

Abstract
The cerebral neuropathology of Type 2 diabetes (CNDM2) has not been positively defined. This review includes a description of CNDM2 research from before the ‘Pubmed Era’. Recent neuroimaging studies have focused on cerebrovascular and white matter pathology. These and prior studies about cerebrovascular histopathology in diabetes are reviewed. Evidence is also described for and against the link between CNDM2 and Alzheimer’s disease pathogenesis. To study this matter directly, we evaluated data from University of Kentucky Alzheimer’s Disease Center (UK ADC) patients recruited while non-demented and followed longitudinally. Of patients who had come to autopsy (N=234), 139 met inclusion criteria. These patients provided the basis for comparing the prevalence of pathological and clinical indices between well-characterized cases with (N=50) or without (N=89) the premortem diagnosis of diabetes. In diabetics, cerebrovascular pathology was more frequent and Alzheimer-type pathology was less frequent than in non-diabetics. Finally, a series of photomicrographs demonstrates histopathological features (including clinical–radiographical correlation) observed in brains of persons that died after a history of diabetes. These preliminary, correlative, and descriptive studies may help develop new hypotheses about CNDM2. We conclude that more work should be performed on human material in the context of CNDM2.
Keywords: Diabetes, Alzheimer’s, Cerebrovascular, Stroke, Cognition, Clinicopathological, Radiographical, Pathology, Review
1. Introduction
More than 90% of diabetes mellitus cases in Western countries correspond to Type 2 diabetes (“non-insulin dependent”, or DM2) [1]. Although technically a misnomer, the term “diabetes” is used herein to encompass clinical signs and symptoms relating to DM2. The central focus of this review is the cerebral neuropathology of DM2 (CNDM2). We will review briefly the scientific literature from studies in epidemiology, neuropsychology, neuroimaging, and histopathology research that are relevant to human CNDM2. Correlative and descriptive data will be analyzed from the University of Kentucky Alzheimer’s Disease Center (UK ADC) autopsy cohort. These studies include a case–control series depicting clinical and pathological indices stratified by the antemortem diagnosis of diabetes. Our study also includes a series of photomicrographs from diabetics’ brains that help depict some of the vascular and white matter changes along with radiographical–neuropathological correlation. A central conclusion is that direct pathological studies of human tissues, despite all their technical challenges, are an important experimental component to the study of CNDM2.
2. Potential confounds
Prior studies have been unable to demonstrate pathognomonic changes that discriminate the brains of humans with diabetes from “non-diabetic” brains. This may reflect the formidable obstacles or potential confounds in studying diabetic brain disease. Below we describe five of the most important potential confounds relevant to research on DM2 and the human brain.
2.1. Cohort effect, case–control pitfalls, and other potential biases
In a historical sense, “diabetes” is a moving target. New treatments emerge each year. Environmental challenges, including dietary changes [2] and medications for other diseases, evolve also. These produce changes that impact entire generations and may affect different groups or cohorts distinctly. Thus, there is no guarantee that a human study performed in 2008 will be relevant directly to diabetics in 2028. Another problem is that in any “case–control” study, most individuals identified as diabetics will have been treated for diabetes, whereas some that were not identified as diabetics will be in fact untreated diabetics. This potential confound can be minimized by stringent inclusion/exclusion criteria and monitoring patients. However, in a study with rigorous clinical documentation and careful case selection, the diabetics may be even better controlled in their medical and diet regimens and their case–control outcome differences may change commensurately. Age effects are also difficult to determine without rigorous controls–the findings of the effects of hyperglycemia on brain tissue in a 30-year old are impossible to project onto those in an 80-year old.
2.2. Distinct group characteristics—comorbidities, environmental factors, and genetic factors
DM2 cases are nonrandomly distributed in populations. Rather, the disease is associated strongly with indices related to medical comorbidities, socioeconomic factors, and genetic factors. Many studies on diabetics have noted systematic abnormalities in blood pressure, atherosclerosis, and blood values for pH, urate, lipids, ketones, and clotting factors [3–7]. Diabetics also tend to have concomitant environmental risks such as obesity (presumably reflecting an altered diet) and smoking [1,2,8,9]. Other potential confounds in diabetes studies are the socioeconomic factors that may induce case-versus-control systematic biases in patients’ trust of, and willingness to participate in, clinical trials [10–12] much less autopsy-based research. Finally, there are hypothesized genetic risk factors that may relate both to the metabolic syndrome and to neurodegeneration, such as the apolipoprotein E allele [13–17]. In summary, it is a challenge to detect whether a brain change is specific to CNDM2 –hyperglycemia and/or insulin resistance per se – rather than a combination of other medical, environmental, and genetic factors, that disproportionately accompany CNDM2. This consideration amounts to multiple potential confounds that are extremely challenging to eliminate completely, irrespective of study design.
