

Abstract
Several models have been proposed for the mechanism of transcript termination by Escherichia coli RNA polymerase at rho-independent terminators. Yager and von Hippel (Yager, T. D. & von Hippel, P. H. (1991) Biochemistry 30, 1097–118) postulated that the transcription complex is stabilized by enzyme–nucleic acid interactions and the favorable free energy of a 12-bp RNA–DNA hybrid but is destabilized by the free energy required to maintain an extended transcription bubble. Termination, by their model, is viewed simply as displacement of the RNA transcript from the hybrid helix by reformation of the DNA helix. We have proposed an alternative model where the RNA transcript is stably bound to RNA polymerase primarily through interactions with two single-strand specific RNA-binding sites; termination is triggered by formation of an RNA hairpin that reduces binding of the RNA to one RNA-binding site and, ultimately, leads to its ejection from the complex. To distinguish between these models, we have tested whether E. coli RNA polymerase can terminate transcription at rho-independent terminators on single-stranded DNA. RNA polymerase cannot form a transcription bubble on these templates; thus, the Yager–von Hippel model predicts that intrinsic termination will not occur. We find that transcript elongation on single-stranded DNA templates is hindered somewhat by DNA secondary structure. However, E. coli RNA polymerase efficiently terminates and releases transcripts at several rho-independent terminators on such templates at the same positions as termination occurs on duplex DNAs. Therefore, neither the nontranscribed DNA strand nor the transcription bubble is essential for rho-independent termination by E. coli RNA polymerase.
Transcription is a cyclic process composed of four main steps: promoter binding and activation, RNA transcript initiation and promoter escape, RNA transcript elongation, and transcript termination and release. Termination, the last step of the transcription cycle, occurs when the RNA polymerase releases the RNA transcript and dissociates from the DNA template. Release of the RNA transcript is irreversible; any subsequent transcription requires reinitiation at a promoter with formation of a new RNA transcript.
Although most terminators are found at the end of genes, they also occur near promoters and between genes in operons. In prokaryotes, many such termination sites serve as targets for regulation of gene expression (1–4). Although the exact mechanism has not been elucidated, regulation of transcript termination also plays an important role in human development and disease (for review, see ref. 5).
Escherichia coli RNA polymerase can recognize rho-independent terminators intrinsically in vitro in the absence of additional factors. The action of terminators usually depends on formation of an RNA hairpin, although not all RNA hairpins lead to termination. RNA transcript release usually occurs 7–9 nucleotides downstream from the base of the RNA hairpin stem in vitro (4, 6). The basic RNA terminator structure can vary considerably yet still facilitate termination. The RNA hairpin is often G+C-rich and RNA sequence 3′ of the hairpin is often a run of uridine residues, but neither are essential elements (7). An extensive comparison of numerous rho-independent terminators revealed that the length of the RNA hairpin varies from 4 to 11 bp, the size of the loop varies from 3 to 8 nucleotides, and the U-rich region is frequently interrupted with other nucleotides (8–11).
The mechanism by which intrinsic terminators cause stable elongation ternary complexes to cease transcription and release their RNA transcripts is not yet established. Yager and von Hippel (12–14) hypothesized that it is the overall thermodynamic stability of the ternary complex that determines whether RNA polymerase continues transcript elongation or terminates. According to this model, when RNA polymerase encounters an intrinsic termination site, it is destabilized both by the reduction of the RNA–DNA hybrid length and by the need to maintain an extended transcription bubble, causing it to spontaneously dissociate. A second mechanistic model for transcript termination places greater emphasis on RNA polymerase interactions with the nascent transcript (7, 11, 15). According to this second model, upon transcription of rho-independent terminators, formation of the RNA terminator hairpin disrupts the interaction of the RNA transcript with an RNA binding site on the RNA polymerase, leading to release of the transcript.
The goal of our experiments was to distinguish between these two models for transcript termination. The Yager–von Hippel model predicts that rho-independent termination would not occur on single-stranded DNA (ssDNA) templates because of the absence of the nontemplate DNA strand and consequent lack of the transcription bubble. However, the second model predicts that termination should be possible in the presence or absence of the nontemplate strand. To distinguish between these two models, we tested whether E. coli RNA polymerase could terminate transcription at rho-independent terminators on ssDNA templates.
MATERIALS AND METHODS
E. coli RNA Polymerase.
His6-tagged E. coli RNA polymerase holoenzyme was purified from E. coli strain RL721 (provided by R. Landick, University of Wisconsin, Madison). Cell extracts were prepared and were fractionated with polyethylenimine (polymin P) by the method of Burgess and Jendrisak (16). The RNA polymerase was then precipitated from the polymin P eluate by addition of ammonium sulfate at 35 g/100 ml. The resulting pellet was extracted twice with equal volumes of 2.0 M ammonium sulfate, and then RNA polymerase was eluted from the pellet with 1.6 M ammonium sulfate. This eluate was fractionated by chromatography on Ni2+-nitrilotriacetic acid (NTA) agarose, and subsequently holoenzyme was separated from core RNA polymerase by phosphocellulose chromatography (17). The holoenzyme preparation used in these studies contained 60–75% active RNA polymerase relative to the amount of protein added, as determined by the quantitative assay for RNA polymerases (18).
Reagents.
