

Abstract
The selective covalent modification of singly protonated peptides in the gas-phase via ion/ion charge inversion reactions is demonstrated. Doubly deprotonated 4-formyl-1,3-benzene disulfonic acid serves as a reagent anion for forming a Schiff base via the reaction of a primary amine on the peptide and the aldehyde functionality of the reagent anion. The process is initiated by the formation of an ion/ion complex comprised to the two reactants. Ion trap collisional activation of the complex results in loss of water from the intermediate that gives rise to Schiff base formation. N-terminally acetylated peptides with no lysine residues do not undergo covalent bond formation upon reaction with the reagent anion. Rather, the adduct species simply loses the reagent either as a neutral species or as a deprotonated species. The ability to modify singly protonated peptide ions covalently and selectively opens up new possibilities for the analysis of peptides, and, possibly, other analyte species with primary amine functionalities.
Keywords: Ion/ion reactions, charge inversion, gas-phase covalent modification
Introduction
The structural characterization of polypeptide ions via tandem mass spectrometry is a central activity in proteomics.1,2 The sequence information that can be generated from a given peptide is determined both by the ion-type (e.g., polarity, charge state, nature of charge bearing site(s), etc.) and the conditions under which the ions are activated. No single ion-type or set of activation conditions is optimal for deriving structural information from all peptides. It is therefore desirable to have the option of generating a variety of ion-types for a given peptide. The ion type is generally determined by the ionization method. Gas-phase ion/ion reactions are proving to be particularly useful in converting ions from one type to another and provide means for decoupling the ionization method from the ion-type subjected to tandem mass spectrometry. Many examples of ion conversions have been demonstrated. These include, inter alia, the reduction of charge via proton transfer,3 the inversion of charge via charge inversion proton transfer,4,5 the increase in charge via double charge inversion,6,7 the replacement of protons with metal ions,8,9 the conversion of polyprotonated peptides to hypervalent radical species via electron transfer,10,11 etc.
By far, most ion/ion reaction transformations have come about via the transfer of charged particles, such as protons, electrons, or metal ions or by the non-covalent attachment of reagent ions to an analyte ion. It has recently been demonstrated that it is also possible to effect selective bond formation within the course of an ion/ion reaction to allow for covalent peptide modification in the gas-phase.12 Specifically, Schiff base formation resulting from the reaction of an aldehyde containing reagent anion (i.e., singly deprotonated 4-formyl-1,3-benzenedisulfonic acid (FBDSA)) with primary amine groups in multiply protonated peptides has been demonstrated. Schiff base formation in small peptide ions has also been observed with ion/molecule reactions involving acetone13 and acetonylacetone.14 In this work, we demonstrate Schiff base formation in polypeptide ions in conjunction with the inversion of charge from positive to negative. The overall process leads exclusively to singly charged anions with the Schiff base modification taking place only with peptides containing a primary amine group. We also demonstrate that collisional activation of the modified peptides generally provides more sequence information than the corresponding singly deprotonated analyte ion. This work demonstrates that gas-phase bio-conjugation via ion/ion reactions can be effected for singly-protonated as well as for more highly protonated species.
Experimental Section
Materials
Methanol, glacial acetic acid, and ammonium hydroxide were obtained from Malinckrodt (Phllipsburg, NJ). KGAILKGAILR was synthesized by SynPep (Dublin, CA). All YGGXXX peptides, where X represents a variable amino acid residue, were purchased from CPC Scientific (San Jose, CA). Angiotensin II and 4-formyl-1,3-benzenedisulfonic acid (FBDSA) were obtained from Sigma (St. Louis, MO). All peptides were used without further purification. The procedure for acetylation of the peptides has been described.15 Peptide ions were produced from a ∼100 μM solution of equal parts of water and methanol with ∼1% acetic acid added for positive nano-electrospray ionization (nano-ESI). Anion reagent was made up of a 3 mM solution of 50/50 (v/v) water/methanol with ∼1% ammonium hydroxide for negative nano-ESI.
Apparatus and Procedures
All experiments were performed on a QqQ tandem mass spectrometer (QTRAP2000, Applied Biosystems/MDS Sciex, Concord, ON, Canada) which was modified for ion/ion reactions.16,17 A home-made ion source allowed for the sequential injections and subsequent reaction of negatively charged reagent ions and positively charged peptide ions.
