

Abstract
LPS stimulation of monocytes/macrophages induces the expression of genes encoding proinflammatory cytokines and the procoagulant protein, tissue factor. Induction of these genes is mediated by various signaling pathways, including mitogen-activated protein kinases, and several transcription factors, including Egr-1, AP-1, ATF-2, and NF-κB. We used a genetic approach to determine the role of the phosphatidylinositol-3-kinase (PI3K)-protein kinase B (Akt) pathway in the regulation of LPS signaling and gene expression in isolated macrophages and in mice. The PI3K-Akt pathway is negatively regulated by the phosphatase and tensin homologue (PTEN). We used peritoneal exudate cells from Pik3r1-deficient mice, which lack the p85α regulatory subunit of PI3K and have reduced PI3K activity, and peritoneal macrophages from PTENflox/flox/LysMCre mice (PTEN−/−), which have increased Akt activity. Analysis of LPS signaling in Pik3r1−/− and PTEN−/− cells indicated that the PI3K-Akt pathway inhibited activation of the ERK1/2, JNK1/2, and p38 mitogen-activated protein kinases and reduced the levels of nuclear Egr-1 protein and phosphorylated ATF-2. Modulating the PI3K-Akt pathway did not affect LPS-induced degradation of IκBα or NF-κB nuclear translocation. LPS induction of TNF-α, IL-6, and tissue factor gene expression was increased in Pik3r1−/− peritoneal exudate cells and decreased in PTEN−/− peritoneal macrophages compared with wild-type (WT) cells. Furthermore, LPS-induced inflammation and coagulation were enhanced in WT mice containing Pik3r1−/− bone marrow compared with WT mice containing WT bone marrow and in mice lacking the p85α subunit in all cells. Taken together, our results indicate that the PI3K-Akt pathway negatively regulates LPS signaling and gene expression in monocytes/macrophages.
Bacterial LPS is a component of the Gram-negative bacterial cell membrane. LPS engagement of the CD14/TLR4/MD2 receptor complex on monocytes/macrophages leads to the phosphorylation of various intracellular kinases, including the IκBβ kinase (IKK)7 and the ERK1/2, p38, and JNK1/2 mitogen-activated protein kinases (MAPKs) (1, 2). Activation of these intracellular signaling pathways results in the activation of a variety of transcription factors, including ATF-2, AP-1, Egr-1, and NF-κB, and the induction of genes encoding proinflammatory cytokines, such as TNF-α and IL-6 (1, 3). In addition, activated monocytes/macrophages express the procoagulant transmembrane protein tissue factor (TF) (4).
Several studies have shown that LPS also activates the phosphatidylinositol-3-kinase (PI3K)-protein kinase B (Akt) pathway in monocytes/macrophages (5–7). The PI3K family is divided into four classes: IA, IB, II and III (8). Recent studies using siRNA silencing of the PI3K catalytic subunit p110α have shown that LPS activation of Akt in monocytic cells is mediated by class IA PI3K (6). This class consists of 85kD regulatory subunits and 110 kD catalytic subunits, which catalyze the phosphorylation of PI(4,5)P2 to the lipid second messenger PI(3,4,5)P3 (PIP3). Binding of PDK-1 and Akt to PIP3 leads to the phosphorylation of Akt. The PI3K-Akt pathway is negatively regulated by the phosphatase and tensin homologue (PTEN), which converts PIP3 to PI(4,5)P2. The PI3K-Akt pathway has been shown to control a variety of cellular processes, including cell survival and proliferation (8). Wortmannin and LY294002 are pharmacologic inhibitors of PI3K. These compounds have been used extensively to analyze the role of PI3K in the regulation of different intracellular pathways (9, 10). However, wortmannin and LY294002 have been reported to inhibit other kinases.
We and others (5, 7, 11–13) have found that inhibition of PI3K with wortmannin and LY294002 enhanced LPS activation of MAPKs (i.e., ERK1/2, JNK1/2, and p38), various transcription factors (i.e., AP-1, Egr-1, and NF-κB), and gene expression (i.e., TNF-α and TF) in monocytic cells. In addition, we found that a dominant-negative mutant of Akt enhanced LPS induction of TNF-α promoter activity in monocytic cells (7). Another study (6) showed that siRNA inhibition of the class I PI3K catalytic subunit p110α enhanced LPS induction of TNF-α and activation of the MAPKs. These studies are consistent with results showing that phosphorylation of Raf-1 and ASK1 by Akt is inhibitory (7, 14 –16). Martin and colleagues (11) have shown that the phosphorylation of GSK-3β by Akt inhibited NF-κB transcriptional activity in monocytes. In addition, inhibition of PI3K enhanced inflammation and coagulation in mouse models of endotoxemia and sepsis (17, 18). These studies indicate that the PI3K-Akt pathway negatively regulates LPS signaling and gene expression in monocytic cells.
In contrast, other studies have shown that the PI3K-Akt pathway is required for LPS induction of gene expression in monocytes/macrophages. For example, one study (19), using wortmannin and LY294002, found that the PI3K-Akt pathway positively regulated LPS induction of TNF-α expression in mouse macrophages. Another study (20) showed that transfection of RAW264.7 cells with a class I PI3K p110α dominant negative mutant inhibited LPS-induced NF-κB activation and the induction of TNF-α expression. Moreover, other studies have shown that pharmacologic inhibition of PI3K with wortmannin or LY294002 prevents LPS-induced IκBα degradation and NF-κB translocation (19, 21, 22). These studies suggest that the PI3K-Akt pathway positively regulates LPS signaling in monocytes/macrophages. The role of the PI3K-Akt pathway in the regulation of LPS signaling has been discussed in two recent reviews (13, 23). Additional studies are required to clarify the role of the PI3K-Akt pathway in LPS activation of intracellular signaling and gene expression in monocytes/macrophages.
We used a genetic approach to examine the role of the PI3K-Akt pathway in LPS-induced cytokine and TF expression in monocytes/macrophages. We determined the effect of either genetically decreasing or increasing the activation of the PI3K-Akt pathway on LPS activation of the MAPKs (i.e., ERK1/2, JNK1/2, and p38), transcription factors (i.e., Egr-1, ATF-2, and NF-κB), and gene expression (i.e., TNF-α, IL-6, and TF). Our results demonstrate that the PI3K-Akt pathway negatively regulates LPS signaling in isolated peritoneal exudate cells and peritoneal macrophages and in endotoxemic mice.
