycling and expansion of the cardiac progenitor cell population. Circ Res. 2006;99:381–388. doi: 10.1161/01.RES.0000236754.21499.1c. [DOI] [PubMed] [Google Scholar]
- 28.Urbanek K, Quaini F, Tasca G, Torella D, Castaldo C, Nadal-Ginard B, Leri A, Kajstura J, Quaini E, Anversa P. Intense myocyte formation from cardiac stem cells in human cardiac hypertrophy. Proc Natl Acad Sci U S A. 2003;100:10440–10445. doi: 10.1073/pnas.1832855100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Laugwitz KL, Moretti A, Lam J, Gruber P, Chen Y, Woodard S, Lin LZ, Cai CL, Lu MM, Reth M, Platoshyn O, Yuan JX, Evans S, Chien KR. Postnatal isl1+ cardioblasts enter fully differentiated cardiomyocyte lineages. Nature. 2005;433:647–653. doi: 10.1038/nature03215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Messina E, De Angelis L, Frati G, Morrone S, Chimenti S, Fiordaliso F, Salio M, Battaglia M, Latronico MV, Coletta M, Vivarelli E, Frati L, Cossu G, Giacomello A. Isolation and expansion of adult cardiac stem cells from human and murine heart. Circ Res. 2004;95:911–921. doi: 10.1161/01.RES.0000147315.71699.51. [DOI] [PubMed] [Google Scholar]
- 31.Oh H, Bradfute SB, Gallardo TD, Nakamura T, Gaussin V, Mishina Y, Pocius J, Michael LH, Behringer RR, Garry DJ, Entman ML, Schneider MD. Cardiac progenitor cells from adult myocardium: homing, differentiation, and fusion after infarction. Proc Natl Acad Sci U S A. 2003;100:12313–12318. doi: 10.1073/pnas.2132126100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chimenti C, Kajstura J, Torella D, Urbanek K, Heleniak H, Colussi C, Di Meglio F, Nadal-Ginard B, Frustaci A, Leri A, Maseri A, Anversa P. Senescence and death of primitive cells and myocytes lead to premature cardiac aging and heart failure. Circ Res. 2003;93:604–613. doi: 10.1161/01.RES.0000093985.76901.AF. [DOI] [PubMed] [Google Scholar]
- 33.Sato H, Shiraishi I, Takamatsu T, Hamaoka K. Detection of TUNEL-positive cardiomyocytes and c-kit-positive progenitor cells in children with congenital heart disease. J Mol Cell Cardiol. 2007;43:254–261. doi: 10.1016/j.yjmcc.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 34.Fazel S, Cimini M, Chen L, Li S, Angoulvant D, Fedak P, Verma S, Weisel RD, Keating A, Li RK. Cardioprotective c-kit+ cells are from the bone marrow and regulate the myocardial balance of angiogenic cytokines. J Clin Invest. 2006;116:1865–1877. doi: 10.1172/JCI27019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aleman BM, van den Belt-Dusebout AW, De Bruin ML, van ’t Veer MB, Baaijens MH, de Boer JP, Hart AA, Klokman WJ, Kuenen MA, Ouwens GM, Bartelink H, van Leeuwen FE. Late cardiotoxicity after treatment for Hodgkin lymphoma. Blood. 2007;109:1878–1886. doi: 10.1182/blood-2006-07-034405. [DOI] [PubMed] [Google Scholar]
- 36.Swerdlow AJ, Higgins CD, Smith P, Cunningham D, Hancock BW, Horwich A, Hoskin PJ, Lister A, Radford JA, Rohatiner AZ, Linch DC. Myocardial infarction mortality risk after treatment for Hodgkin disease: a collaborative British cohort study. J Natl Cancer Inst. 2007;99:206–214. doi: 10.1093/jnci/djk029. [DOI] [PubMed] [Google Scholar]
- 37.Maejima Y, Adachi S, Ito H, Hirao K, Isobe M. Induction of premature senescence in cardiomyocytes by doxorubicin as a novel mechanism of myocardial damage. Aging Cell. 2008;7:125–136. doi: 10.1111/j.1474-9726.2007.00358.x. [DOI] [PubMed] [Google Scholar]
- 38.Elmore LW, Rehder CW, Di X, McChesney PA, Jackson-Cook CK, Gewirtz DA, Holt SE. Adriamycin-induced senescence in breast tumor cells involves functional p53 and telomere dysfunction. J Biol Chem. 2002;277:35509–35515. doi: 10.1074/jbc.M205477200. [DOI] [PubMed] [Google Scholar]
- 39.Morrison SJ, Prowse KR, Ho P, Weissman IL. Telomerase activity in hematopoietic cells is associated with self-renewal potential. Immunity. 1996;5:207–216. doi: 10.1016/s1074-7613(00)80316-7. [DOI] [PubMed] [Google Scholar]
- 40.Ferron S, Mira H, Franco S, Cano-Jaimez M, Bellmunt E, Ramirez C, Farinas I, Blasco MA. Telomere shortening and chromosomal instability abrogates proliferation of adult but not embryonic neural stem cells. Development. 2004;131:4059–4070. doi: 10.1242/dev.01215. [DOI] [PubMed] [Google Scholar]
- 41.Cimini M, Fazel S, Zhuo S, Xaymardan M, Fujii H, Weisel RD, Li RK. c-kit dysfunction impairs myocardial healing after infarction. Circulation. 2007;116:I77–82. doi: 10.1161/CIRCULATIONAHA.107.708107. [DOI] [PubMed] [Google Scholar]
- 42.Factor SM, Robinson TF, Dominitz R, Cho SH. Alterations of the myocardial skeletal framework in acute myocardial infarction with and without ventricular rupture. A preliminary report. Am J Cardiovasc Pathol. 1987;1:91–97. [PubMed] [Google Scholar]
- 43.Aicher A, Heeschen C, Mildner-Rihm C, Urbich C, Ihling C, Technau-Ihling K, Zeiher AM, Dimmeler S. Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. Nat Med. 2003;9:1370–1376. doi: 10.1038/nm948. [DOI] [PubMed] [Google Scholar]
- 44.Schuleri KH, Amado LC, Boyle AJ, Centola M, Saliaris AP, Gutman MR, Hatzistergos KE, Oskouei BN, Zimmet JM, Young RG, Heldman AW, Lardo AC, Hare JM. Early improvement in cardiac tissue perfusion due to mesenchymal stem cells. Am J Physiol Heart Circ Physiol. 2008;294:H2002–2011. doi: 10.1152/ajpheart.00762.2007. [DOI] [PubMed] [Google Scholar]
- 45.Orlic D, Kajstura J, Chimenti S, Limana F, Jakoniuk I, Quaini F, Nadal-Ginard B, Bodine DM, Leri A, Anversa P. Mobilized bone marrow cells repair the infarcted heart, improving function and survival. Proc Natl Acad Sci U S A. 2001;98:10344–10349. doi: 10.1073/pnas.181177898. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.