

Abstract
The large potential of redox enzymes to carry out formation of high value organic compounds motivates the search for innovative strategies to regenerate the cofactors needed by their biocatalytic cycles. Here, we describe a bioreactor where the reducing power to the cycle is supplied directly to purified cytochrome CYP101 (P450cam; EC 1.14.15.1) through its natural redox partner (putidaredoxin) using an antimony-doped tin oxide working electrode. Required oxygen was produced at a Pt counter electrode by water electrolysis. A continuous catalytic cycle was sustained for more than 5 h and 2,600 enzyme turnovers. The maximum product formation rate was 36 nmol of 5-exo-hydroxycamphor/nmol of CYP101 per min.
Interest in biocatalytic hydroxylation derives from its ability to transform organic substrates with no functional groups into oxygen-bearing compounds with high regio- or stereoselectivity. The cytochrome P450 enzyme class is widely distributed in various forms of life and participates in diversified metabolic reactions as the oxygen-activating component of monooxygenase systems (1, 2). Its unique oxygenation chemistry and its substrate specificity offer the opportunity to develop enzymatic systems for synthesizing fine chemicals, sensing biochemically active compounds, and detoxifying environmental contamination. A major hurdle to implementation of P450 catalysis for commercial syntheses is the requirement for stoichiometric amounts of freely dissociated cofactors, such as NADH and/or redox partner proteins, which supply necessary reducing equivalents. This requirement is met currently either as an additional nutrient cost during in vivo whole cell biocatalysis (fermentation) or as a cost of supplying fresh cofactor along with target substrate during in vitro biocatalysis. Efficient and stable methods for cofactor regeneration are being pursued (3) to make these systems economical.
A particular member of the P450s, cytochrome CYP101 (P450cam; EC 1.14.15.1) monooxygenase, is found in the bacterium Pseudomonas putida PpG786 when it is cultured on camphor. The enzyme catalyzes camphor hydroxylation at the 5-exo position and requires NADH as well as two protein cofactors. Stereospecific hydroxylation of other substrates has been investigated (4, 5). As shown in Fig. 1 (Upper), the overall biocatalytic cycle is composed of many individual reactions (1, 2, 6). The two electrons necessary for the reaction are supplied by reduced putidaredoxin Pdxr, a 2Fe-2S protein (11.6 kDa), which mediates the transfer of these electrons from NADH and the FAD-containing putidaredoxin reductase PdR to the heme active center of the cytochrome CYP101 (45 kDa). In addition to the electron mediation function, Pdx is also an “effector” for product release in the final step of the cycle (7). The overall reaction is thermodynamically controlled; redox potential and substrate binding are modulated by the cytochrome spin-state equilibrium (8).
Figure 1.
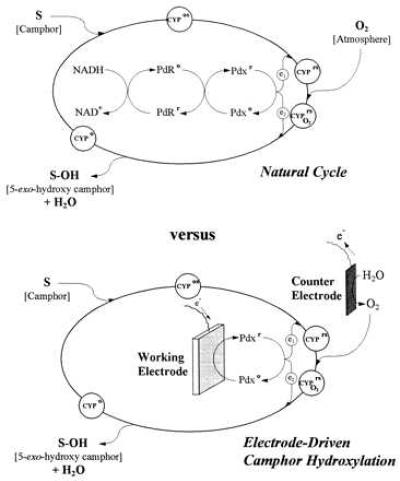
Natural (Upper) and direct-electrode-driven (Lower) catalytic cycles for substrate (camphor) hydroxylation by the P450cam monooxygenase system. Substrate binds with the oxidized form of cytochrome hemoprotein CYP101 (P450cam) to form the protein complex CYPos, which receives the first electron e1 from reduced putidaredoxin Pdxr. Dissolved oxygen is bound in the natural cycle to form the complex CYPO2rs, which receives a second electron e2 from Pdxr, whereupon the complex decomposes to give the stereospecifically hydroxylated product (5-exo-hydroxy-camphor). In the electrode-driven process, a semiconductor working electrode supplies reducing power to oxidized putidaredoxin Pdxo, replacing NADH and putidaredoxin reductase PdR as reducing components. Also, oxygen is generated electrochemically at a controlled surface area counter electrode in an anaerobic bioreactor.