The following reagents were purchased from the sources noted: α-32P-labeled nucleoside triphosphates (NEN or Amersham); HPLC grade nucleoside triphosphates and deoxynucleoside triphosphates (Pharmacia); Hybond N+ nylon membrane (Amersham); dinucleotide ApU, BSA, yeast Torula RNA, polymin P, and rifampicin (Sigma); M-280 streptavidin Dynabeads (Dynal, Oslo); Seq-Light (Tropix, Bedford MA); exonuclease III (Exo III) (Boehringer Mannheim); AmpliTaq (Perkin–Elmer); Ni2+-NTA agarose (Qiagen, Chatsworth CA); phosphocellulose (Whatman); X-Omat scientific imaging film (Kodak); and 5′ biotin phosphoramidite (Glen Research, Sterling, VA).
Buffers.
Buffer composition is as follows: general transcription buffer, TKG-B40M4 [44 mM Tris⋅HCl, pH 8.0/30 mM KCl/4 mM MgCl2/2% (vol/vol) glycerol/acetylated BSA (Ac-BSA; 40 μg/ml; preparation of Ac-BSA from ref.17], TBE buffer (89 mM Tris base/89 mM boric acid/2.5 mM EDTA), and urea/SDS load buffer (10 M urea/0.5% SDS/1× TBE/10 mM EDTA/0.025% xylene cyanol/0.025% bromophenol blue). The variable components of the general transcription buffer are 0, 1, or 4 mM MgCl2 (M1 or M4, respectively), 0 or 350 mM NaCl (N), and Ac-BSA at 40 or 500 μg/ml (B40 or B500, respectively).
DNA Templates.
Oligonucleotides were synthesized by the solid-phase phosphoramidite method on a Biosearch 8750 DNA synthesizer and are designated with the suffix-BT to indicate 5′ biotinylation. SMU2-BT is 5′-BT-CACTATAAGGAGAGACAACT, SMU3 is 5′-CGCGCAAGGAATTTACCAAC, SMU4 is 5′-TTCGCAACGTTCAAATCCGC, END-BT is 5′-BT-CGCCAGGGTTTTCCCAGTCACGAC, and ENUS2 is 5′-ATGCCTGCAGGTCGACTCTAGA. 5′ biotinylated DNA templates were prepared by PCR using one 5′ biotinylated oligonucleotide and one nonbiotinylated oligonucleotide based upon conditions suggested by Higuchi et al. (19). The following plasmid DNAs encoding the indicated terminators were the starting materials for PCR: pA T7 (T7Te, phage T7 early termination site; ref. 20), pSU9–84 (rrnBT1, ribosomal RNA operon T1 terminator; ref. 21), pENtR2 (λ tR2, phage λ termination site R2; a gift from E. Nudler and A. Goldfarb, ref. 22). Transcription was initiated at the bacteriophage T7 A1 promoter for all three terminators.
Semi-Solid State in Vitro Transcription Reactions.
We used two methods to affix ternary complexes to a solid matrix: either immobilization of biotinylated DNA template to M-280 streptavidin Dynabeads or chelation of His6-tagged RNA polymerase to Ni2+-NTA agarose. Biotinylated DNA templates were bound to streptavidin-coated paramagnetic beads (3–4 pmol of DNA template per 100 μg of beads) by using conditions suggested by the manufacturer. The bead-bound DNA template was then used immediately to form open promoter complexes (EPo). To form EPo, 40 nM His6-tagged RNA polymerase was incubated with 20 nM Dynabead-bound DNA template for 10 min at 30°C in TGK-B500M4. Alternatively, 40 nM His6-RNA polymerase was bound to 10 μl of preequilibrated 1:1 Ni2+-NTA agarose slurry per 4 pmol of RNA polymerase in the presence of 80 nM 5′ biotinylated DNA template in TGK-B40M4 for 15 min at room temperature.
Once the EPo was formed, transcription was initiated by addition of the appropriate nucleotides. A typical reaction volume was 100 μl. Ternary complexes are referred to by the transcript length and the 3′ terminal RNA nucleotide; for example, A20 complex has a 20-nucleotide transcript with an AMP as the 3′ terminal nucleotide. To form A20 on either the λ tR2 or rrnBT1 templates, transcription was initiated by adding 50 μM of the dinucleotide ApU, 2 μM ATP, 2 μM GTP, and 2 μM CTP for 5 min at 30°C. To form A11 on the T7Te template, transcription was initiated by adding 250 μM ApU, 10 μM ATP, 10 μM CTP, and 200 μM ITP for 5 min at 30°C (23). To facilitate RNA detection, all reactions also included one α-32P-radiolabeled NTP with a specific activity of 50,000 cpm/pmol. For the λ tR2 and rrnBT1 terminator templates, A20 was walked to G23 by addition of 2 μM ATP, 2 μM GTP, and 2 μM UTP; then to C34 by addition of 2 μM ATP, 2 μM CTP, and 2 μM UTP; then to G44 by addition of 2 μM ATP, 2 μM GTP, and 2 μM CTP; and finally to C47 by addition of 2 μM CTP and 2 μM UTP. C46 was formed on the T7Te template by walking from A11 to U19 by addition of 2 μM GTP, 2 μM CTP, and 2 μM UTP; then to U25 by addition of 2 μM ATP, 2 μM GTP, and 2 μM UTP; then to C32 by addition of 2 μM GTP, 2 μM CTP, and 2 μM UTP; and finally to C46 by addition of 2 μM ATP, 2 μM GTP, and 2 μM CTP. Each of these steps was performed at 30°C for 5 min. Unincorporated NTPs were removed during walking by pelleting the resin either with a magnetic field (Dynabeads) or by gentle centrifugation at 2,000 × g (Ni2+-NTA agarose), withdrawing the supernatant fluid and resuspending the beads in an appropriate buffer. When Dynabeads were used, complexes were washed with three times the original reaction volume of TGK-B500 twice and then resuspended in 1 vol of TGK-B500M4. When Ni2+-NTA agarose was used, the complexes were washed five times with 1 ml of TGK-B40 and then resuspended in 1 vol of TGK-B40M4.