Reactions were performed by accumulating doubly deprotonated FBDSA in the Q2 cell. The analyte peptide cations, formed via positive nano-ESI, were then transmitted into Q2 where they were then stored with the FBDSA anions for 50-500 ms.18 The product ions were subsequently transferred to the Q3 LIT where further stages of ion isolation and activation could be performed. Finally, mass analysis was performed via mass-selective axial ejection (MSAE).19
Results and Discussion
The ability to form a Schiff base derivative of an ion in the gas phase opens the possibility that various functional groups can be added selectively to analyte ions. Selective gas-phase modification via an ion/ion reaction results in the loss of at least one charge from the analyte ion. This fact may have little consequence for analysis when the analyte ions are multiply-charged but can be problematic when the analyte ions are singly charged due to the fact that they can be neutralized in the modification step. However, singly-charged analyte ions can be modified without being neutralized if the reagent anion is multiply charged. The overall process described here uses doubly deprotonated 4-formyl-1,3-benzenedisulfonic acid (FBDSA) to modify protonated peptides, represented generically as [M+H]+, as summarized in Equation 1 and Scheme 1.
Scheme 1.

Summary of the steps involved in formation of a Schiff base via charge inversion of a singly protonated peptide, with subsequent collisional activation.
| (1) |
The first step in the process is the formation of a binary complex comprised of the two reactants. (Note that some of the analyte ions may be neutralized from a single proton transfer that takes place either via a proton transfer at a crossing point in the interaction potential or via a long-lived complex that serves as an intermediate for proton transfer.4,5) A long-lived complex formed by the attachment of the reagent anion to the peptide may undergo a rearrangement to an intermediate that, upon collisional activation, loses a water molecule to generate the Schiff base (see Scheme 1). This can occur when a primary amine of the peptide, which can be the ε-amino group of a lysine residue or the N-terminus, can interact with the aldehyde group of FBDSA. The (◆) symbol associated with the label of an ion indicates the addition of the expected mass of the Schiff base modification.
In previous charge inversion reaction studies with doubly deprotonated reagent anions, [R-2H]2-, collisional activation of the long-lived complexes observed resulted in loss of the neutral reagent,4 as shown in Equation 2, or loss of the neutral peptide, as shown in Equation 3:
| (2) |
| (3) |
The long-lived complex in this case is an intermediate for proton transfer. However, the observation of the loss of a water molecule, as indicated in Equation (1), cannot be taken as definitive and sufficient evidence for Schiff base formation due to the common observation of water loss from peptide anions and cations. Dissociation of the water loss product provides a much more definitive test. Figure 1a shows the results of an MS3 experiment resulting from the reaction of protonated KGAILKGAILR with doubly-deprotonated FBDSA, isolation of the ion/ion complex, ion trap collision-induced dissociation (CID) to generate a first generation water loss product, and subsequent isolation and ion trap collisional activation of the water loss product. For comparison, Figure 1b shows the ion trap CID spectrum of the Schiff base derivative of KGAILKGAILR formed in solution and subjected to negative nano-ESI. The two spectra are indistinguishable within experimental reproducibility. The spectra indicate that the modification apparently takes place largely at the N-terminus, as suggested by the appearance of b◆ product ions from almost every amide bond cleavage and the absence of any abundant y◆ ions. The strong similarity of the two spectra provides good evidence that the gas-phase process proceeds similarly to the process that takes place in solution.
Figure 1.
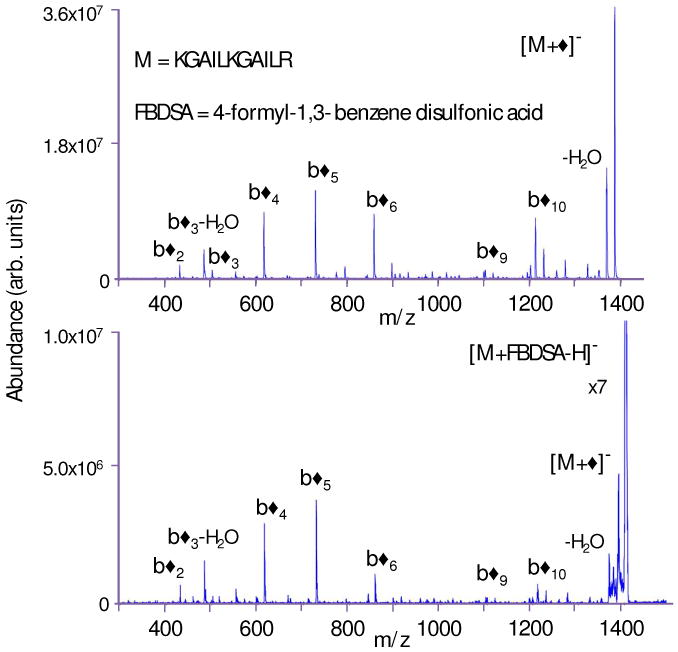
(a) Product ion spectrum from the MS3 experiment involving [M+H]+ + [FBDSA-2H]2- → [M+FBDSA-H]- → [M+◆]-→ products for M= KGAILKGAILR. (b) Ion trap CID MS/MS of the Schiff base product, [M+◆]-, formed in solution. (Note that the [M+FBDSA-H]- ion in Figure 1(b) was present after the isolation step but was not subjected to ion trap CID.)