Research into the use of electrodes to supply the reducing power for driving P450 catalytic cycles is being actively pursued (9–13). Estabrook and coworkers (9) (University of Texas Southwestern Medical Center at Dallas) have shown mediated electrode-driven biocatalysis using cobalt(III) sepulchrate to transfer electrons to a rat recombinant liver P450 fusion protein, whereas Kazlauskaite et al. (10) and Zhang et al. (11) have demonstrated direct electron transfer from carbon-based electrodes to CYP101 (P450cam). In the mediated biocatalysis studies, the electrolysis enzyme turnover rate was comparable with the NADPH-driven cycle for a number of recombinant fusion microsomal P450 enzymes (12). This report also showed a turnover rate of more than 100 nmol of product/min/nmol of P450 when lauric acid was hydroxylated by organocobalt mediation of the only known single protein P450, CYP102 (P450BM3). The mechanism of the organocobalt mediator interaction with the P450 heme, or its effect on the natural system’s thermodynamic balance, was not characterized. The direct electron transfer study of Kazlauskaite et al. (10) showed reversible electrochemical response from glassy carbon electrodes corresponding to the first of the two electron transfers to CYP101 that are required to initiate the catalytic cycle (see step e1 of Fig. 1), whereas Zhang et al. (11) showed direct electron transfer from lipid-modified pyrolytic graphite electrodes to CYP101.
In the present report, the CYP101 (P450cam) enzymatic cycle is used in a bioelectrochemical reactor, without synthetic mediators, to catalyze the generation of a stereochemical hydroxylation product. Our strategy has been to use an electrode to reduce the intermediate redox protein Pdx rather than attempt turnover of the catalytic cycle by direct reduction of the terminal hydroxylase, CYP101. This was done, in part, because of the difficulty of interacting directly with the interior heme of the large CYP101 protein and in part because of the important role Pdx plays in maintaining the viability of the natural catalytic cycle (i.e., product stereochemistry, turnover rate, minimization of peroxide formation) (7, 15). We recently showed reversible oxidation/reduction cycling of Pdx on bare and modified metal electrodes (13, 14). Now, we show how biocatalysis by direct electron transfer from a semiconductor electrode to Pdx can be used in a bioreactor (see Fig. 1 Lower) to achieve rapid and persistent electron transfer to CYP101 (P450cam), resulting in near-stoichiometric conversion of camphor to 5-exo-hydroxycamphor. The need for NADH and the flavin protein, PdR, has been eliminated, and the necessary dissolved oxygen requirement is supplied in this anaerobic reactor by local generation at a platinum counter electrode.
METHODS
Protein Preparation.
In all of our work, the proteins that make up the P450cam system are derived from recombinant Escherichia coli clones (DH5-α), each containing a single plasmid for one of the three P450cam subunit proteins (5). On the basis of absorbance ratios, the mole fraction purity × 100 of each subunit protein was greater than 90. In addition, several preparations of the proteins were examined by high resolution two-dimensional gel electrophoresis, and from imaging analysis, the mole fraction purity × 100 was estimated to be greater than 97 for each protein. The full NADH-driven in vitro assay for camphor hydroxylation under saturating Pdx conditions gave a turnover rate of 17 s−1.
Tin Oxide Electrode Preparation.