As one measure of ternary complex stability, before and after digestion with Exo III, an aliquot of the reaction was withdrawn and all four nucleotides were added to a final concentration of 500 μM and incubated for 10 min at either 37°C or 42°C (a chase reaction, see figures). Reinitiation and nonspecific transcription was prevented by the addition of rifampicin at 20 μg/ml. The chase reactions were performed in either TGK-B40M1 or TGK-B40M4 and when Ni2+-NTA agarose was used, the buffer also included yeast Torula RNA at 0.8 mg/ml. Terminated RNA transcripts released into the supernatant fluid were precipitated by addition of ammonium acetate to 1 M, yeast Torula RNA to 40 μg/ml, and ethanol to 70% (vol/vol). Individual aliquots of the transcription reactions were stopped by the addition of an equal volume of urea/SDS load buffer, heated to 95°C for 3 min, chilled on ice, and then loaded directly onto a 7 M urea/15% polyacrylamide [38:2 acrylamide/N,N′-methylene-bisacrylamide] gel. Electrophoresis was carried out at 1,800 V, until the xylene cyanol migrated 26 cm from the wells (about 4.5 h). The gel was then exposed to Kodak X-Omat scientific imaging film with an intensifying screen at −80°C overnight. Termination efficiency (%T) was calculated as described (ref. 11, see also legend to Table 1) by using a Molecular Dynamics Storm Phosphorimaging system and imagequant software.
Table 1.
Quantitative comparison of termination efficiencies on ssDNA and dsDNA templates
| Terminator | % reaches terminator
|
% termination
|
||
|---|---|---|---|---|
| dsDNA | ssDNA | dsDNA | ssDNA | |
| λtR2 | 97.9 ± 0.5 (2) | 25.9 (1) | 59.0 ± 0.6 (2) | 69.5 ± 7.3 (4) |
| T7Te | 97.6 (1) | 7.1 (1) | 57.5 (1) | 65.2 (1) |
| rrnBT1 | 86.4 ± 2.2 (3) | 11.9 ± 4.1 (3) | 81.8 ± 6.2 (3) | 59.5 ± 5.8 (3) |
In vitro termination efficiencies for each terminator on ssDNA or dsDNA templates were determined by using a Molecular Dynamics Storm Phosphorimaging system and imagequant software. The calculation of termination efficiency was based upon quantitation of transcripts longer than expected for termination (run-off) compared to the quantitation of the 104- and 105-nucleotide transcripts of λtR2, the 95- and 96-nucleotide transcripts of T7Te, and the 132- to 134-nucleotide transcripts of rrnBT1. % reaches terminator is the percentage of RNA polymerase that have transcripts shorter than the length expected for the termination site after the chase reaction is completed. Data were taken from analysis of the number of independent reactions indicated within parenthesis and presented as the mean ± SD, where indicated. Errors were calculated as sample standard deviation. Large standard deviations of the rrnBT1 data reflect difficulty in resolution of slightly shorter RNA transcripts, which were inefficiently released from the efficiently released 132- to 134-nucleotide terminated transcripts.
Exo III Digestion.
Exo III digestion was usually performed at 2,000–5,000 units/ml, in TGK-B40M1 or TGK-B40M4 at either 30°C or 37°C, as indicated. Digestion was quenched by washing the ternary complexes with TGK-B40 or TGK-B40M4. Residual EPo were disrupted by high salt treatment (either TGK-B40N or TGK-B500N) immediately prior to digestion by Exo III. To generate and then determine the extent of ssDNA template downstream of the halted ternary complex, the following steps were always performed in sequence: (i) formation of ternary complexes; (ii) Exo III digestion; (iii) separation of the DNA fragments by 6% polyacrylamide [38:2 acrylamide/N,N′-methylene-bisacrylamide]/7 M urea PAGE gels at 45 W until the xylene cyanol had migrated from 28 to 34 cm, depending upon the length of the DNA template; and (iv) detection of DNA fragments using chemiluminescence.
Chemiluminescence Detection.
To determine the extent Exo III digested the DNA template, we used the Seq-Light kit (Tropix) and followed the manufacturer’s instructions. Briefly, this involved transferring the DNA fragments from the 6% urea PAGE gel to Hybond N+ nylon membrane by capillary transfer. The biotinylated DNA fragments were then UV cross-linked to the membrane (120 mJ/cm2). Detection of the biotinylated DNA was facilitated by binding a streptavidin-alkaline phosphatase conjugate to it, followed by exposure to the alkaline phosphatase substrate CSPD (Tropix). Upon dephosphorylation by alkaline phosphatase, CSPD decomposes generating visible light that can be detected with Kodak X-Omat scientific imaging film. A typical exposure time was 10 min, between 4 and 6 h after addition of CSPD.