2.3. Hypoglycemia and other treatment effects
Insulin or other agents can induce hypoglycemia iatrogenically. Hypoglycemia induces seizures, coma, and widespread cerebral cortical neuronal loss when glucose levels fall below ~1–1. 5 mM (18–27 mg/dl) [18–24]. The neuropathology that is associated with this devastating condition is not identical to that of widespread ischemia or hypoxia [23,24]. The mechanism of this special type of neuropathology is apparently excitotoxicity via the neurotransmitter aspartate [23,24]. More relevant to many diabetics are the effects of episodic, short stretches of hypoglycemia, aggravated perhaps by chronic recurrence with cycles of hyperglycemia and/or respiratory depression [25]. Depending on many factors, the net effects may be subtle and idiosyncratic. In human studies, teasing out the specific importance of hypoglycemia, versus hyperglycemia, is not a trivial challenge. Further, hypoglycemic agents have effects on the brain other than those that are involved in glucose regulation [26,27]. Medicinal preparations intended to lower blood glucose differentially affect albumin binding, inflammation, blood-brain transport, and other brain and liver indices [26–29]. Additional treatment effects may also be important. Most diabetics take many drugs concomitantly, many of which relate to metabolism, electrolytes, lipids, platelets, hormones, immunomodulation, and/or blood pressure. For example, in the data presented below, the diabetic patients (N=50, average age at death, 84 years) had an average intake of over 12 different medications daily. The biological effects of these drugs, in isolation or together, may alter observed brain pathology.
2.4. Glucose—one sugar, many pathways, and complicated curves
Most organisms use glucose as a transportable energy source. Glucose is also a moiety that can be attached – enzymatically or non-enzymatically – to proteins, nucleic acids, and lipids [30–32]. Highly amenable to molecular modifications itself, glucose is a potential “player” in many biochemical pathways [30]. Exactly how these pathways are stimulated and inhibited in vivo is currently poorly understood; there is still debate about the basic fundamentals of cellular glucose metabolism in the brain [33–37]. The brain glucose and insulin pathways involve complicated, interacting ripples of effects and counter-effects. The impact on blood vessels by hyperglycemia is thought to be partly mediated through the polyol sorbitol pathway, myoinositol depletion, diacylglycerol pathway, platelet regulation, and many others [18,20,38–49]. Furthermore, cardiovascular factors can have complicated and non-linear dynamics. For example, in compelling mammalian models of cerebral infarction, hyperglycemia can be either neuroprotective, or by contrast neurotoxic, depending on the model parameters [50–54]. An additional example of complex non-linear cerebrovascular dynamics in humans is the well-documented but unexplained epidemiological “J-shaped curve” effect of alcohol intake upon stroke risk [55–61]. Given these considerations, data can be difficult to extrapolate from an experimental model to the human brain. For example, how can we know if the effects of hyperglycemia at blood levels of 100 mg/dl, 200 mg/dl, and 300 mg/dl on a given brain cell parameter are linear, exponential, or opposite from each other?
2.5. Pitfalls of animal models and comparative disease-related neurobiology
Historically, diabetes research provides outstanding examples of how animal models can be used to inform and improve treatment of human diseases [62,63]. In the context of CNDM2, a number of hypotheses have been developed and tested based largely on animal models [64–70]. On the other hand, there are also drawbacks in using animals to model human cognition and human-specific pathology. Because there is not a known, specific cerebral pathological substrate for diabetes in humans, it is instructive to review the experience in animal models of diabetic kidney disease. A pathognomonic diabetic nephropathy lesion is known, so a mouse model should theoretically help to understand the disease mechanisms and work toward a cure. Unfortunately, although kidney failure is commonplace in humans [71–73], this change has been extremely difficult to reproduce in mouse diabetes models [74,75]. There is considerable mouse strain-specific variation in diabetes-related nephropathy [74,75]. Commonly used strains such as C57BL/6 are almost entirely resistant to diabetic changes in the kidney, and in fact less than 5% of mouse strains have reported such pathology [74,75].