The working electrodes are antimony-doped tin oxide-coated (0.3-μm thickness) glass slides (4 × 2 cm, Delta Technologies, Stillwater, MN)‡ with donor concentration as determined from Mott-Schottky plots Nd = 5 × 1020 cm−3. Before biocatalysis experiments, electrodes were sonicated in 1 M NaOH for 2 h followed by multiple rinsing with distilled water. They were activated by cycling between −0.5 and 1.5 V for 5 min in 15% HCl and stored in distilled water. Film electrode resistance as measured by a four-point probe (Cascade Microtech, Inc., model C4S-44, Beaverton, OR) was 40–50 Ω/sq. The counter electrode was made as a coil from Pt 0.5-mm wire. The reference electrode was a standard Ag/AgCl (3 M KCl) reference microelectrode (Abtech Inc., RE 803).
Electron Transfer Rate Measurements.
Absorbance during the electrolysis was monitored using a thin layer (0.2 mm) cell comprising two tin oxide electrodes. Spectra were recorded with a fiber optic-based CCD spectrophotometer (Ocean Optics, Inc.) in 0.7-s intervals. For determination of the heterogeneous rate constant for Pdx reduction on the electrode, khet, a potential pulse was applied starting from open circuit to a final voltage in the range from −0.45 to −0.9 V. The decay in absorbance at 458 nm was used to obtain khet values according to the correlations developed by Albertson et al. (16). The homogeneous (observed) second order rate constant for Pdxr and oxidized, camphor-bound CYP101 (CYPos) conversion to Pdxo + CYPrs was determined in an anaerobic solution consisting of a Tris/KCl buffer (50 mM Tris⋅HCl/0.18 M KCl/16 mM MgCl2, pH ≈ 7.4) and an initial protein concentration ratio of [CYPos]/[Pdxr] = 0.1. A potential pulse was applied starting from open circuit to a final voltage −0.9 V, and absorbance at 455 nm was used to calculate kobs (see ref. 17). Comparable results were obtained from the exponential decay of the 392 nm absorbance (after subtracting the Pdx contribution at that wavelength) giving a pseudo-first order rate constant of 0.017 s−1 for the conversion of CYPos to CYPrs.
Bioelectrochemical Reactor Design and Operation.
Fig. 2 is a schematic of the reactor used to generate the data shown in Fig. 3 corresponding to the 4.2 nmol product/min-nmol CYP101 turnover rate listed in Table 1. The bioreactor working solution volume in this design was about 20 ml and incorporated two tin oxide working electrodes (16 cm2 reaction surface). The large solution volume was needed to accommodate the combination oxygen/temperature electrode (Orion, no. 084010) used in some experiments. Other experiments were performed in a smaller reactor (1 ml solution volume), which retained the most important features of the larger reactor design: electrode surface area/solution volume, Pt counter-electrode and Pt grid shielding of the working electrode, stir-bar mixing, solution purging, and head space gas handling. During electrolysis, the potential of the working electrode was maintained at −0.7 V vs. the Ag/AgCl reference electrode, whereas the potential on the counter electrode was adjusted to +1.0 V by controlling the extent of submergence (which directly affected the counter electrode current density) at the fixed working electrode potential. An additional reference electrode was inserted during the adjustment. Although the buffer solution contained relatively high chloride concentration, chlorine evolution was minimized by holding the counter electrode potential less noble than the reversible Cl−/Cl2 potential (E0 = +1.17 V). Temperature was maintained at 21°C by air cooling, and a water bath was used to control temperature at 37°C in some experiments.
Figure 2.