RESULTS
Experimental Design.
To study rho-independent termination of E. coli RNA polymerase on ssDNA templates, we took the approach diagrammed in Fig. 1A. These experiments used PCR-generated duplex DNA templates containing a promoter and an intrinsic termination site. First, RNA polymerase elongation complexes were walked 46 or 47 nucleotides from the start site by sequentially adding and removing different subsets of nucleotides. This forms a stable ternary complex positioned from 50 to 90 nucleotides from the actual site of termination. Next, we added E. coli Exo III, a processive 3′ to 5′ double-strand specific exonuclease (26) to digest both the nontemplate and template strands from the 3′ ends up to the halted RNA polymerase (27). We then washed away the exonuclease and added all four nucleotides (a chase reaction) to enable RNA polymerase to resume transcript elongation on the remaining single-stranded template DNA strand. Because we used linear DNA templates, those elongation complexes that did not release at the intrinsic termination signal would be expected to run off the end of the remaining DNA template. %T at the single-stranded intrinsic termination site was compared with termination on double-stranded DNA (dsDNA) templates by using denaturing gel electrophoresis. Note that in all cases, RNA polymerase had already moved entirely downstream of the remaining duplex DNA region, upon transcribing the termination signal on the ssDNA template.
Figure 1.
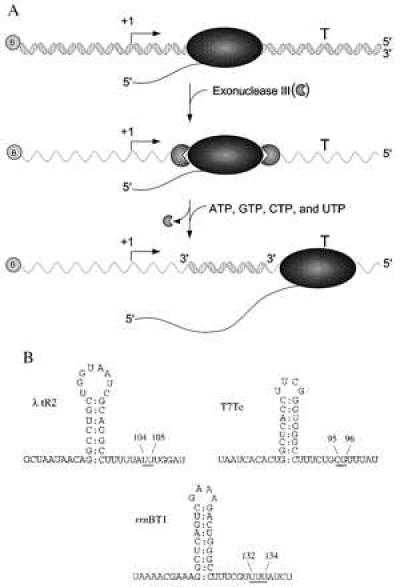
Diagram of experimental design and intrinsic terminator RNA secondary structures. (A) Transcription was initiated at the promoter (bent rightward arrow) and the ternary complexes were walked proximal to the termination site (T) on double-stranded PCR-generated templates that were biotinylated (shaded circle containing the letter B) at one of the 5′ ends. Next, the stalled elongation complexes (shaded ellipse) were digested with Exo III (shaded circles, missing a piece) and then all four NTPs were added, allowing the stalled elongation complex to resume transcript elongation. 5′ and 3′ ends of the DNA are as indicated. (B) The RNA secondary structures of the three intrinsic terminators, λ tR2, T7Te, and rrnBT1, are shown. The underlined bases indicate the positions of transcript release and the length of the RNA product [λ tR2 and rrnBT1 (ref. 24, T. Kerppola and M.J.C., unpublished results) and T7Te (25)]. The RNA secondary structures are drawn to conform to the classical model of transcript termination, with a G+C-rich stem and U-rich region immediately downstream. It should be noted, however, that for the λ tR2 and the rrnBT1 terminators, the hairpin can be extended to include upstream adenosines and downstream uridines.
E. coli RNA Polymerase Terminates Transcription at the Bacteriophage λ tR2 Terminator on ssDNA Templates.
We initially examined termination at the bacteriophage λ tR2 intrinsic termination site on dsDNA (Fig. 2A). The λ tR2 template was designed so that termination at the λ tR2 site generated two RNAs, 104 and 105 nucleotides long (Fig. 1B and refs. 22 and 29). Any ternary complexes that failed to terminate at λ tR2 continued elongation until they reached the end of the DNA template, 177 nucleotides from the T7 A1 promoter. We formed C47 by sequentially walking the RNA polymerase from A20 to C47 (Fig. 2A, lane 1) by using streptavidin-coated paramagnetic beads. When all four nucleotides were added to C47 before Exo III digestion, three RNAs, 104, 105, and 177 nucleotides long, were generated (Fig. 2A, lane 2), corresponding to the expected lengths of the two terminated transcripts and the run-off transcript, respectively.
Figure 2.