Materials and Methods
Mice
All studies were approved by The Scripps Research Institute Animal Care and Use Committee and comply with National Institutes of Health guidelines. Male mice used for these studies were between 6 and 8 wk of age. Wild-type (WT) C57BL/6J mice were obtained from The Scripps Research Institute Breeding Colony. The Pik3r1 gene encodes the PI3K regulatory subunit p85α and the two splice variants, p55α and p50α. Mice deficient in p85α, p55α, and p50α (hereafter referred to as Pik3r1−/− mice) were provided by Dr. L. C. Cantley (Harvard Medical School, Boston, MA) (24) and were maintained on a mixed 129Sv/C57BL/6J background. Pik3r1−/− mice develop hypoglycemia, liver necrosis, and exhibit a high rate of perinatal lethality. We generated 18% of the expected number of mice (total of 20 mice) from intercrosses between Pik3r1−/− mice. Mice deficient in p85α, which retain p50α and p55α (hereafter referred to as p85α−/− mice) were obtained from the RIKEN Bioresource Center and were deposited by Dr. Shigeo Koyasu, Keio University, Tokyo, Japan (25). p85α−/− mice were maintained on a mixed 129Sv/C57BL/6J background and were generated at the expected frequency from intercrosses between p85α+/− mice. PTENflox/flox mice were provided by Dr. J.M. Penninger (University Health Network, Toronto, Canada) (26). To selectively reduce PTEN expression in myeloid cells, PTENflox/flox mice were crossed with mice expressing the Cre recombinase under the control of the Lysozyme M (LysM) promoter (provided by Dr. R. Johnson, University of California San Diego, La Jolla, CA) (27). PTENflox/flox/LysMCre mice and PTENflox/flox mice were backcrossed 6 times onto the C57BL/6J background. PTENflox/flox/LysMCre mice and PTENflox/flox mice were generated by breeding male PTENflox/flox/LysMCre mice with female PTENflox/flox mice.
Bone marrow transplantation
WT C57BL/6J mice (8-wk-old) were irradiated with 13 Gy (1300 rad) using a cesium 137 irradiator (Gammacell 40; Atomic Energy of Canada) to ablate endogenous bone marrow-derived cells. Irradiated mice were injected via the retro-orbital sinus with 2 × 106 bone marrow cells isolated from male or female Pik3r1−/− mice or WT littermate mice. Mice were allowed to recover for 6 wk. PBMC were used for genotyping to demonstrate reconstitution with the donor bone marrow (24).
Mouse endotoxemia
The mouse model of endotoxemia used in these studies consisted of a single intraperitoneal injection of 5 mg/kg Escherichia coli LPS of serotype 0111:B4 (Sigma-Aldrich) (17, 28). Blood was collected from the retro-orbital sinus into sodium citrate (final concentration, 0.38%) at various times after LPS administration (0 – 8 h). For analysis of plasma thrombin-antithrombin (TAT) levels, blood was collected from the inferior vena cava into sodium citrate (final concentration, 0.38%). Plasma was collected and stored at −80°C until analyzed.
Isolation and culture of peritoneal exudate cells (PECs) and peritoneal macrophages (PMs)
PECs were isolated by peritoneal lavage 3 days after intraperitoneal injection of mice with 2 ml of a 3% thioglycollate solution. Nonadherent cells were removed by washing, and the adherent PMs were allowed to recover overnight in fresh medium. PMs were isolated from PTENflox/flox mice and PTENflox/flox/LysMCre mice. Loss of Pik3r1 causes a defect in macrophage adherence (29). Therefore, for studies with Pik3r1−/− mice we used PECs that contained both adherent and nonadherent cells. PECs were isolated from WT and Pik3r1−/− mice, as well as WT mice containing WT or Pik3r1−/− bone marrow. Cells were adjusted to a density of 1 × 106 cells/ml in RPMI 1640 supplemented with 2 mM L-glutamine, 10 mM HEPES, 50 U/ml penicillin 50 μg/ml streptomycin, and 8% heat-inactivated FCS, and cultured at 37°C in 5% CO2. Cells were stimulated with 1 μg/ml LPS. This concentration has been shown to elicit induction of TNF-α through TLR4 signaling (30).
Isolation of cytosolic and nuclear extracts
Whole cell extracts were prepared by lysis of cell pellets in 2× SDS sample buffer (Invitrogen). For isolation of nuclear proteins, 3 × 106 cells were collected from culture plates, resuspended in 1 ml of ice-cold PBS, and cells centrifuged at 2000 × g for 2 min. The cell pellet was resuspended in 50 μl of a buffer containing 10 mM HEPES, 10 mM KCl, 300 mM sucrose, 1.5 mM MgCl2, 0.5 mM DTT, 0.5 mM PMSF, 0.1% Nonidet P-40, and protease inhibitors (Roche Applied Science), incubated for 5 min on ice, and centrifuged for 10 min at 3500 × g. Supernatant (cytosolic fraction) was collected and the resulting pellet was resuspended in 50 μl of a buffer containing 20 mM HEPES, 100 mM KCl, 100 mM NaCl, 0.5 mM DTT, 0.5 mM PMSF, 20% glycerol, and protease inhibitors (Roche). After a 30 min incubation on ice, samples were centrifuged for 2 min at 21,000 × g, and the supernatants (nuclear proteins) collected, aliquoted, and stored at −80°C before analysis.
Western blotting and densitometry
Proteins were separated by SDS-PAGE on 8–16% Tris-glycine gels (Invitrogen Life Technologies) and transferred to Immobilon-P membrane (Millipore). The phosphorylation of Akt (Ser473), ERK1/2 (Thr202/Tyr204), p38 (Thr180/Tyr182), JNK1/2 (Thr183/Tyr185), and ATF-2 (Thr69/71) was determined by overnight incubation at 4°C with a 1/1000 dilution of anti-phosphospecific primary Abs (Cell Signaling Technology), followed by incubation for 1 h at room temperature with a secondary anti-rabbit IgG-HRP conjugated Ab diluted at 1/1000 (Amersham Biosciences). Membranes were washed and incubated with Supersignal West Pico substrate (Pierce Biotechnology) solution and exposed to Hyblot CL film (Denville Scientific). Blots were stripped and reprobed using a 1/1000 dilution of Abs against the nonphosphorylated forms of each protein (Cell Signaling Technology) to monitor protein loading. Levels of IκBα and actin levels were determined using Abs from Santa Cruz Biotechnology. p85α protein levels were determined using an anti-p85α Ab (Millipore). Films were scanned and band intensity was determined using a Bio-Rad densitometer and Quantity One software (Bio-Rad).
EMSA
Nuclear extracts were incubated with radiolabeled double-stranded oligonucleotide probes (Operon Technologies) containing either the murine Ig Ig κB site (underlined), 5′-CAGAGGGGACTTTCCGAGA-3′; an Egr-1 site (underlined), 5′-CCCGGCGCGGGGGCGATTTCGAGTCA-3′; or a Sp1 site (underlined), 5′-ATTCGATCGGGGCGGGGCGAGC-3′ (31). Protein-DNA complexes were separated from free DNA probe by electrophoresis through 6% nondenaturing tris-borate-EDTA gels (Invitrogen Life Technologies) in 0.5× tris-borate-EDTA buffer (Invitrogen Life Technologies). Gels were dried and protein-DNA complexes were visualized by autoradiography.