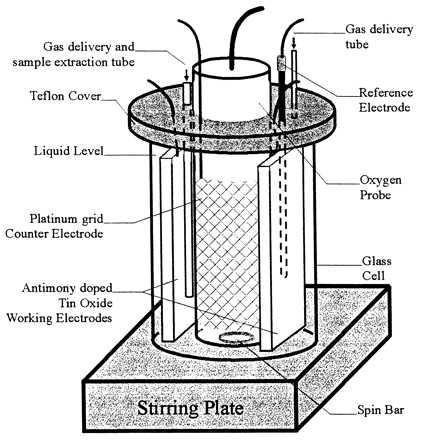
Bioelectrochemical reactor for the electrode-driven P450cam biocatalytic cycle. The reactor is modified from a 40-ml cylindrical glass vessel (6 cm high). Two tin oxide working electrodes with a total surface area of 16 cm2 were wrapped in Pt mesh (not shown) that had no electric contact and was at open circuit potential during electrolysis. At the point of minimum separation, the working electrodes were about 1 mm from the Pt grid counter electrode. The extent of Pt grid counter electrode submergence was varied to maximize electrochemical oxygen evolution without causing chlorine evolution, which is severely detrimental to activity. Details concerning operation of the reactor during electrolysis are given in the text.
Figure 3.
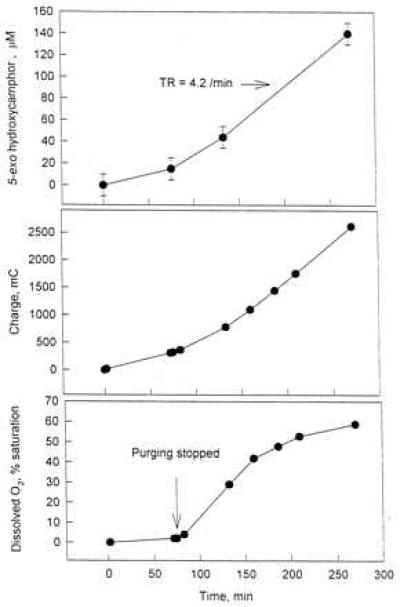
Measurements of product evolution, charge consumption, and dissolved oxygen during electrolysis in the bioreactor (see Table 1; 4.2 nmol of 5-exo-camphor/nmol of CYP101/min of entry). Total consumed charge was evaluated by integrating the current to 270 min. An initial 30-min Ar gas purge (by gas bubbling) reduced dissolved O2 to less than 2% saturation. Electrolysis was initiated, and Ar purging continued for another 70 min; then, purging was stopped and an Ar gas blanket was maintained above the electrolyte for the remainder of the electrolysis period.
Table 1.
Camphor hydroxylation for several bioelectrochemical reactor conditions
| Reactor solution composition* | Oxygen condition | Product turnover rate, nmol of 5-exo-camphor/min per nmol of CYP101 |
|---|---|---|
| 41 μM Pdx | Ar purge | 0 |
| 12.8 μM CYP101 | Ar purge | 0 |
| 41 μM Pdx + 0.41 μM CYP101 | Air saturated | 0.9 |
| 55 μM Pdx + 0.18 μM CYP101 | Ar purge | 4.2 |
| 108 μM Pdx + 0.23 μM CYP101 | Ar purge | 6.4 |
| 1030 μM Pdx + 0.13 μM CYP101† | Ar purge | 34 |
| 1030 μM Pdx + 0.13 μM CYP101‡ | Ar purge | 36 |
Standard experimental electrolysis reaction conditions: buffer was 50 mM Tris⋅HCl, 0.18 M KCl. 16 mM MgCl2; 800 μM initial camphor concentration; Ewe = −0.7 V on the tin oxide working electrode; temperature was 21–22°C.
Standard condition except initial camphor concentration was 2 mM.
Standard condition except initial camphor concentration was 2 mM and temperature was 37°C.
Electrolysis experiments were initiated by combining the enzyme components in the Tris/KCl buffer and purging the stirred solution with argon for about 30 min at open circuit potential. After extraction of an initial sample (100 μl), the potential of −0.7 V (vs. Ag/AgCl) was applied, and the Ar purge was redirected to reactor headspace. Solution samples for product analysis were extracted through one of the 2-mm diameter Teflon tubes that also served as purging ports.
Product (5-exo-Hydroxycamphor) Measurement.