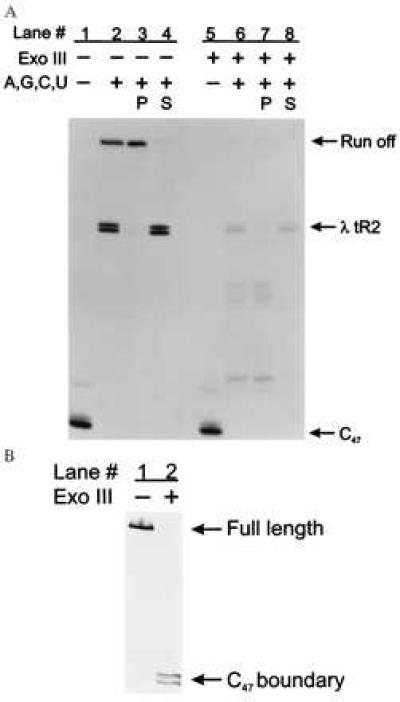
Assaying for intrinsic termination at the λ tR2 terminator on both ss- and dsDNA templates. (A) Stalled C47 complexes (lanes 1 and 5) in TGK-B40M1 were chased with 500 μM ATP, 500 μM GTP, 500 μM CTP, and 500 μM UTP for 10 min at 42°C in the presence of rifampicin at 20 μg/ml (lanes 2–4 and 6–8). Lanes 2 and 6 are aliquots of the chase reactions before the pellet was separated from the supernatant fluid, lanes 3 and 7 are the RNAs associated with the Dynabeads (P), and lanes 4 and 8 are the RNAs released into the supernatant fluid (S). The λ tR2 terminator is at positions +104 to +105 relative to the transcription start site, whereas the end of the DNA template or run-off occurs at position +177. Lanes: 1–4, no Exo III; 5–8, digested with Exo III (2,000 units/ml) for 5 min at 30°C. Quantitation of the total counts within a lane between the C47 complex and the run-off transcript revealed that equivalent counts were present in lanes 1 and 2 and in lanes 5 and 6. One interesting observation we made was that the majority of the run-off transcripts, particularly in the case of the double-stranded chase reaction, remained associated with the pellet (compare lanes 3 and 4). A plausible explanation for this is that the RNAs associated with the pellet are part of ternary complexes that have arrested proximal to the end of the linear DNA template. This type of phenomenon has also been reported for mammalian RNA polymerase II (28). (B) Exo III-digested DNA was analyzed on a 6% denaturing PAGE gel and detected by chemiluminescence. The nontemplate strand was end-labeled with on the 5′ end with a biotin moiety. Lanes: 1, no Exo III; 2, Exo III at 2,000 units/ml for 5 min at 30°C. The initial DNA template, 315 nucleotides long was reduced to approximately 204 nucleotides after Exo III digestion.
To determine whether the 104- and 105-nucleotide RNAs were due to transcript termination and release at the λ tR2 site, we took advantage of the properties of the solid-phase transcription system. We separated any remaining bead-bound ternary complexes from the RNA transcripts released into the supernatant fluid and then loaded the RNAs present in the pellet (P, lane 3) and the supernatant fluid (S, lane 4) in separate lanes for denaturing electrophoresis. As expected, the 104- and 105-nucleotide RNAs were found in the supernatant fluid (compare lanes 3 and 4), indicating they were terminated and released at the λ tR2 terminator.
We then tested the ability of Exo III to quantitatively remove the nontemplate DNA strand downstream of the C47 complex. We followed the extent of Exo III digestion by determining the length of the individual biotinylated DNA strands before and after digestion. The initial DNA template containing the λ tR2 termination site was 315 nucleotides long (Fig. 2B, lane 1). However, after Exo III treatment, the 315-nucleotide nontemplate DNA was quantitatively shortened to approximately 204 nucleotides, the size expected if further digestion was inhibited by the halted C47 RNA polymerase elongation complexes (Fig. 2B, lane 2).
When C47 complexes resumed elongation on the remaining ssDNA (Fig. 2A, lane 6), the 104- and 105-nucleotide RNAs characteristic of termination at λ tR2 were clearly seen, whereas very little of the 177-nucleotide run-off product was produced (Fig. 2A, lane 6). As was the case for termination on dsDNA, the majority of the 104- and 105-nucleotide RNAs were present in the supernatant fluid (Fig. 2A, compare lanes 3 and 4 with lanes 7 and 8). Quantitative measurement of the relative levels of terminated products compared with the run-off product reveals that %T at λ tR2 on ssDNA is about 70% whereas %T on dsDNA is about 60% under these reaction conditions (Table 1). %T was calculated as described (11) by using a Molecular Dynamics Storm Phosphorimaging system and imagequant software.
We observed one significant difference between the dsDNA and ssDNA chase reactions. In contrast to the results seen with dsDNA templates, a substantial fraction of the elongation complexes failed to reach the λ tR2 termination site during the 10-min chase on ssDNA (Fig. 2A, compare lanes 2 and 6, and Table 1, compare % reaches terminator for dsDNA and ssDNA reactions). These shorter RNAs were found in the bead pellet rather than the supernatant fluid (Fig. 2A, compare lanes 7 and 8), and we attribute these RNAs to either paused or arrested elongation complexes. The fraction of these prematurely blocked elongation complexes increased when the distance between the halted ternary complex and the terminator was increased. This effect could be lessened by increasing the temperature during the chase (data not shown). Therefore, we believe that these blocks to elongation were caused in part by DNA secondary structure impeding the progress of RNA polymerase. As an additional test of the specificity of the release of the 104- and 105-nucleotide RNAs, we performed transcription on a DNA template that was identical to the λ tR2 fragment except that 22 nucleotides encoding the stem loop of the terminator had been deleted. Elimination of the terminator hairpin abolished intrinsic termination on both dsDNA and ssDNA templates but did not affect the increased incidence of elongation blocks on ssDNA templates (data not shown).
