

Myocardial infarction is a major clinical problem [1]. Thrombotic occlusion of coronary arteries leads to tissue ischemia and loss of myocardium [2,3]. Restoration of blood flow is necessary to reduce myocardial necrosis, but this causes additional injury [2]. This so-called ‘reperfusion injury’ is mediated by the release of oxygen radicals and inflammatory mediators from infiltrating leukocytes, and by the activation of the complement and coagulation cascades [2,4].
Inhibition of tissue factor (TF), the primary initiator of coagulation cascade, has been shown to reduce infarct size in animal models of ischemia–reperfusion (I/R) injury [5,6]. In this issue of the Journal, Loubele et al. extend these studies by showing that inhibition of the TF–factor VIIa (FVIIa) complex with active site-inhibited mouse FVIIa (ASIS) attenuates activation of nuclear factor kappaB (NF-κB) and the expression of inflammatory mediators in a mousemodel of I/R injury [7]. The question is, does the TF–FVIIa complex enhance inflammation in cardiac I/R injury via its coagulation and/or signaling activity?
I/R injury damages the endothelial barrier and allows leakage of clotting factors into themyocardium [6]. Subsequent contact with TF on cardiomyocytes activates the clotting cascade and leads to the generation of coagulation proteases, such as FVIIa, FXa and thrombin, and ultimately to fibrin deposition within the myocardium [6]. There are several pathways downstream of the TF–FVIIa complex that may explain how this complex enhances inflammation. These include a pathway mediated by a fibrin degradation product called E1, and two other pathways driven by activation of protease-activated receptors (PARs) (Fig. 1). Indeed, PAR1 and PAR2 are widely expressed by cells in the heart [8].
Fig. 1.
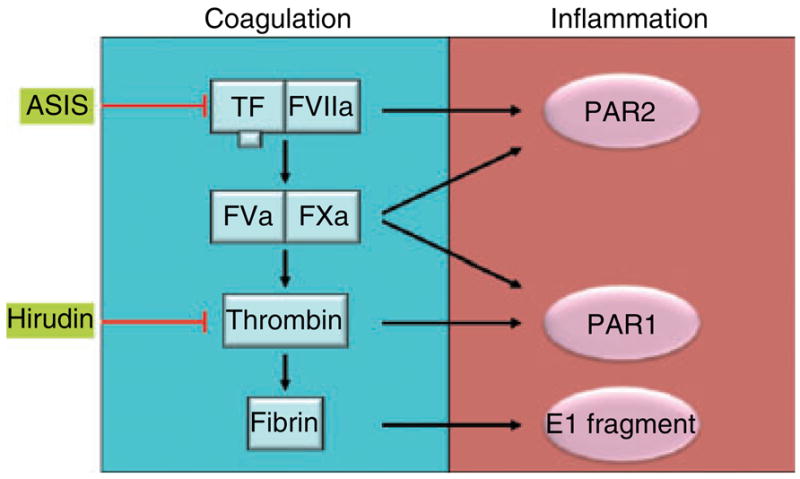
Cross-talk between coagulation and inflammation in cardiac I/R injury may be mediated by multiple pathways.
The E1 fibrin degradation fragment has been shown to facilitate neutrophil infiltration into the myocardium after I/R injury by forming a bridge between leukocytes and endothelial cells [9]. Specifically, the N-terminus of the α-chain of E1 interacts with CD11c on leukocytes, and the N-terminus of the β-chain of E1 interacts with VE-cadherin located at cell–cell junctions between endothelial cells. Inhibition of the TF–FVIIa complex would reduce formation of the E1 fragment and may explain, in part, the anti-inflammatory effects of ASIS. Notably, Loubele et al. observed a reduction in the number of neutrophils in themyocardium of mice treated with ASIS [7].
Inhibition of thrombin with hirudin also reduces myocardial infarction [6,10]. Hirudin would block the generation of the E1 fragment and its inflammatory activity, as well as reduce PAR1 signaling. Strande et al. reported that a PAR1 antagonist called SCH 79797 reduces the infarct size in a rat model of myocardial I/R injury [11]. In contrast, we did not observe any difference in infarct size between wild-type and PAR1 knockout mice, but we observed a reduction in cardiac remodeling in these mice [10]. These results indicate that additional studies using other PAR1 inhibitors are necessary to clarify the role of PAR1 in cardiac I/R injury and to determine whether other members of PAR family, particularly PAR4, can compensate for a lack of PAR1.