RNA isolation, cDNA synthesis, and real-time PCR
RNA was isolated from 3 × 106 cells using TRI reagent (Applied Bio-systems) according to the manufacturer’s protocol. cDNA was synthesized from 1 μg of RNA according to the manufacturer’s protocol using a High Capacity cDNA Reverse Transcription kit (Applied Biosystems) and MyCycler themal cycler (Bio-Rad). Levels of TNF-α, IL-6, TF, Egr-1, and GAPDH were determined using TaqMan gene expression assays from Applied Biosystems (Mm99999915_g1, Mm00656724_m1, Mm00443258_m1, Mm00446190_m1, Mm00438855_m1) and TaqMan gene expression master mix (Applied Biosystems) on an ABI Prism 7300 sequence detection system (Applied Biosystems). The expression of each gene was normalized relative to GAPDH expression levels, and relative expression levels were determined using the comparative Ct method.
Procoagulant activity
Cell pellets (3 × 106 cells) were solubilized at 37°C for 15 min using 15 mM n-Octyl-β-D-glucopyranoside. The procoagulant activity of cell lysates was measured using a one-stage clotting assay as described previously (32) using a Start4 clotting machine (Diagnostica Stago). Clotting times were converted to procoagulant activity by comparison with a standard curve established with mouse brain extract. The procoagulant activity of each sample was normalized to total protein concentration determined using a Bio-Rad DC protein assay (Bio-Rad).
Cytokine and TAT
Levels of IL-6 and TNF-α in cell culture supernatant and plasma were determined using commercially available ELISA kits (R&D Systems). Levels of TAT in plasma were determined using a commercially available ELISA (Dade-Behring).
Determination of white blood cell counts
Blood leukocyte counts were determined using a Unopette white blood cell determination kit (BD Biosciences) and a hemocytometer. Slides were prepared from whole blood anticoagulated with EDTA and stained using the Hema 3 staining system (Fisher Diagnostics). Differential counting was performed in a blinded manner.
Measurement of LDH activity
Cells were pelleted by centrifugation and the culture supernatant removed. Cells were lysed in 1% Triton X-100. The activity of LDH in the supernatant and pellet fractions was determined spectrophotometrically and the percentage LDH release determined.
Statistical analysis
Data are presented as mean ± SEM. Unpaired Student’s t test was used when two groups were compared. The criterion for significance for all experiments was p < 0.05.
Results
Genetic modulation of the PI3K-Akt pathway alters LPS activation of the MAPK pathways in macrophages
First, we determined the effect of reducing PTEN expression on LPS activation of Akt and the MAPK pathways. Both basal and LPS activation of Akt were enhanced in PTEN−/− PMs compared with WT PMs (Fig. 1A). In contrast, LPS activation of ERK1/2, JNK1/2, and p38 was significantly reduced in PTEN−/− PMs compared with WT cells (Fig. 1, B–D). Next, we evaluated the effect of Pik3r1 deficiency on LPS activation of Akt and the MAPK pathways. LPS activation of Akt was significantly reduced in Pik3r1−/− PECs compared with WT PECs (Fig. 2A). In contrast, LPS activation of ERK1/2 and JNK1/2 was significantly enhanced in Pik3r1−/− PECs (Fig. 2, B and C). LPS activation of p38 was slightly enhanced in Pik3r1−/− PECs 20 min after LPS, although this increase did not achieve statistical significance (Fig. 2D). These results indicate that the PI3K-Akt pathway inhibits LPS activation of the MAPK pathways.
FIGURE 1.

LPS activation of Akt and MAPK pathways in WT and PTEN−/− PMs. WT and PTEN−/− PMs were stimulated with LPS (1 μg/ml) for 0 –20 min. The activation of Akt (A), ERK1/2 (B), JNK1/2 (C), and p38 (D) was determined in whole cell extracts by western blotting using anti-phospho specific Abs. Each blot was stripped and reprobed for the nonphosphorylated form of each protein to assess loading. Representative western blots are shown. Normalized levels of phosphorylated proteins are shown as mean ±SEM (n = 3– 6) relative to WT cells stimulated with LPS for 20 min (defined as 100%).*, p < 0.05.
FIGURE 2.

LPS activation of Akt and MAPK pathways in WT and Pik3r1−/− PECs. WT and Pik3r1−/− PECs were stimulated with LPS (1 μg/ml) for 0 –20 min. The activation of Akt (A), ERK1/2 (B), JNK1/2 (C), and p38 (D) was determined in whole cell protein extracts by Western blotting using anti-phospho specific Abs. Each blot was stripped and reprobed for the non-phosphorylated form of each protein to assess loading. Representative western blots are shown. Normalized levels of phosphorylated proteins are shown as mean ± SEM (n = 3– 6) relative to WT cells stimulated with LPS for 20 min (defined as 100%).*, p < 0.05.
Role of the PI3K-Akt pathway in LPS activation of ATF-2 in macrophages
LPS activation of p38 and JNK1/2 induces the phosphorylation and activation of various transcription factors, including ATF-2 (33). ATF-2 is a member of the AP-1 family of transcription factors and ATF-2/c-Jun heterodimers have been shown to mediate induction of TNF-α expression in LPS-stimulated monocytes (3, 34, 35). We found that the nuclear levels of phosphorylated ATF-2 were reduced in LPS-stimulated PTEN−/− PMs compared with WT PMs (Fig. 3, A and B). In contrast, nuclear levels of phosphorylated ATF-2 were increased in LPS-stimulated Pik3r1−/− PECs compared with WT PECs (Fig. 3, C and D). These results indicate that the PI3K-Akt pathway negatively regulates LPS activation of ATF-2.
FIGURE 3.

LPS activation of ATF-2 in WT and PTEN−/− PMs, and WT and Pik3r1−/− PECs. WT and PTEN−/− PMs (A and B), and WT and Pik3r1−/− PECs (C and D) were stimulated with LPS (1 μg/ml) for 30 min. Levels of phosphorylated ATF-2 in nuclear extracts were determined by western blotting using an anti-phospho specific Ab. Each blot was stripped and reprobed for the nonphosphorylated form of ATF-2 to assess loading. Representative western blots are shown. Normalized levels of phosphorylated ATF-2 are shown as mean ± SEM (n = 3– 6) relative to WT cells (defined as 100%). *, p < 0.05.
Role of the PI3K-Akt pathway in LPS induction of Egr-1 in macrophages
LPS activation of the MEK1-ERK1/2 pathway leads to the induction of Egr-1 gene expression by phosphorylation of the transcription factor Elk1 (4). Egr-1 mediates LPS induction of TNF-α and TF gene expression in monocytes (4, 36, 37). We found that LPS induction of Egr-1 mRNA and protein was significantly reduced in PTEN−/− PMs compared with WT PMs (Fig. 4, A–C). In contrast, the levels of Egr-1 mRNA and protein were increased in LPS-stimulated Pik3r1−/− PECs compared with WT PECs (Fig. 4, D–F). These results indicate that the PI3K-Akt pathway negatively regulates LPS induction of Egr-1 gene expression.