E. coli RNA Polymerase Terminates Transcription at the Bacteriophage T7Te and the rrnBT1 Terminators on ssDNA Templates.
To test the generality of termination on ssDNA templates, we examined two additional well-characterized intrinsic terminators, the bacteriophage T7 early terminator (T7Te, see Fig. 1B) and the ribosomal RNA operon T1 terminator (rrnBT1, see Fig. 1B). Comparisons of intrinsic termination at the T7Te and the rrnBT1 termination sites on dsDNA and ssDNA templates are shown in Figs. 3 and 4, respectively. Both terminators induce RNA polymerase to release RNA transcripts on dsDNA templates (Figs. 3 and 4, lanes 2–4) and ssDNA templates (Figs. 3 and 4, lanes 6–8). Thus, rho-independent termination on ssDNA is not limited to the λ tR2 terminator but is a more general phenomenon. Additionally, the %T of T7Te and rrnBT1 when single-stranded was about 65% and 60%, respectively, compared with 58% and 82% when double-stranded (Table 1). Therefore, as was observed for λ tR2, termination at T7Te on ssDNA templates was just as great or greater than the efficiency on dsDNA templates.
Figure 3.
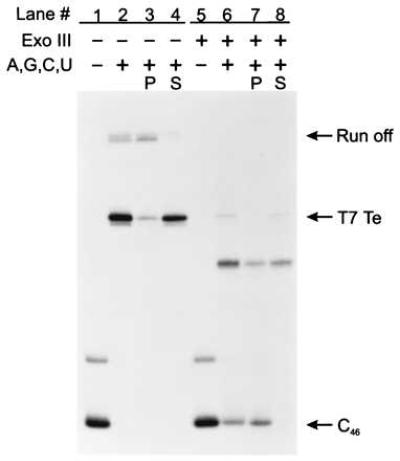
Assaying for intrinsic transcript termination at T7Te on ssDNA. C46 complexes bound to Ni2+-NTA agarose in TGK-B40M4 were chased with 500 μM ATP, 500 μM GTP, 500 μM CTP, and 500 μM UTP for 10 min at 37°C in the presence of rifampicin at 20 μg/ml and yeast Torula RNA at 0.8 mg/ml. The lanes are assigned as for Fig. 2A. The T7Te terminator is at positions +95 and +96, whereas transcription to the end of the DNA template generates a run-off RNA of +146 nucleotides. C46 complexes in lanes 5–8 were digested with Exo III at 5,000 units/ml for 5 min at 37°C. Unlike C47, some of the C46 complexes failed to resume elongation after treatment with Exo III (see lanes 6 and 7). The mechanism of this inactivation is unknown but similar observations have been made by others (29, 30).
Figure 4.
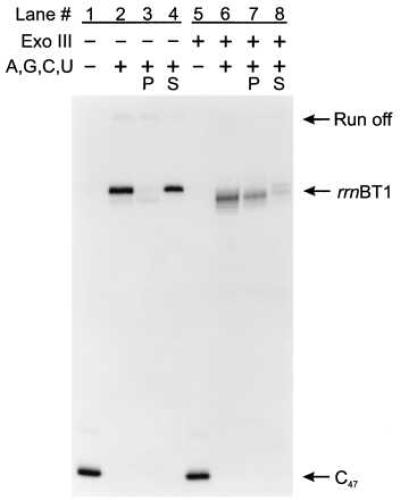
Assaying for intrinsic transcript termination at rrnBT1 on ssDNA. C47 complexes bound to Ni2+-NTA agarose in TGK-B40M4 were chased with 500 μM ATP, 500 μM GTP, 500 μM CTP, and 500 μM UTP for 10 min at 37°C in the presence of rifampicin at 20 μg/ml and yeast Torula RNA at 0.8 mg/ml. The lanes are assigned as for Fig. 2A. The rrnBT1 terminator is at positions +132, +133, and +134, whereas transcription to the end of the DNA template generates a run-off RNA of 205 nucleotides. C47 complexes in lanes 5–8 were digested with Exo III at 5,000 units/ml for 5 min at 37°C.
There were two noticeable differences between termination at the T7Te and rrnBT1 sites and termination at the λ tR2 site in the absence of the nontemplate strand. (i) We observed an additional site of partial transcript release upstream of the T7Te terminator. On the basis of the transcript length, we have mapped this site to position G83 and U84 (data not shown), both of which occur within the stem of the terminator hairpin. Unlike the quantitative transcript release observed at the test terminators on ssDNA, transcript release at G83 and U84 was inefficient. (ii) We observed an increase in the distribution of RNA sizes produced during the ssDNA rrnBT1 chase reaction as compared with the dsDNA chase reaction (Fig. 4, compare lanes 2 and 6). Whereas termination on dsDNA rrnBT1 templates resulted in three released RNAs, 132–134 nucleotides long (Fig. 4, lanes 2–4), termination on ssDNA templates generated RNAs ranging in size from 111 to 134 nucleotides (Fig. 4, lanes 6–8). The majority of the 132- to 134-nucleotide transcripts were released into the supernatant fluid but most of the shorter 111- to 131-nucleotide RNA transcripts remained associated with the Ni2+-NTA agarose pellet (Fig. 4, compare lanes 7 and 8).
DISCUSSION
To study the mechanism of rho-independent transcript termination by E. coli RNA polymerase, we have examined termination at the λ tR2, T7Te, and rrnBT1 intrinsic termination sites. At each of these rho-independent termination sites E. coli RNA polymerase terminated and released specific transcripts on ssDNA and dsDNA templates. Termination by E. coli RNA polymerase on ssDNA templates behaved very similarly to termination on dsDNA templates: (i) the RNA transcripts released from the ssDNA λ tR2 and T7Te terminators were identical to those released on dsDNA templates and (ii) termination at two of these rho-independent sites was somewhat more efficient on ssDNA templates. There were some notable differences between termination on dsDNA and ssDNA templates, as well: (i) less RNA polymerase reached the terminator and (ii) the positions of transcript release at the rrnBT1 site were more heterogeneous on ssDNA than on dsDNA templates.