Product 5-exo-hydroxycamphor was identified by GC-MS and quantified by GC with flame ionization detection. GC injection samples of 1–2 μl were prepared from approximately equal volumes of reaction mixture and an extraction mixture (0.3 g methylene chloride + 0.02 g KCl with 300 μM n-decane as internal standard). The GC was operated with a DB-5 column (30 m) with temperature programming (2 min at 60°C, ramp at 10°C/min to 120°C; 2 min at 120°C, ramp at 15°C/min to 160°C). Retention times (He carrier gas at 2.8 bar gauge pressure) were: n-decane, 4.7 min; camphor, 7.0 min; and 5-exo-hydroxycamphor, 10.9 min. Chromatograms showed peaks of only internal standard, substrate (camphor), and product (5-exo-hydroxycamphor, 1 μM minimum detectability). Identification of 5-exo-hydroxycamphor as product was done by comparing m/e pattern (VG analytical mass spectrometer, 70 eV electron impact energy) with published spectra (18).
RESULTS AND DISCUSSION
Metal Electrode-Driven Putidaredoxin Turnover.
Our strategy in obtaining electrode-driven multiple turnovers of the P450cam system, as shown in Fig. 1 (Lower), is based on partial reconstitution using purified recombinant putidaredoxin and cytochrome CYP101. Previous studies of single and partial turnover of P450 cycles using nonprotein electron sources focused on elucidation of the natural charge-transfer sequence (19, 20). As discussed earlier, multiple turnovers in electrochemical cells have been achieved with cobalt(III) sepulchrate (12) or phenazine methosulfate (15) as mediators. Our goal was to develop an electrode that eliminated NADH and simplified the catalytic cycle as much as possible while still retaining the high product specificity found in the native cycle.
We eliminated putidaredoxin reductase (PdR) because of its sole function as electron mediator between NADH and Pdx with no apparent effect on thermodynamic equilibria (8). Pdx was retained because of its ability to perform the difficult selective electron transfer to CYP101 (P450cam) and because of its role as a structure modifier upon binding to CYP, thereby facilitating product release (see ref. 2). Also, Pdx was felt to be a viable candidate for direct heterogeneous reduction due to its relatively small size and a redox-active center that is close to the surface (21). Direct electron transfer to ferredoxins by overcoming unfavorable electrostatics so as to ensure reversible protein-electrode association has been reported (22, 23).
Our initial results showed reversible oxidation/reduction cycling of Pdx on polycrystalline gold or silver electrodes that were modified by immobilizing ionizable organic molecules to overcome the repulsive electrostatic interaction that arises when trying to reduce the negatively charged Pdx (pI 4.5) at the electrode (13, 14). This was a hopeful indication that direct electrolysis of the P450cam catalytic cycle might be achieved via Pdx as the natural mediator; however, the response was short-lived. Both in situ surface-enhanced Raman spectroscopic and ellipsometric measurements demonstrated rapid deterioration of the organic electrode modifier while the electrode was held at potentials less than −0.7 V. It also remained to establish that an electrode could be made that would rapidly and continuously reduce solution Pdx while having a minimal effect on other reaction components. The successful electrode must not be irreversibly coated by other components, including CYP101.
Metal (Tin) Oxide Electrode-Driven Turnover.