FIGURE 4.

LPS induction of Egr-1 in WT and PTEN−/− PMs, and WT and Pik3r1−/− PECs. WT and PTEN−/− PMs (A–C) or WT and Pik3r1−/− PECs (D–F) were stimulated with LPS (1 μg/ml) and the induction of Egr-1 mRNA was determined (A and D). Levels of Egr-1 and Sp1 in nuclear extracts of PMs stimulated for 120 min were determined by EMSA. Sp1 was used to assess loading. Representative EMSAs are shown. Egr-1 mRNA levels normalized to levels of GAPDH are shown as mean ± SEM (n = 5– 6) relative to WT cells stimulated with LPS (defined as 100%). Egr-1 levels normalized to levels of Sp1 are shown as mean ± SEM (n = 3– 6) relative to WT cells stimulated with LPS (defined as 100%). *, p <0.05.
Genetic modulation of the PI3K-Akt pathway does not affect LPS-induced IκBα degradation or nuclear translocation of NF-κB in macrophages
LPS induces IKK-dependent phosphorylation of IκBα and subsequent degradation. This is required for nuclear translocation of NF-κB. We found that LPS-induced IκBα degradation and resynthesis were not affected by either reducing PTEN levels in PMs or deleting Pik3r1 in PECs (Fig. 5, A, B, D, and E). In addition, changes in the levels of the PI3K-Akt pathway did not affect nuclear translocation of NF-κB (Fig. 5, C and F). These results indicate that the PI3K-Akt pathway does not modulate LPS-induced IκBα degradation or nuclear translocation of NF-κB in mouse peritoneal exudate cells or peritoneal macrophages.
FIGURE 5.

LPS-induced IκBα degradation and resynthesis and NF-κB nuclear translocation in WT and PTEN−/− PMs, and WT and Pik3r1−/− PECs. A–C, Data from WT or PTEN−/− PMs and data from WT or Pik3r1−/− PECs is also shown (D–F). Cells were stimulated with LPS (1 μg/ml) for 0 –120 min. The degradation (0 –20 min) and resynthesis of IκBα (0, 120 min) was determined in whole cell extracts by Western blotting using an anti-IκBα Ab (A, B, D, and E). A and D, Representative Western blots from three to six independent experiments. B and E, Results from three independent samples. Levels of NF-κB in nuclear extracts were determined by EMSA 30 min after LPS stimulation (C and F).
Genetic modulation of the PI3K-Akt pathway alters LPS induction of TNF-α, IL-6, and TF expression in macrophages
To determine whether PTEN or Pik3r1 deficiency alters LPS induction of gene expression, we evaluated the expression of genes encoding both proinflammatory and procoagulant proteins in PTEN−/− PMs and Pik3r1−/− PECs. LPS induction of TNF-α, IL-6, and TF mRNA expression was reduced in PTEN−/− PMs compared with WT PMs (Fig. 6, A–C). In contrast, LPS induction of TNF-α, IL-6, and TF mRNA expression was increased in Pik3r1−/− PECs compared with WT PECs (Fig. 6, D–F). However, the increase in TNF-α mRNA in the Pik3r1−/− PECs did not achieve statistical significance (Fig. 6D). Next, we determined whether PTEN or Pik3r1 deficiency altered the levels of TNF-α, IL-6, and TF protein expression. LPS induction of all three proteins was reduced in PTEN−/− PMs compared with WT PMs (Fig. 7, A–C). In contrast, LPS induction of TNF-α, IL-6, and TF expression was significantly increased in Pik3r1−/− PECs compared with WT PECs (Fig. 7, D–F). Cell viability was determined by the release of LDH into the culture medium after 6 h of LPS stimulation. Cell viability was not affected by either deletion of Pik3r1 in PECs or reducing PTEN levels in PMs (data not shown). These results indicate that modulation of the PI3K-Akt signaling pathway affects LPS induction of gene expression in macrophages.
FIGURE 6.

LPS-induced expression of TNF-α, IL-6, and TF mRNA in WT and PTEN−/− PMs, and WT and Pik3r1−/− PECs. Levels of TNF-α (A), IL-6 (B), and TF (C) mRNA in WT and PTEN−/− PMs were determined 60 and 120 min after LPS stimulation. Levels of TNF-α (D), IL-6 (E), and TF (F) mRNA in WT and Pik3r1−/− PECs were determined 60 and 120 min after LPS stimulation. mRNA levels normalized to GAPDH are expressed as mean ± SEM (n = 5– 6) relative to WT cells stimulated with LPS for 120 min (defined as 100%).*, p <0.05.
FIGURE 7.

LPS-induced expression of TNF-α and IL-6 protein, and TF activity in WT and PTEN−/− PMs, and WT and Pik3r1−/− PECs. Levels of TNF-α (A), IL-6 (B), and TF (C) protein in WT and PTEN−/− PMs were determined 6 h after LPS stimulation. Levels of TNF-α (D), IL-6 (E), and TF (F) protein in WT and Pik3r1−/− PECs were determined 6 h after LPS stimulation. Data are expressed as mean ± SEM (n = 8 –10). *, p < 0.05.
Regulation of inflammation and coagulation by the PI3K-Akt pathway in endotoxemic mice
We found that the loss of PTEN in myeloid cells led to a 2.5-fold increase in the number of circulating monocytes (Table I). This increase in the number of circulating inflammatory cells precluded studies on the effect of increasing Akt activity on LPS-induced inflammation and coagulation in vivo.
Table I.
White blood cell counts in PTENflox/flox/LysMCre micea
| No. of Cells/μL of Blood |
||||
|---|---|---|---|---|
| Mouse | WBCs | Lymphocytes | Neutrophils | Monocytes |
| PTENflox/flox | 9358 ± 438 | 8069 ± 360 | 1065 ± 94 | 208 ± 44 |
| PTENflox/flox/LysMCre | 11367 ± 736b | 9341 ± 644 | 1482 ± 186b | 494 ± 66b |
Determination of white blood cell counts in untreated PTENflox/flox and PTENflox/flox/LysMCre mice. Data are expressed as mean ± SEM.
Significantly different from WT mice. p < 0.05. n = 9 –12 mice per group.