Finally, inhibition of the TF–FVIIa complex may reduce pathologic PAR2 signaling [12]. In general, PAR2 signaling has been shown to be proinflammatory. For instance, PAR2 activation on endothelial cells and macrophages induces the expression of proinflammatory cytokines, including interleukin (IL)-1β, tumor necrosis factor-alpha and IL-6 [13–15]. Furthermore, leukocyte rolling and adhesion is reduced in PAR2 knockout mice [16]. These data would support the hypothesis that TF–FVIIa-dependent PAR2 signaling contributes to myocardial infarction during I/R injury by increasing the inflammatory response. Consistently with this notion, our preliminary results indicate that infarct size is significantly reduced in PAR2-deficient mice as compared to wild-type controls (Pawlinski and Mackman, unpublished data).
Loubele et al. found that ASIS attenuated the activation of NF-κB and IL-6 expression [7]. The observed reduction in Toll-like receptor 4 (TLR4) expression led to the suggestion that TF–FVIIa signaling may be linked to TLR4 activation. Indeed, the TLR4 pathway has been shown to contribute to infarction after cardiac I/R injury [17]. Furthermore, activation of TLR4 and PAR2 produces a synergistic induction of cytokines from endothelial cells [13]. The question is, how do TLR4 and PAR2 cooperate? Two recent studies provide a model to explain this cooperation [18,19]. One study demonstrated that responses to PAR2 agonist peptide were significantly diminished in TLR4-deficient macrophages [18]. A second study identified a putative TLR/IL-1 receptor-like domain in the cytoplasmic C-terminus of PAR2 that interacts with TLR4 and showed that mutation of this domain abolished PAR2-dependent activation of NF-κB [19]. Further studies are required to determine whether TLR4 and PAR2 cooperate in a similar manner in the heart after I/R injury.
In conclusion, Loubele et al. provide additional evidence that there is cross-talk between coagulation and inflammation. A limitation of the study is that it does not elucidate the pathways downstream of the TF–FVIIa complex that enhance inflammation. As with all other anticoagulants, the major side-effect of using ASIS would be bleeding, but this was not observed in the study. This issue is particularly important because we and others have shown that TF is required for heart hemostasis [20,21]. Nevertheless, the current study may stimulate the development of new therapies to reduce inflammation after myocardial injury.
Acknowledgments
This publication was supported by a grant from the National Institutes of Health (HL084087-01 to N. Mackman).
Footnotes
Disclosure of Conflict of Interests
The authors state that they have no conflict of interest.
References
- 1.White HD, Chew DP. Acute myocardial infarction. Lancet. 2008;372:570–84. doi: 10.1016/S0140-6736(08)61237-4. [DOI] [PubMed] [Google Scholar]
- 2.Braunwald E, Kloner RA. Myocardial reperfusion: a double-edged sword? J Clin Invest. 1985;76:1713–19. doi: 10.1172/JCI112160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luther T, Mackman N. Tissue factor in the heart. Multiple roles in hemostasis, thrombosis, and inflammation. Trends Cardiovasc Med. 2001;11:307–12. doi: 10.1016/s1050-1738(01)00129-3. [DOI] [PubMed] [Google Scholar]
- 4.Zacharowski K, Zacharowski P, Reingruber S, Petzelbauer P. Fibrin(ogen) and its fragments in the pathophysiology and treatment of myocardial infarction. J Mol Med. 2006;84:469–77. doi: 10.1007/s00109-006-0051-7. [DOI] [PubMed] [Google Scholar]
- 5.Golino P, Ragni M, Cirillo P, Scognamiglio A, Ravera A, Buono C, Guarino A, Piro O, Lambiase C, Botticella F, Ezban M, Condorelli M, Chiariello M. Recombinant human, active site-blocked factor VIIa reduces infarct size and no-reflow phenomenon in rabbits. Am J Physiol Heart Circ Physiol. 2000;278:H1507–16. doi: 10.1152/ajpheart.2000.278.5.H1507. [DOI] [PubMed] [Google Scholar]
- 6.Erlich JH, Boyle EM, Labriola J, Kovacich JC, Santucci RA, Fearns C, Morgan EN, Yun W, Luther T, Kojikawa O, Martin TR, Pohlman TH, Verrier ED, Mackman N. Inhibition of the tissue factor–thrombin pathway limits infarct size after myocardial ischemia–reperfusion injury by reducing inflammation. Am J Pathol. 2000;157:1849–62. doi: 10.1016/S0002-9440(10)64824-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Loubele STBG, Spek CA, Leenders P, van Oerle R, Aberson HL, van der Voort D, Hamulyák K, Petersen LC, Spronk HMH, ten Cate H. Active site inhibited factor VIIa attenuates myocardial ischemia/reperfusion injury in mice. J Thromb Haemost. 2009;7:290–8. doi: 10.1111/j.1538-7836.2008.03232.x. [DOI] [PubMed] [Google Scholar]