The choice of the antimony-doped tin oxide was suggested by its rather negative flat band potential (Efb= −1.4 V vs. Ag/AgCl) ensuring the excess positive charge in the potential range of interest (−0.5 to −0.9 V). Another important consideration was its reported resistance toward the adsorption of organic surfactants (24). We used traditional electrochemical (cyclic voltammetry, AC voltammetry) and spectroelectrochemical techniques (16, 17) to characterize the interaction of Pdx with the tin oxide electrode in both the presence and absence of its CYP101 (P450cam) redox partner. For Pdxo reduction, the E1/2 value was determined to be −0.43 V (vs. Ag/AgCl), which is in good agreement with the reported titration value −0.42 V (25). The response was stable for at least the lifetime of Pdx under enzymatic cycle conditions (up to 60 h). The heterogeneous electron-transfer rate constant determined from the decay in absorbance at 455 nm was khet = 1.1 × 10−4 cm s−1, which is about an order of magnitude lower than that determined on a modified gold electrode (13). Absorbance measurements of Soret band shift from 392 to 408 nm after adding camphor, and CYPos gave evidence of electron transfer from reduced Pdxr to the camphor-bound CYP101 (P450cam) species. The observed apparent second order rate constant (kobs) was determined to be 1.3 × 104 M−1 s−1. The formal interprotein electron transfer rate constant for the first electron transfer in the P450cam enzymatic cycle (step e1), ket, can be obtained by taking account of the rapid Pdx-CYP complex formation. Dividing kobs by the Pdxr-CYPos charge-transfer complex equilibrium constant (Keq = 2 μM−1) (18, 19) gives ket = 0.0065 s−1. This value is much lower than that obtained from single turnover kinetic studies of the first electron transfer between Pdx and CYP101 (P450cam) using stopped-flow measurements (26) (ket = 10–15 s−1). We believe this is indicative of the low concentration ratio, [Pdxr]/[CYPos] = 10, that was needed to see simultaneously the absorption maxima of both species and due to an insufficient rate of production and transport of Pdxr species from the electrode to the bulk solution.
Bioelectrochemical Reactor Design and Performance.
After failing to find significant product under aerobic operation, we realized that the molecular oxygen co-reagent must be introduced in a way that minimizes both the rapid reoxidation of putidaredoxin (before Pdxr can deliver its reducing power to CYP) and the excessive cathodic reduction to hydrogen peroxide (which can diminish protein stability and stereochemical purity of product). We addressed this oxygen delivery dilemma by operating the reactor in an anaerobic environment using an argon purge while generating the necessary oxygen at a controlled surface-area platinum counter electrode through water oxidation (2H2O → 4e− + 4H+ + O2; E0 = 0.6 V vs. Ag/AgCl at pH 7.4). In addition, the tin oxide working electrode was screened by a Pt mesh to catalyze the decomposition of trace hydrogen peroxide, which could originate during oxygen reduction at this electrode.
Initial reactor trials involved exploring the effects of dissolved oxygen and working electrode potential over the range −0.5 to −0.9 V. Results shown in Table 1 are compared against control electrolysis experiments in which all components were present except one of the proteins, Pdx or CYP101 (P450cam). Little or no product formation was detected when the electrolysis reaction was run under oxygen saturation conditions or before the working electrodes were shielded by a platinum mesh coil. An argon purge before electrolysis, followed by electrolysis under an Ar blanket, was found to lower the dissolved oxygen in the bioreactor to less than 2% of air saturation levels. This anaerobic environment was sustainable indefinitely under open circuit conditions. Variation of the working electrode potential showed that initial product formation rate increased with overvoltage but at the expense of the bioreactor system sustainability. Therefore, the optimum potential of Ewe= −0.7 V was held for the remaining biocatalysis experiments.
The time dependence of product formation together with electrolysis-derived dissolved oxygen concentration and charge consumption are shown in Fig. 3 for the reaction that displayed the product turnover rate of 4.2 nmol of 5-exo-hydroxycamphor/nmol of CYP101 (P450cam)/min. The rates of product formation and charge consumption increased with the onset of electrochemical oxygen generation (t > 70 min). The average current efficiency, based on product formation, was about 25% in this deaerated incubation (100% efficiency = 2 mol of electrons/mole of product as per the natural cycle). Oxygen reduction was the dominant side reaction accounting for the remaining current consumption. Oxygen concentration increased linearly after cessation of Ar purging and leveled off at about 60% saturation.