Hemopoietic cells are a major source of cytokine and TF expression in endotoxemic mice (28, 38). To determine the effect of selectively reducing activation of the PI3K-Akt pathway in hemopoietic cells on LPS-induced inflammation and coagulation in vivo, we used bone marrow transplantation to generate WT C57BL/6J mice containing either Pik3r1−/− or WT bone marrow. Reconstitution of the bone marrow in WT recipient mice was confirmed by PCR analysis of peripheral blood cell DNA and the level of p85α protein in blood cells (Fig. 8, A and B). Endotoxemic WT mice containing Pik3r1−/− bone marrow had significantly higher plasma levels of TNF-α, IL-6, and TAT, a biomarker of coagulation, compared with WT mice containing WT bone marrow (Fig. 8, C–E). We also analyzed LPS induction of inflammation and coagulation in p85α−/− mice, which lack the p85α regulatory subunit in all cells. Plasma levels of TNF-α, IL-6, and TAT were significantly increased in endotoxemic p85α−/− mice compared with WT mice (Fig. 8, F–H). These results indicate that genetically reducing the activation of the PI3K-Akt pathway either selectively in hemopoietic cells or in all cells enhances LPS-induced inflammation and coagulation in mice.
FIGURE 8.

Effect of Pik3r1 or p85α deficiency on inflammation and coagulation in endotoxemic mice. A, Genotyping of blood cells from WT mice containing either WT or Pik3r1−/− bone marrow (4 mice per group). B, p85α protein levels in blood cells isolated from WT mice containing either WT or Pik3r1−/− bone marrow. WT mice with either WT or Pik3r1−/− bone marrow were injected with LPS (5 mg/kg) and the plasma levels of TNF-α (C), IL-6 (D), and TAT (E) (8 h) were determined. n = 20 –30 mice for each group. WT and p85α−/− mice were injected with LPS (5 mg/kg) and the plasma levels of TNF-α (F), IL-6 (G), and TAT (H) (8 h) were determined. n = 10 mice for each group. Data are expressed as mean ± SEM.*, p < 0.05.
Discussion
We used a genetic approach to determine the role of the PI3K-Akt pathway in LPS signaling and gene expression in monocytes/macrophages. We show that Pik3r1-deficient macrophages have reduced LPS activation of Akt, whereas PTEN-deficient macrophages have enhanced LPS activation of Akt. Using these models, we found that the PI3K-Akt pathway negatively regulates LPS activation of intracellular signaling pathways, transcription factors and induction of gene expression in monocytes/macrophages.
LPS activation of various MAP3Ks leads to phosphorylation of JNK1/2, ERK1/2, and p38 (1, 2). Akt has been shown to phosphorylate and inhibit the MAP3Ks ASK1 and Raf-1, which are required for LPS activation of ERK1/2, p38, and JNK1/2 (14 –16). We show that genetically increasing levels of phosphorylated Akt results in reduced LPS activation of the MAPK pathways. In contrast, decreasing PI3K-Akt pathway activation enhances LPS activation of ERK1/2 and JNK1/2. The kinetics of Akt activation relative to MAPK inhibition were consistent with inhibitory phosphorylation of upstream MAP3Ks by Akt and feedback inhibition of the MAPKs. This result is consistent with our previous studies showing that pharmacologic inhibition of PI3K enhanced LPS activation of the MAPKs (7). LPS activation of p38 was slightly enhanced in Pik3r1-deficient PECs, although this pathway was less sensitive to the effects of decreasing PI3K activity. Importantly, we show that the levels of Egr-1 mRNA and nuclear Egr-1, a transcription factor induced by the MEK1-ERK1/2 pathway (4, 36), and the phosphorylation of ATF-2, which is activated by the JNK1/2 and p38 MAPK pathways (33, 39), are decreased in PTEN−/− PMs and increased in Pik3r1-deficient PECs. ATF-2 and Egr-1 coordinate the expression of various proinflammatory genes including TNF-α, IL-6, and TF (4, 34). Taken together, these results indicate that the PI3K-Akt pathway inhibits LPS-induced gene expression by inhibiting activation of the MAPK pathways and the transcription factors Egr-1 and ATF-2.
The transcription factor NF-κB is required for the induction of TNF-α, IL-6 and TF gene expression in monocytic cells. We and others (5, 7) have previously shown that pharmacologic inhibition of PI3K enhances LPS-induced IκBα degradation and the nuclear translocation of NF-κB. However, contradictory findings have been published and exactly how the PI3K-Akt pathway regulates nuclear translocation of NF-κB remains unclear (19, 21). This may be due, in part, to nonspecific effects of the PI3K inhibitors, or other factors including species, cell type, agonist, and the concentration of the inhibitor. For example, neither LY294002 nor wortmannin affected LPS-induced NF-κB nuclear translocation in RAW264.7 cells (19). In another study, LY294002, but not wortmannin, inhibited NF-κB nuclear translocation in LPS-treated RAW264.7 cells (21). Interestingly, an inactive analog of LY294002 also inhibited LPS-induced NF-κB nuclear translocation, suggesting an effect of LY294002 independent of PI3K (21). We show here using a genetic approach that modulation of the PI3K-Akt pathway does not affect LPS-induced degradation of IκBα and NF-κB nuclear translocation in mouse macrophages. This result suggests that the PI3K-Akt pathway does not inhibit LPS-induced gene expression in mouse macrophages by reducing NF-κB nuclear translocation. However, a recent study showed that the PI3K-Akt pathway does regulate NF-κB nuclear activity by affecting its interaction with CREB and CBP (11).
Importantly, we show that genetically reducing the activity of the PI3K-Akt pathway by deleting the Pik3r1 gene enhances LPS-induced TNF-α, IL-6 and TF expression in PECs. These results are consistent with previous studies, which showed that pharmacologic inhibition of PI3K enhanced LPS induction of TF and TNF-α in monocytic cells (7, 40). In contrast, reducing the level of PTEN increased Akt activation and inhibited LPS induction of TNF-α, IL-6 and TF expression. Our results are consistent with previous studies showing reduced TNF-α expression in LPS-stimulated macrophages from PTENflox/flox/LysMCre mice, and macrophages from mice deficient in SH2 domain-containing inositol phosphatase, another phosphatase that negatively regulates Akt activation (41, 42). Taken together, our results indicate that activation of the PI3K-Akt pathway inhibits LPS induction of TNF-α, IL-6 and TF expression in macrophages.
Wortmannin and LY294002 enhanced inflammation in mouse models of endotoxemia and cecalligation and puncture (17, 18). However, the potential for effects of these compounds independent of PI3K inhibition may be increased in vivo due to distribution or pharmacokinetics. These experiments also do not address the relative contribution of the PI3K-Akt pathway in different cell types. Our in vitro results indicate that the PI3K-Akt pathway modulates LPS induction of proinflammatory and procoagulant mediators in macrophages. Hemopoietic cells, including monocytes/macrophages, contribute significantly to inflammation and coagulation in endotoxemic mice (28, 38). Using a bone marrow transplantation strategy, we show that genetically decreasing PI3K-Akt pathway activity in hemopoietic cells significantly increases the plasma levels of TNF-α, IL-6, and TAT in endotoxemic mice. These results identify the PI3K-Akt pathway in hemopoietic cells as an important anti-inflammatory signaling pathway in endotoxemia. We also found that the plasma levels of TNF-α, IL-6 and TAT were enhanced in endotoxemic p85α−/− mice compared with WT mice. Interestingly, the enhancement in TNF-α, IL-6, and TAT levels in endotoxemic p85α−/− mice was greater than that observed in WT mice with Pik3r1−/− bone marrow. This suggests that the PI3K-Akt pathway in both hemopoietic and nonhemopoietic cells inhibits inflammation and coagulation in endotoxemic mice. Consistent with these findings, a recent study showed that inflammation induced by the TLR5 agonist flagellin was increased in p85α−/− mice (43).