Schiff base formation involves the reaction of a primary amine with either an aldehyde or ketone. When there are no primary amines in the peptide sequence, Schiff base formation is not expected. Therefore, water loss from the ion/ion complex that is associated with Schiff base formation should not occur, although water loss from other mechanisms is not precluded. Rather, loss of the reagent, either as a neutral species (Equation 2) or as an anion (Equation 3) would be expected to dominate. Control experiments comparing the ion trap CID behavior of the [M+FBDSA-H]- ions of several six residue peptides of the form YGGXXX with their fully acetylated versions were conducted. In all cases, a water loss product from the acetylated versions of the [M+FBDSA-H]- ions, if observed, was at least several times smaller, relative to the product from either process (2) or process (3), than was noted for the non-acetylated versions. Subsequent ion trap CID of the water loss products from the non-acetylated products all showed evidence for Schiff base formation. Several examples of such spectra are provided in Supplementary Information.
It is of interest to consider the efficiency with which Schiff base formation occurs in an ion/ion charge inversion experiment (i.e., the percentage of [M+H]+ ions that are converted to [M+◆]- ions). A number of factors go into the overall efficiency, including the fraction of ions that undergoes an ion/ion reaction, the fraction that undergoes charge inversion (as opposed to neutralization), the fraction that result in the formation of a long-lived complex, the fraction that undergo Schiff base formation within the complex, and the fraction of the complex that undergoes CID. It is possible to ensure that all cations undergo an ion/ion reaction and it is possible to fragment all of the complex ions via ion trap CID. The keys to the maximum achievable efficiency are the fraction of ions that undergo charge inversion to a complex and the fraction of ions in the complex that undergo Schiff base chemistry. For a given reagent dianion, it is possible that these factors could be highly dependent on the size, composition, and sequence of the peptide reactant. We have not yet made systematic studies to explore this possibility. However, we have made several repeated measurements for the ion signals associated with each step for the YGGFLK system. Roughly 6-12% of the signal associated with the [YGGFLK+H]+ ions that underwent an ion/ion reaction was converted to [YGGFLK+ ◆]- signal. This is regarded as an “operational” efficiency because the relative detection efficiencies for positive and negative ions in this instrument are not known. Furthermore, the reagent anion selection window was sufficiently wide to allow significant numbers of other anions to enter the ion trap, which likely reduced the reaction efficiency because the other anions likely resulted in neutralization of the analyte ion. The reagent ion, analyte ion, and post-ion/ion reaction product spectra for singly protonated angiotensin II are provided as Supplementary Information to illustrate typical ion/ion reaction data.
At this point, applications for gas-phase covalent modification of analyte ions have not been developed. Rather, the focus has been on means for selective coupling between reagent ions and analyte ions. However, even at this early stage of development, it is clear that gas-phase modification of analyte ions can lead to improved structural characterization of analyte ions. An example is provided in Figure 2, which compares the ion trap CID spectrum of the deprotonated Schiff base modified version of angiotensin II (DRVYIHPF) (Figure 2a) with that of unmodified deprotonated (Figure 2b) and protonated (Figure 2c) angiotensin II. It is apparent that modification takes place at the N-terminus from the appearance of the series of b◆-ions, which covers much of the sequence of the peptide. The unmodified version of the peptide, on the other hand, shows much less sequence coverage with virtually all the product ion signal concentrated in the y7 product, in the case of the [M-H]- precursor, or the y7 and b6 products in the case of the [M+H]+ precursor. The y7 species are presumably generated via the well-known C-terminal aspartic acid cleavage and the b6 product can arise from the well-known N-terminal proline fragmentation. The appearance of a y◆7-ion in Figure 2a might be interpreted as evidence for Schiff base modification at some location other than the N-terminus. However, it is plausible that some of the precursor ion population consists of adduct species that do not undergo Schiff base formation but lose water upon activation of the [M+FBDSA-H]- ion to give a [M+FBDSA-H2O-H]- complex that does not engage in Schiff base formation. Some of these ions may give rise to a [y7+FBDSA-H2O]- product via the facile process observed for the [M-H]- species. Regardless of the origin of the nominal y◆7-ion, it is clear that the Schiff base modification exerts major influence over the fragmentation of the peptide anion. Further examples of rich primary structure information obtained from fragmentation of the [M+◆]- species derived from several model dodecapeptides are provided in Supplementary Information.