As displayed in Table 1, the absence of either Pdx or CYP101 (P450cam) in the reaction mixture resulted in negligible product formation (1 μM detection limit). This also confirms the role of Pdx as an effector of product release in addition to electrochemical mediator (7). In other trials with both Pdx and CYP101 (P450cam) present, and of longer duration, camphor conversion to the 5-exo-hydroxycamphor product exceeded 95%. This result is close to the same product fidelity that is achieved in the natural cycle.
The highest total number of turnovers for CYP101 (P450cam) observed in our experiments was equal to 2,600 with an average rate (turnover rate) of about 0.5 s−1. As seen in Fig. 3, there is no sign of diminishing activity up to 5 h, and in other experiments at 4°C, the catalytic cycle was still functioning at 16 h. Initial turnover rates were found to increase with [Pdx]/[CYP101] ratio. The maximum sustained product turnover rate we measured was 36 nmol of 5-exo-hydroxycamphor/min/nmol of CYP101 at [Pdx]/[CYP101] = 7,800. This rate is comparable with the turnover rates achieved in organocobalt-mediated P450-catalyzed conversions (12), some of which were run at 37°C. Increasing temperature to 37°C had only a modest effect on our turnover rate and had no observable effect on the stability of the system. The trend of increasing product formation rate with increasing [Pdx]/[CYP101] ratio is interpreted as another indication of insufficient Pdxr production from the working electrode because of slow heterogeneous electron transfer, slow bulk solution transport rates, or molecular oxygen reoxidation limitations. The greater part of the enzyme, which is in the bulk of the solution far from the electrode surface, is totally inefficient; therefore, confinement of the reaction layer close to the working electrode surface may be required to optimize the turnover rate. Further, improved bioreactor design could also include a means of separating oxygen from direct interaction with reduced Pdx.
This present study demonstrates the feasibility of direct electrochemically driven enzymology for generating practical quantities of a chiral hydroxylation product. There may be opportunities to apply this engineered redox enzymology to related systems such as the methane monooxygenase, cytochrome P450s from other bacterial or mammalian mitochondrial sources, or dioxygenases of similar molecular architecture (toluene dioxygenase) for the production of regio- or stereospecific hydroxylated compounds. In each of these cases, a dual function redox protein like putidaredoxin is required in the catalytic cycle for carrying out electron transfer and for acting as an effector of product specificity and high turnover rates. Extensions of this approach to microsomal, plant, or yeast P450s that have only a single reductase redox partner would require other electrode and/or bioreactor design strategies to accommodate the direct reduction of the reductase partner proteins. The alternative approach of trying to reduce the hydroxylase protein directly at an electrode could result in loss of product specificity (where it was a feature of the natural cycle) and/or a reduction in catalytic turnover rates.
Acknowledgments
This work was supported by the National Science Foundation, the Du Pont Company, and the National Institute of Standards and Technology.
ABBREVIATIONS
- Pdx
putidaredoxin
- PdR
putidaredoxin reductase
Footnotes
Certain commercial equipment, instruments, and materials are identified in this paper to specify adequately the experimental procedure. In no case does such identification imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the material or equipment is necessarily the best available for the purpose.