Based on our in vitro results, we hypothesized that the selective activation of Akt in monocytes/macrophages would reduce inflammation and coagulation in endotoxemic mice. Importantly, PTEN is a tumor suppressor gene and its deletion is associated with many cancers. Recent studies (44, 45) have shown that deletion of the PTEN gene in hemopoietic cells causes the proliferation of myeloid cells. We found that reducing PTEN expression in myeloid cells using Cre-LoxP technology selectively increased the number of circulating monocytes and neutrophils. Although genetic activation of the PI3K-Akt pathway in monocytes may yield confounding results due to altered cellular proliferation, several studies have suggested that acute, pharmacologic activation of the PI3K-Akt pathway inhibits inflammation in endotoxemic mice. The anti-inflammatory effects of some agents, including glucan and α-lipoteichoic acid are mediated by activation of the PI3K-Akt pathway (18, 46). Some peroxovanadium compounds have been shown to inhibit PTEN (47). However, these compounds are highly unstable in an aqueous environment and have a short half-life in vivo. At present, stable and selective small m.w. inhibitors of PTEN are not yet available.
In summary, we show using a genetic approach that the PI3K-Akt pathway negatively regulates LPS induction of TF and inflammatory cytokines in macrophages by inhibiting activation of the MAPK pathways and the transcription factors ATF-2 and Egr-1 (Fig. 9). Moreover, we show that reducing PI3K-Akt pathway activity either selectively in hemopoietic cells or in all cell types enhances inflammation and coagulation in endotoxemic mice. Taken together, our results suggest that pharmacologic activation of the PI3K-Akt pathway or inhibition of PTEN may be novel strategies to limit inflammation and coagulation and improve survival in endotoxemia and other inflammatory diseases.
FIGURE 9.
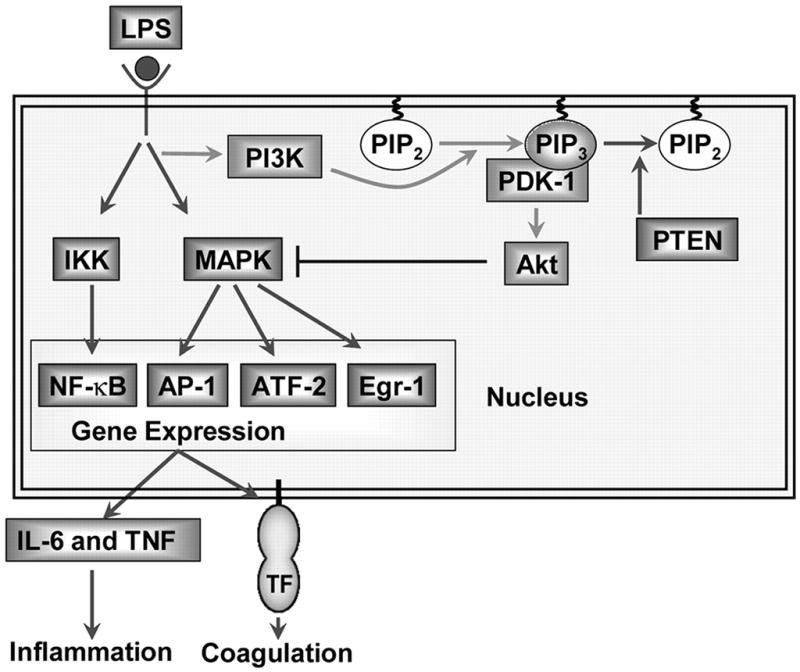
The PI3K-Akt pathway negatively regulates LPS signaling and gene expression in mouse macrophages by inhibiting the activation of MAPKs. Deleting the p85α regulatory subunit reduces PI3K activity and Akt phosphorylation, which increases LPS activation of the MAPKs and gene expression. In contrast, a deficiency in PTEN leads to an increase in Akt phosphorylation and a reduction in LPS activation of the MAPKs and gene expression. Changes in the levels of phosphorylated Akt do not alter LPS-induced nuclear translocation of NF-κB.
Acknowledgments
We thank Cheryl Johnson for assistance in preparation of the manuscript and Dr. Linda Kidd and Dr. Ulla Knaus for helpful discussion and critique of the manuscript.
Footnotes
Abbreviations used in this paper: IKK, IκBβ kinase; MAPK, mitogen-activated protein kinase; TF, tissue factor; PI3K, the phosphatidylinositol-3-kinase; PTEN, phosphatase and tensin homologue; WT, wild type; TAT, thrombin-antithrombin; PEC, peritoneal exudate cell; PM, peritoneal macrophage.
Disclosures
The authors have no financial conflict of interest.
This work was supported by National Institutes of Health Grant HL48872 (to N.M.), National Institutes of Health National Research Service Award F32 HL085983 (to J.P.L), Austrian Science Fund FWF P19850 (to G.A.S.), COBRE Grant P20 RR021940, and a Kansas IDeA Network of Biomedical Research Excellence Recruitment Package 5 P20 RR016475 (to J.P.L).