Figure 2.

Ion trap CID product ion spectra of (a) the [M+◆]-, (b) [M-H]-, and (c) [M+H]+ species derived from M=DRVYIHPF (angiotensin II).
Conclusions
Singly protonated peptides with a primary amine group can be selectively modified via ion/ion reaction with doubly deprotonated FBDSA. One of the sulfate groups carries an excess charge while the other serves to deprotonate the peptide. A rearrangement reaction can occur in the negatively charged ion/ion adduct species to give an intermediate that generates a Schiff base adduct on the peptide upon water loss. This reaction occurs between the aldehyde group of the reagent and a primary amine of the peptide. The net result is selective modification of the peptide cation to yield a peptide anion. Subsequent CID of the modified peptide can give greater sequence information than CID of the unmodified version of the anion. Perhaps more significant is that these results suggest that other functionalities might be attached to polypeptide analyte ions via selective Schiff base formation in the gas-phase. As these experiments are conducted within the context of MS3 experiments, they provide a high degree of control over the identities of the reactants and avoid complications that may arise from attempts at covalent modification in solution.
Supplementary Material
Acknowledgments
Research supported by the Office of Basic Energy Sciences, Office of Sciences, U.S. Department of Energy, under Award No. DE-FG02-00ER15105 and the National Institutes of Health under Grant GM 45372.
References
- 1.Yates JR. J Mass Spectrom. 1998;33:1–19. doi: 10.1002/(SICI)1096-9888(199801)33:1<1::AID-JMS624>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 2.Peng JM, Gygi SP. J Mass Spectrom. 2001;36:1083–1091. doi: 10.1002/jms.229. [DOI] [PubMed] [Google Scholar]
- 3.Stephenson JL, Jr, McLuckey SA. J Am Chem Soc. 1996;118:7390–7397. [Google Scholar]
- 4.He M, Emory JF, McLuckey SA. Anal Chem. 2005;77:3173–3182. doi: 10.1021/ac0482312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Emory JF, McLuckey SA. Int J Mass Spectrom. 2008;276:102–109. [Google Scholar]
- 6.He M, McLuckey SA. J Am Chem Soc. 2003;125:7756–7757. doi: 10.1021/ja0354521. [DOI] [PubMed] [Google Scholar]
- 7.He M, McLuckey SA. Anal Chem. 2004;76:4189–4192. doi: 10.1021/ac496087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Newton KA, McLuckey SA. J Am Chem Soc. 2003;125:12404–12405. doi: 10.1021/ja036924e. [DOI] [PubMed] [Google Scholar]
- 9.Gunawardena HP, O'Hair RAJ, McLuckey SA. J Proteome Res. 2006;5:2087–2092. doi: 10.1021/pr0602794. [DOI] [PubMed] [Google Scholar]
- 10.Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Proc Natl Acad Sci USA. 2004;101:9528–9533. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coon JJ, Shabanowitz J, Hunt DF, Syka JEP. J Am Soc Mass Spectrom. 2005;16:880–882. doi: 10.1016/j.jasms.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 12.Han H, McLuckey SA. J Am Chem Soc. 2009;131:12884–12885. doi: 10.1021/ja904812d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O'Hair RAJ, Reid GE. J Am Soc Mass Spectrom. 2000;11:244–256. doi: 10.1016/S1044-0305(99)00142-7. [DOI] [PubMed] [Google Scholar]
- 14.Gur EH, de Koning LJ, Nibbering NMM. Int J Mass Spectrom Ion Processes. 1997;167:135–147. [Google Scholar]
- 15.Reid GE, Simpson RJ, O'Hair RAJ. J Am Soc Mass Spectrom. 1998;9:945–956. [Google Scholar]
- 16.Shevchenko A, Chernushevich I, Ens W, Standing KG, Thomson B, Wilm M, Mann M. Rapid Commun Mass Spectrom. 1997;11:1015–1024. doi: 10.1002/(SICI)1097-0231(19970615)11:9<1015::AID-RCM958>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 17.Xia Y, Chrisman PA, Erickson DE, Liu J, Liang X, Londry FA, Yang MJ, McLuckey SA. Anal Chem. 2006;78:4146–4154. doi: 10.1021/ac0606296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xia Y, Wu J, Londry FA, Hager JW, McLuckey SA. J Am Soc Mass Spectrom. 2005;16:71–81. doi: 10.1016/j.jasms.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 19.Londry FA, Hager JW. J Am Soc Mass Spectrom. 2003;14:1130–1147. doi: 10.1016/S1044-0305(03)00446-X. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.