References
- 1.Ortiz de Montellano P R, editor. Cytochrome P450: Structure, Mechanism and Biochemistry. New York: Plenum; 1995. [Google Scholar]
- 2.Omura T, editor. Cytochrome P450. New York: VCH; 1993. [Google Scholar]
- 3.Steckhan E. In: Topics in Current Chemistry: Electrochemistry V. Steckhan E, editor. Berlin: Springer; 1994. pp. 84–111. [Google Scholar]
- 4.Fruetel J A, Collins J R, Camper D L, Loew G H, Ortiz de Montellano P R. J Am Chem Soc. 1992;114:6987–6993. [Google Scholar]
- 5.Grayson D A, Tewari Y B, Mayhew M P, Vilker V L, Goldberg R N. Arch Biochem Biophys. 1996;332:239–247. doi: 10.1006/abbi.1996.0338. [DOI] [PubMed] [Google Scholar]
- 6.Brewer C B, Peterson J A. Arch Biochem Biophys. 1986;249:515–521. doi: 10.1016/0003-9861(86)90029-9. [DOI] [PubMed] [Google Scholar]
- 7.Lipscomb J D, Sligar S G, Namtvedt M J, Gunsalus I C. J Biol Chem. 1976;251:1116–1124. [PubMed] [Google Scholar]
- 8.Sligar S G, Gunsalus I C. Proc Natl Acad Sci USA. 1976;73:1078–1082. doi: 10.1073/pnas.73.4.1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Faulkner K M, Shet M S, Fisher C W, Estabrook R W. Proc Natl Acad Sci USA. 1995;92:7705–7709. doi: 10.1073/pnas.92.17.7705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kazlauskaite, J., Westlake, A. C. G., Wong, L.-L. & Hill, H. A. O. (1996) Chem. Commun. 2189–2190.
- 11.Zhang Z, Nasser A E F, Schenkman J B, Rusling J F. J Chem Soc Faraday Trans 2. 1997;93:1769–1774. [Google Scholar]
- 12.Estabrook R W, Faulkner K M, Shet M S, Fisher C W. Methods Enzymol. 1996;272:44–50. doi: 10.1016/s0076-6879(96)72007-4. [DOI] [PubMed] [Google Scholar]
- 13.Wong L S, Vilker V L, Yap W T, Reipa V. Langmuir. 1995;11:4818–4822. [Google Scholar]
- 14.Reipa V, Gaigalas A K, Edwards J J, Vilker V L. J Electroanal Chem. 1995;395:299–303. [Google Scholar]
- 15.Eble K S, Dawson J H. Biochemistry. 1984;23:2068–2073. doi: 10.1021/bi00304a029. [DOI] [PubMed] [Google Scholar]
- 16.Albertson D E, Blount H N, Hawkridge F M. Anal Chem. 1979;51:556–560. [Google Scholar]
- 17.Ryan M D, Wilson G S. Anal Chem. 1975;47:885–890. doi: 10.1021/ac60356a045. [DOI] [PubMed] [Google Scholar]
- 18.White R E, McCarthy M-B, Egeberg K D, Sligar S G. Arch Biochem Biophys. 1984;228:493–502. doi: 10.1016/0003-9861(84)90015-8. [DOI] [PubMed] [Google Scholar]
- 19.Pederson T C, Austin R H, Gunsalus I C. In: Microsomes and Drug Oxidations. Ullrich V, Roots I, Hildebrandt A, Estabrook R, editors. New York: Pergamon; 1976. pp. 275–283. [Google Scholar]
- 20.Sligar S G, Kennedy K A, Pearson D C. Proc Natl Acad Sci USA. 1980;77:1240–1244. doi: 10.1073/pnas.77.3.1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pochapsky T C, Ye X M, Ratnaswamy G, Lyons T A. Biochemistry. 1994;33:6424–6432. doi: 10.1021/bi00187a006. [DOI] [PubMed] [Google Scholar]
- 22.Armstrong F A. Struct Bonding (Berlin) 1990;72:139–221. [Google Scholar]
- 23.Nishiyama K, Ishida H, Taniguchi I. J Electroanal Chem. 1994;373:255–258. [Google Scholar]
- 24.Elliot D, Zellmer D L, Laitinen H A. J Electrochem Soc. 1970;117:1343–1348. [Google Scholar]
- 25.Gunsalus I C, Meeks J R, Lipscomb J D. Ann N Y Acad Sci. 1973;212:107–117. doi: 10.1111/j.1749-6632.1973.tb47590.x. [DOI] [PubMed] [Google Scholar]
- 26.Brewer C, Peterson J A. J Biol Chem. 1988;263:791–798. [PubMed] [Google Scholar]