References
- 1.Guha M, Mackman N. LPS induction of gene expression in human monocytes. Cell Signal. 2001;13:85–94. doi: 10.1016/s0898-6568(00)00149-2. [DOI] [PubMed] [Google Scholar]
- 2.Sweet MJ, Hume DA. Endotoxin signal transduction in macrophages. J Leukocyte Biol. 1996;60:8–26. doi: 10.1002/jlb.60.1.8. [DOI] [PubMed] [Google Scholar]
- 3.Foletta VC, Segal DH, Cohen DR. Transcriptional regulation in the immune system: all roads lead to AP-1. J Leukocyte Biol. 1998;63:139–152. doi: 10.1002/jlb.63.2.139. [DOI] [PubMed] [Google Scholar]
- 4.Guha M, O’Connell MA, Pawlinski R, Yan SF, Stern D, Mackman N. Lipopolysaccharide activation of the MEK-ERK1/2 pathway in human monocytic cells mediates tissue factor and tumor necrosis factor α expression by inducing Elk-1 phosphorylation and Egr-1 expression. Blood. 2001;98:1429–1439. doi: 10.1182/blood.v98.5.1429. [DOI] [PubMed] [Google Scholar]
- 5.Diaz-Guerra MJ, Castrillo A, Martin-Sanz P, Bosca L. Negative regulation by phosphatidylinositol 3-kinase of inducible nitric oxide synthase expression in macrophages. J Immunol. 1999;162:6184–6190. [PubMed] [Google Scholar]
- 6.Lee JS, Nauseef WM, Moeenrezakhanlou A, Sly LM, Noubir S, Leidal KG, Schlomann JM, Krystal G, Reiner NE. Monocyte p110α phosphatidylinositol 3-kinase regulates phagocytosis, the phagocyte oxidase, and cytokine production. J Leukocyte Biol. 2007;81:1548–1561. doi: 10.1189/jlb.0906564. [DOI] [PubMed] [Google Scholar]
- 7.Guha M, Mackman N. The phosphatidylinositol 3-kinase-akt pathway limits lipopolysaccharide activation of signaling pathways and expression of inflammatory mediators in human monocytic cells. J Biol Chem. 2002;277:32124–32132. doi: 10.1074/jbc.M203298200. [DOI] [PubMed] [Google Scholar]
- 8.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 9.Ward SG, Finan P. Isoform-specific phosphoinositide 3-kinase inhibitors as therapeutic agents. Curr Opin Pharmacol. 2003;3:426–434. doi: 10.1016/s1471-4892(03)00078-x. [DOI] [PubMed] [Google Scholar]
- 10.Gharbi SI, Zvelebil MJ, Shuttleworth SJ, Hancox T, Saghir N, Timms JF, Waterfield MD. Exploring the specificity of the PI3K family inhibitor LY294002. Biochem J. 2007;404:15–21. doi: 10.1042/BJ20061489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol. 2005;6:777–784. doi: 10.1038/ni1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martin M, Schifferle RE, Cuesta N, Vogel SN, Katz J, Michalek SM. Role of the phosphatidylinositol 3 kinase-Akt pathway in the regulation of IL-10 and IL-12 by Porphyromonas gingivalis lipopolysaccharide. J Immunol. 2003;171:717–725. doi: 10.4049/jimmunol.171.2.717. [DOI] [PubMed] [Google Scholar]
- 13.Fukao T, Koyasu S. PI3K and negative regulation of TLR signaling. Trends Immunol. 2003;24:358–363. doi: 10.1016/s1471-4906(03)00139-x. [DOI] [PubMed] [Google Scholar]
- 14.Matsuzawa A, Saegusa K, Noguchi T, Sadamitsu C, Nishitoh H, Nagai S, Koyasu S, Matsumoto K, Takeda K, Ichijo H. ROS-dependent activation of the TRAF6-ASK1–p38 pathway is selectively required for TLR4-mediated innate immunity. Nat Immunol. 2005;6:587–592. doi: 10.1038/ni1200. [DOI] [PubMed] [Google Scholar]
- 15.Kim AH, Khursigara G, Sun X, Franke TF, Chao MV. Akt phosphorylates and negatively regulates apoptosis signal-regulating kinase 1. Mol Cell Biol. 2001;21:893–901. doi: 10.1128/MCB.21.3.893-901.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rommel C, Clarke BA, Zimmermann S, Nunez L, Rossman R, Reid K, Moelling K, Yancopoulos GD, Glass DJ. Differentiation stage-specific inhibition of the Raf-MEK-ERK pathway by Akt. Science. 1999;286:1738–1741. doi: 10.1126/science.286.5445.1738. [DOI] [PubMed] [Google Scholar]
- 17.Schabbauer G, Tencati M, Pedersen B, Pawlinski R, Mackman N. PI3K-Akt pathway suppresses coagulation and inflammation in endotoxemic mice. Arterioscler Thromb Vasc Biol. 2004;24:1963–1969. doi: 10.1161/01.ATV.0000143096.15099.ce. [DOI] [PubMed] [Google Scholar]
- 18.Williams DL, Li C, Ha T, Ozment-Skelton T, Kalbfleisch JH, Preiszner J, Brooks L, Breuel K, Schweitzer JB. Modulation of the phosphoinositide 3-kinase pathway alters innate resistance to polymicrobial sepsis. J Immunol. 2004;172:449–456. doi: 10.4049/jimmunol.172.1.449. [DOI] [PubMed] [Google Scholar]
- 19.Ojaniemi M, Glumoff V, Harju K, Liljeroos M, Vuori K, Hallman M. Phosphatidylinositol 3-kinase is involved in Toll-like receptor 4-mediated cytokine expression in mouse macrophages. Eur J Immunol. 2003;33:597–605. doi: 10.1002/eji.200323376. [DOI] [PubMed] [Google Scholar]
- 20.Kuo CC, Lin WT, Liang CM, Liang SM. Class I and III phosphatidylinositol 3′-kinase play distinct roles in TLR signaling pathway. J Immunol. 2006;176:5943–5949. doi: 10.4049/jimmunol.176.10.5943. [DOI] [PubMed] [Google Scholar]
- 21.Kim YH, Choi KH, Park JW, Kwon TK. LY294002 inhibits LPS-induced NO production through a inhibition of NF-κB activation: independent mechanism of phosphatidylinositol 3-kinase. Immunol Lett. 2005;99:45–50. doi: 10.1016/j.imlet.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 22.Park SJ, Lee SC, Hong SH, Kim HM. Degradation of IκBα in activated RAW264.7 cells is blocked by the phosphatidylinositol 3-kinase inhibitor LY294002. Cell Biol Toxicol. 2002;18:121–130. doi: 10.1023/a:1015384201785. [DOI] [PubMed] [Google Scholar]
- 23.Ruse M, Knaus UG. New players in TLR-mediated innate immunity: PI3K and small ρ GTPases. Immunol Res. 2006;34:33–48. doi: 10.1385/IR:34:1:33. [DOI] [PubMed] [Google Scholar]
- 24.Fruman DA, Mauvais-Jarvis F, Pollard DA, Yballe CM, Brazil D, Bronson RT, Kahn CR, Cantley LC. Hypoglycaemia, liver necrosis, and perinatal death in mice lacking all isoforms of phosphoinositide 3-kinase p85α. Nat Genet. 2000;26:379–382. doi: 10.1038/81715. [DOI] [PubMed] [Google Scholar]
- 25.Terauchi Y, Tsuji Y, Satoh S, Minoura H, Murakami K, Okuno A, Inukai K, Asano T, Kaburagi Y, Ueki K, et al. Increased insulin sensitivity and hypoglycaemia in mice lacking the p85α subunit of phosphoinositide 3-kinase. Nat Genet. 1999;21:230–235. doi: 10.1038/6023. [DOI] [PubMed] [Google Scholar]