- 8.Steinberg SF. The cardiovascular actions of protease-activated receptors. Mol Pharmacol. 2005;67:2–11. doi: 10.1124/mol.104.003103. [DOI] [PubMed] [Google Scholar]
- 9.Petzelbauer P, Zacharowski PA, Miyazaki Y, Friedl P, Wickenhauser G, Castellino FJ, Groger M, Wol K, Zacharowski K. The fibrin-derived peptide Bbeta15-42 protects the myocardium against ischemia–reperfusion injury. Nat Med. 2005;11:298–304. doi: 10.1038/nm1198. [DOI] [PubMed] [Google Scholar]
- 10.Pawlinski R, Tencati M, Hampton CR, Shishido T, Bullard TA, Casey LM, Andrade-Gordon P, Kotzsch M, Spring D, Luther T, Abe J, Pohlman TH, Verrier ED, Blaxall BC, Mackman N. Protease-activated receptor-1 contributes to cardiac remodeling and hypertrophy. Circulation. 2007;116:2298–306. doi: 10.1161/CIRCULATIONAHA.107.692764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Strande JL, Hsu A, Su J, Fu X, Gross GJ, Baker JE. SCH 79797, a selective PAR1 antagonist, limits myocardial ischemia/reperfusion injury in rat hearts. Basic Res Cardiol. 2007;102:350–8. doi: 10.1007/s00395-007-0653-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cottrell GS, Amadesi S, Schmidlin F, Bunnett N. Protease-activated receptor 2: activation, signalling and function. Biochem Soc Trans. 2003;31:1191–7. doi: 10.1042/bst0311191. [DOI] [PubMed] [Google Scholar]
- 13.Chi L, Li Y, Stehno-Bittel L, Gao J, Morrison DC, Stechschulte DJ, Dileepan KN. Interleukin-6 production by endothelial cells via stimulation of protease-activated receptors is amplified by endotoxin and tumor necrosis factor-alpha. J Interferon Cytokine Res. 2001;21:231–40. doi: 10.1089/107999001750169871. [DOI] [PubMed] [Google Scholar]
- 14.Johansson U, Lawson C, Dabare M, Syndercombe-Court D, Newland AC, Howells GL, Macey MG. Human peripheral blood monocytes express protease receptor-2 and respond to receptor activation by production of IL-6, IL-8, and IL-1{beta} J Leukoc Biol. 2005;78:967–75. doi: 10.1189/jlb.0704422. [DOI] [PubMed] [Google Scholar]
- 15.Noorbakhsh F, Vergnolle N, McArthur JC, Silva C, Vodjgani M, Andrade-Gordon P, Hollenberg MD, Power C. Proteinase-activated receptor-2 induction by neuroinflammation prevents neuronal death during HIV infection. J Immunol. 2005;174:7320–9. doi: 10.4049/jimmunol.174.11.7320. [DOI] [PubMed] [Google Scholar]
- 16.Lindner JR, Kahn ML, Coughlin SR, Sambrano GR, Schauble E, Bernstein D, Foy D, Hafezi-Moghadam A, Ley K. Delayed onset of inflammation in protease-activated receptor-2-deficient mice. J Immunol. 2000;165:6504–10. doi: 10.4049/jimmunol.165.11.6504. [DOI] [PubMed] [Google Scholar]
- 17.Frantz S, Ertl G, Bauersachs J. Toll-like receptor signaling in the ischemic heart. Front Biosci. 2008;13:5772–9. doi: 10.2741/3114. [DOI] [PubMed] [Google Scholar]
- 18.Rallabhandi P, Nhu QM, Toshchakov VY, Piao W, Medvedev AE, Hollenberg MD, Fasano A, Vogel SN. Analysis of proteinase-activated receptor 2 and TLR4 signal transduction: a novel paradigm for receptor cooperativity. J Biol Chem. 2008;283:24314–25. doi: 10.1074/jbc.M804800200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramachandran R, Hollenberg MD. Proteinases and signalling: pathophysiological and therapeutic implications via PARs and more. Br J Pharmacol. 2008;153 (Suppl 1):S263–82. doi: 10.1038/sj.bjp.0707507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pawlinski R, Tencati M, Holscher T, Pedersen B, Voet T, Tilley RE, Marynen P, Mackman N. Role of cardiac myocyte tissue factor in heart hemostasis. J Thromb Haemost. 2007;5:1693–700. doi: 10.1111/j.1538-7836.2007.02649.x. [DOI] [PubMed] [Google Scholar]
- 21.Snyder LA, Rudnick KA, Tawadros R, Volk A, Tam SH, Anderson GM, Bugelski PJ, Yang J. Expression of human tissue factor under the control of the mouse tissue factor promoter mediates normal hemostasis in knock-in mice. J Thromb Haemost. 2008;6:306–14. doi: 10.1111/j.1538-7836.2008.02833.x. [DOI] [PubMed] [Google Scholar]