Recently, it was reported that T7 RNA polymerase terminates transcription intrinsically at the rrnBT1 termination site on both ssDNA and dsDNA templates (31). In addition, the N4 virion RNA polymerase terminates transcription at rho-independent terminators on ssDNA templates (32, 33). Therefore, the ability of ternary complexes to terminate transcription at rho-independent terminators on ssDNA appears to be a general characteristic of RNA polymerases.
The Yager–von Hippel transcript termination model proposes that the overall thermodynamic stability of the ternary complex governs whether RNA polymerase continues transcript elongation or terminates (12–14). In this model, the stability of a ternary complex (standard Gibbs free energy of the elongation complex formation, ΔG°f,complex) is described by the following thermodynamic stability function:
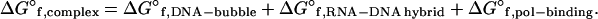 |
During transcript elongation when the transcription complex is very stable, the unfavorable free energy of maintaining a 17-bp transcription bubble is outweighed by the favorable free energies of the proposed 12-bp RNA–DNA hybrid, ΔG°f,RNA-DNA hybrid, and the interaction of the RNA polymerase with the nucleic acid components of the ternary complex, ΔG°f,pol-binding. Termination is caused when terminator hairpin formation reduces the length of the RNA–DNA hybrid, leaving only the 3′ proximal U-rich region within the RNA–DNA hybrid. Because rU⋅dA base pairs are particularly unstable (34), the unfavorable energy of maintaining the transcription bubble becomes dominant. This dramatically reduces the stability of the ternary complex (ΔG°f,complex ∼ 0) and causes the RNA polymerase to release its transcript. Because the only unfavorable free energy source is ΔG°f,DNA-bubble, this model requires that rho-independent termination could not occur on ssDNA templates.
Our results demonstrate that the nontemplate strand and ΔG°f,DNA-bubble are not essential parts of the intrinsic termination signal or its action. Therefore, the multipartite intrinsic signal that induces RNA polymerase to terminate transcription must be mediated by RNA polymerase interacting with the nascent RNA transcript and the DNA template strand. How might the components of the rho-independent terminator signal the elongating RNA polymerase to release its RNA transcript? Recent evidence suggests that E. coli RNA polymerase contains two nonionic single-strand specific RNA binding sites (15, 35). Moreover, studies of binary complexes between RNA polymerase and RNA have determined that RNAs containing secondary structure bind more poorly to RNA polymerase than RNAs without hairpins. However, hairpin containing RNAs can be bound by RNA polymerase if there is a region of unstructured RNA on either side of the hairpin (35, 36). Because of the specificity for unstructured RNA of the RNA binding sites, it seems reasonable that the terminator RNA hairpin serves to extricate or impede binding of the RNA transcript to one or both of these RNA binding sites.
Acknowledgments
This work was supported by an National Institutes of Health training grant (GM 07232) and a research grant (GM 12010) from National Institute of General Medical Sciences to M.J.C.
ABBREVIATIONS
- dsDNA
double-stranded DNA
- ssDNA
single-stranded DNA
- rrnBT1
ribosomal RNA operon T1 terminator
- T7Te
phage T7 early termination site
- λ tR2
phage λ termination site R2
- Exo III
exonuclease III
- %T
termination efficiency
- ΔG°f,complex
standard Gibbs free energy of the elongation complex formation
- NTA
nitrilotriacetic acid
References
- 1.Henkin T M. Annu Rev Genet. 1996;30:35–57. doi: 10.1146/annurev.genet.30.1.35. [DOI] [PubMed] [Google Scholar]
- 2.Landick R, Turnbough C L, Jr, Yanofsky C. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. Neidhardt F C, editor. Vol. 1. Washington, DC: Am. Soc. Microbiol.; 1996. pp. 1263–1286. [Google Scholar]
- 3.Friedman D I, Court D L. Mol Microbiol. 1995;18:191–200. doi: 10.1111/j.1365-2958.1995.mmi_18020191.x. [DOI] [PubMed] [Google Scholar]
- 4.Richardson J P, Greenblatt J. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. Neidhardt F C, editor. Vol. 1. Washington, DC: Am. Soc. Microbiol.; 1996. pp. 822–848. [Google Scholar]