- 26.Suzuki A, Yamaguchi MT, Ohteki T, Sasaki T, Kaisho T, Kimura Y, Yoshida R, Wakeham A, Higuchi T, Fukumoto M, et al. T cell-specific loss of Pten leads to defects in central and peripheral tolerance. Immunity. 2001;14:523–534. doi: 10.1016/s1074-7613(01)00134-0. [DOI] [PubMed] [Google Scholar]
- 27.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- 28.Pawlinski R, Pedersen B, Schabbauer G, Tencati M, Holscher T, Boisvert W, Andrade-Gordon P, Frank RD, Mackman N. Role of tissue factor and protease activated receptors in a mouse model of endotoxemia. Blood. 2004;103:1342–1347. doi: 10.1182/blood-2003-09-3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Munugalavadla V, Borneo J, Ingram DA, Kapur R. p85α subunit of class IA PI-3 kinase is crucial for macrophage growth and migration. Blood. 2005;106:103–109. doi: 10.1182/blood-2004-10-4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lehner MD, Morath S, Michelsen KS, Schumann RR, Hartung T. Induction of cross-tolerance by lipopolysaccharide and highly purified lipoteichoic acid via different Toll-like receptors independent of paracrine mediators. J Immunol. 2001;166:5161–5167. doi: 10.4049/jimmunol.166.8.5161. [DOI] [PubMed] [Google Scholar]
- 31.Oeth PA, Parry GCN, Kunsch C, Nantermet P, Rosen CA, Mackman N. Lipopolysaccharide induction of tissue factor gene expression in monocytic cells is mediated by binding of c-Rel/p65 heterodimers to a κB-like site. Mol Cell Biol. 1994;14:3772–3781. doi: 10.1128/mcb.14.6.3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morrissey JH, Fair DS, Edgington TS. Monoclonal antibody analysis of purified and cell-associated tissue factor. Thromb Res. 1988;52:247–261. doi: 10.1016/0049-3848(88)90084-9. [DOI] [PubMed] [Google Scholar]
- 33.Ardeshna KM, Pizzey AR, Devereux S, Khwaja A. The PI3 kinase, p38 SAP kinase, and NF-κB signal transduction pathways are involved in the survival and maturation of lipopolysaccharide-stimulated human monocyte-derived dendritic cells. Blood. 2000;96:1039–1046. [PubMed] [Google Scholar]
- 34.Reimold AM, Kim J, Finberg R, Glimcher LH. Decreased immediate inflammatory gene induction in activating transcription factor-2 mutant mice. Int Immunol. 2001;13:241–248. doi: 10.1093/intimm/13.2.241. [DOI] [PubMed] [Google Scholar]
- 35.Tsai EY, Falvo JV, Tsytsykova AV, Barczak AK, Reimold AM, Glimcher LH, Fenton MJ, Gordon DC, Dunn IF, Goldfeld AE. A lipopolysaccharide-specific enhancer complex involving Ets, Elk-1, Sp1, and CREB binding protein and p300 is recruited to the tumor necrosis factor α promoter in vivo. Mol Cell Biol. 2000;20:6084–6094. doi: 10.1128/mcb.20.16.6084-6094.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shi L, Kishore R, McMullen MR, Nagy LE. Lipopolysaccharide stimulation of ERK1/2 increases TNF-α production via Egr-1. Am J Physiol. 2002;282:C1205–C1211. doi: 10.1152/ajpcell.00511.2001. [DOI] [PubMed] [Google Scholar]
- 37.Yao J, Mackman N, Edgington TS, Fan ST. Lipopolysaccharide induction of the tumor necrosis factor-α promoter in human monocytic cells: regulation by Egr-1, c-Jun, and NF-κB transcription factors. J Biol Chem. 1997;272:17795–17801. doi: 10.1074/jbc.272.28.17795. [DOI] [PubMed] [Google Scholar]
- 38.Bultinck J, Brouckaert P, Cauwels A. The in vivo contribution of hematopoietic cells to systemic TNF and IL-6 production during endotoxemia. Cytokine. 2006;36:160–166. doi: 10.1016/j.cyto.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 39.Morton S, Davis RJ, Cohen P. Signalling pathways involved in multisite phosphorylation of the transcription factor ATF-2. FEBS Lett. 2004;572:177–183. doi: 10.1016/j.febslet.2004.07.031. [DOI] [PubMed] [Google Scholar]
- 40.Park YC, Lee CH, Kang HS, Chung HT, Kim HD. Wortmannin, a specific inhibitor of phosphatidylinositol-3-kinase, enhances LPS-induced NO production from murine peritoneal macrophages. Biochem Biophys Res Commun. 1997;240:692–696. doi: 10.1006/bbrc.1997.7722. [DOI] [PubMed] [Google Scholar]
- 41.Cao X, Wei G, Fang H, Guo J, Weinstein M, Marsh CB, Ostrowski MC, Tridandapani S. The inositol 3-phosphatase PTEN negatively regulates Fcγ receptor signaling, but supports Toll-like receptor 4 signaling in murine peritoneal macrophages. J Immunol. 2004;172:4851–4857. doi: 10.4049/jimmunol.172.8.4851. [DOI] [PubMed] [Google Scholar]
- 42.Fang H, Pengal RA, Cao X, Ganesan LP, Wewers MD, Marsh CB, Tridandapani S. Lipopolysaccharide-induced macrophage inflammatory response is regulated by SHIP. J Immunol. 2004;173:360–366. doi: 10.4049/jimmunol.173.1.360. [DOI] [PubMed] [Google Scholar]
- 43.Yu Y, Nagai S, Wu H, Neish AS, Koyasu S, Gewirtz AT. TLR5-mediated phosphoinositide 3-kinase activation negatively regulates flagellin-induced proinflammatory gene expression. J Immunol. 2006;176:00. doi: 10.4049/jimmunol.176.10.6194. [DOI] [PubMed] [Google Scholar]
- 44.Zhang J, Grindley JC, Yin T, Jayasinghe S, He XC, Ross JT, Haug JS, Rupp D, Porter-Westpfahl KS, Wiedemann LM, et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature. 2006;441:518–522. doi: 10.1038/nature04747. [DOI] [PubMed] [Google Scholar]
- 45.Zhu D, Hattori H, Jo H, Jia Y, Subramanian KK, Loison F, You J, Le Y, Honczarenko M, Silberstein L, Luo HR. Deactivation of phosphatidylinositol 3,4,5-trisphosphate/Akt signaling mediates neutrophil spontaneous death. Proc Natl Acad Sci USA. 2006;103:14836–14841. doi: 10.1073/pnas.0605722103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang WJ, Wei H, Hagen T, Frei B. α-lipoic acid attenuates LPS-induced inflammatory responses by activating the phosphoinositide 3-kinase/Akt signaling pathway. Proc Natl Acad Sci USA. 2007;104:4077–4082. doi: 10.1073/pnas.0700305104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schmid AC, Byrne RD, Vilar R, Woscholski R. Bisperoxovanadium compounds are potent PTEN inhibitors. FEBS Lett. 2004;566:35–38. doi: 10.1016/j.febslet.2004.03.102. [DOI] [PubMed] [Google Scholar]