- 5.Uptain S M, Kane C M, Chamberlin M J. Annu Rev Biochem. 1997;66:117–172. doi: 10.1146/annurev.biochem.66.1.117. [DOI] [PubMed] [Google Scholar]
- 6.Roberts J W. In: Regulation of Gene Expression in Escherichia coli. Lin E C C, Lynch A S, editors. Austin, TX: R. G. Landes; 1996. pp. 27–45. [Google Scholar]
- 7.Reynolds R, Chamberlin M J. J Mol Biol. 1992;224:53–63. doi: 10.1016/0022-2836(92)90575-5. [DOI] [PubMed] [Google Scholar]
- 8.Brendel V, Trifonov E N. Nucleic Acids Res. 1984;12:4411–4427. doi: 10.1093/nar/12.10.4411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brendel V, Hamm G H, Trifonov E N. J Biomol Struct Dyn. 1986;3:705–723. doi: 10.1080/07391102.1986.10508457. [DOI] [PubMed] [Google Scholar]
- 10.d’Aubenton Carafa Y, Brody E, Thermes C. J Mol Biol. 1990;216:835–858. doi: 10.1016/s0022-2836(99)80005-9. [DOI] [PubMed] [Google Scholar]
- 11.Reynolds R, Bermudez-Cruz R M, Chamberlin M J. J Mol Biol. 1992;224:31–51. doi: 10.1016/0022-2836(92)90574-4. [DOI] [PubMed] [Google Scholar]
- 12.Yager T D, von Hippel P H. Biochemistry. 1991;30:1097–1118. doi: 10.1021/bi00218a032. [DOI] [PubMed] [Google Scholar]
- 13.von Hippel P H, Yager T D. Proc Natl Acad Sci USA. 1991;88:2307–2311. doi: 10.1073/pnas.88.6.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.von Hippel P H, Yager T D. Science. 1992;255:809–812. doi: 10.1126/science.1536005. [DOI] [PubMed] [Google Scholar]
- 15.Chamberlin M J. Harvey Lect. 1995;88:1–21. [PubMed] [Google Scholar]
- 16.Burgess R R, Jendrisak J J. Biochemistry. 1975;14:4634–4638. doi: 10.1021/bi00692a011. [DOI] [PubMed] [Google Scholar]
- 17.Gonzalez N, Wiggs J, Chamberlin M J. Arch Biochem Biophys. 1977;182:404–408. doi: 10.1016/0003-9861(77)90521-5. [DOI] [PubMed] [Google Scholar]
- 18.Chamberlin M J, Nierman W C, Wiggs J, Neff N. J Biol Chem. 1979;254:10061–10069. [PubMed] [Google Scholar]
- 19.Higuchi R, Krummel B, Saiki R K. Nucleic Acids Res. 1988;16:7351–7367. doi: 10.1093/nar/16.15.7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Telesnitsky A P W, Chamberlin M J. J Mol Biol. 1989;205:315–330. doi: 10.1016/0022-2836(89)90343-4. [DOI] [PubMed] [Google Scholar]
- 21.Arndt K M, Chamberlin M J. J Mol Biol. 1988;202:271–285. doi: 10.1016/0022-2836(88)90457-3. [DOI] [PubMed] [Google Scholar]
- 22.Nudler E, Goldfarb A, Kashlev M. Science. 1994;265:793–796. doi: 10.1126/science.8047884. [DOI] [PubMed] [Google Scholar]
- 23.Krummel B, Chamberlin M J. J Mol Biol. 1992;225:221–237. doi: 10.1016/0022-2836(92)90917-9. [DOI] [PubMed] [Google Scholar]
- 24.Schmidt M C, Chamberlin M J. J Mol Biol. 1987;195:809–818. doi: 10.1016/0022-2836(87)90486-4. [DOI] [PubMed] [Google Scholar]
- 25.Briat J F, Chamberlin M J. Proc Natl Acad Sci USA. 1984;81:7373–7377. doi: 10.1073/pnas.81.23.7373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rogers S G, Weiss B. Methods Enzymol. 1980;65:201–211. doi: 10.1016/s0076-6879(80)65028-9. [DOI] [PubMed] [Google Scholar]
- 27.Landick R, Yanofsky C. J Mol Biol. 1987;196:363–377. doi: 10.1016/0022-2836(87)90697-8. [DOI] [PubMed] [Google Scholar]
- 28.Izban M G, Samkurashvili I, Luse D S. J Biol Chem. 1995;270:2290–2297. doi: 10.1074/jbc.270.5.2290. [DOI] [PubMed] [Google Scholar]
- 29.Nudler E, Kashlev M, Nikiforov V, Goldfarb A. Cell. 1995;81:351–357. doi: 10.1016/0092-8674(95)90388-7. [DOI] [PubMed] [Google Scholar]
- 30.Wang D, Meier T I, Chan C L, Feng G, Lee D N, Landick R. Cell. 1995;81:341–350. doi: 10.1016/0092-8674(95)90387-9. [DOI] [PubMed] [Google Scholar]
- 31.Hartvig L, Christiansen J. EMBO J. 1996;15:4767–4774. [PMC free article] [PubMed] [Google Scholar]
- 32.Markiewicz P, Malone C, Chase J W, Rothman-Denes L B. Genes Dev. 1992;6:2010–2019. doi: 10.1101/gad.6.10.2010. [DOI] [PubMed] [Google Scholar]
- 33.Dai X, Greizerstein M B, Nadas-Chinni K, Rothman-Denes L B. Proc Natl Acad Sci USA. 1997;94:2174–2179. doi: 10.1073/pnas.94.6.2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martin F H, Tinoco I., Jr Nucleic Acids Res. 1980;8:2295–2299. doi: 10.1093/nar/8.10.2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Altmann C R, Solow-Cordero D E, Chamberlin M J. Proc Natl Acad Sci USA. 1994;91:3784–3788. doi: 10.1073/pnas.91.9.3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Altmann C R. Ph.D. thesis. Berkeley, CA: Univ. of California; 1994. pp. 1–171. [Google Scholar]