What about rodent models of human brain disease in diabetes? The human brain is unique, and there are crucial differences between rodent and human aging trajectories. As such it is not surprising that rodent models of CNDM2 have shortcomings [68,76]. Transgenic or treated rodents have been unable to model the best-established substrate for cognitive loss in diabetic humans, namely atherogenic cerebrovascular changes [64,68,76]. Models of other diabetes-related changes on cognition also have varied results. These studies are compounded by the challenges to test subtypes of cognitive domains in rodents. Some diabetes-related studies have produced no cognitive changes in the presence of hyperglycemia, and others show abnormalities in rodent cognitive/behavioral indices even without hyperglycemia [64]. Furthermore, the perturbations that accompany diabetes studies in animals bear consideration. For example, intracerebral or intraperitoneal injection of streptozotocin (SZT) leads to pathological changes analogous to Alzheimer’s disease (AD), namely increased phosphorylated tau [69,77–81]. It should be borne in mind that SZT has direct CNS toxicity beyond the known effects of diabetes per se [82–84], and some of the effect of SZT on tau proteins is mediated through hypothermia [69]. The animal studies are biologically interesting and may prove their relevance to humans. However, reciprocal validation is important because animal research and human studies have both complementary strengths as well as complementary weaknesses.
3. Effects of diabetes on cognition
Due partly to the challenges described above, there has been some variability in the results of studies about changes in cognition linked to diabetes. The neurological dysfunction associated with this disease has been designated “diabetic encephalopathy” [45,85,86], yet this term has not been rigorously defined by a consensus of experts. Specific cognitive disturbances associated with diabetes have been described (for reviews see [5,17,46,87–90]). Deficits have been repeatedly observed in particular cognitive domains, including memory and psychomotor speed [46,91]. There is some overlap between the findings of cognitive changes in DM1 and DM2 [43,46,92–94] and an increased risk for dementia or mild cognitive impairment in DM2 and some of this literature is described below.
Research about cognitive changes in diabetics demonstrates the importance of study design. Many experiments assessing the effects of diabetes on cognition and/or pathology (including the research described below) have employed a case–control research design. This study format has important advantages in that cases and controls can be carefully monitored, compared, and described; however, these studies are prone to selection and recruitment biases. A study design involving fewer such biases is a population-based study. These studies typically have more patients and are more representative of a large and heterogeneous cohort. Almost one-half of the published population studies have been interpreted to show no effect of diabetes on cognition [17]. However, these results also may have methodological problems including difficulty with accurate identification of cases and controls.
Future studies may overcome the many confounds and discriminate in fine details which specific aspects of DM2 correspond to which subdomains of cognitive dysfunction. In the meantime, the literature on the cognitive deficits related to diabetic encephalopathy produces a general consensus that multiple cognitive domains are affected adversely in diabetics. There is general, but by no means universal, agreement about “direction of effect”–hyperglycemia is associated with mild cognitive dysfunction in many studies. However, there is uncertainty about the precise relationships to biological mechanisms. Thus, specific aspects of prior studies should be interpreted with critical scrutiny.
4. Recent neuroimaging studies related to diabetes-linked anatomic and cognitive changes
To the extent that diabetes can be documented to affect cognition, neuroimaging can correlate structural brain parameters with cognitive changes. Unlike neuropathological studies, brain scans can be used to monitor quantitatively the three dimensional effects of a disease over time in an individual. Further, MRIs can assess neuroanatomical areas suspected of involvement in the diabetic brain, namely the cerebrovascular and white matter disease, which are problematic to study routinely in the context of tissue-based neuropathology. For these reasons, neuroimaging studies are practically tailor-made to surmount many of the obstacles in assessing the cerebral neuropathology of diabetes.
Van Harten et al. (2006) provided an outstanding meta-analysis summarizing critically the literature on neuroimaging of diabetes [95]. This study included formal analysis of 46 studies (including population-based, case–control, and clinical studies with various vascular risk factors) using MRI and/or CT. All of these included at least 20 diabetics with specified criteria for diabetes. According to this meta-analysis, the following three structural changes are described consistently in the brains of diabetic patients:
White matter lesions (WMLs; 27 studies analyzed) – Some but not all studies show a positive correlation between the presence of WMLs and DM2. When studying cohorts with many vascular risk factors in addition to diabetes, there were weak or no correlations between diabetes and WMLs. In outpatient case–control studies, there was a weak association between WMLs and DM2. A typical depiction of WMLs in the brain of a diabetic is shown (Fig. 1).
Lacunar infarcts (LIs; 20 studies analyzed) – A significant association was found between the presence of lacunar infarcts and DM2 across different study designs.
Cortical atrophy (CA; 10 studies analyzed) – Studies assessing cortical atrophy were too heterogeneous methodologically to summarize; however, nine of the ten publications in the meta-analysis showed a positive correlation.
Fig. 1.
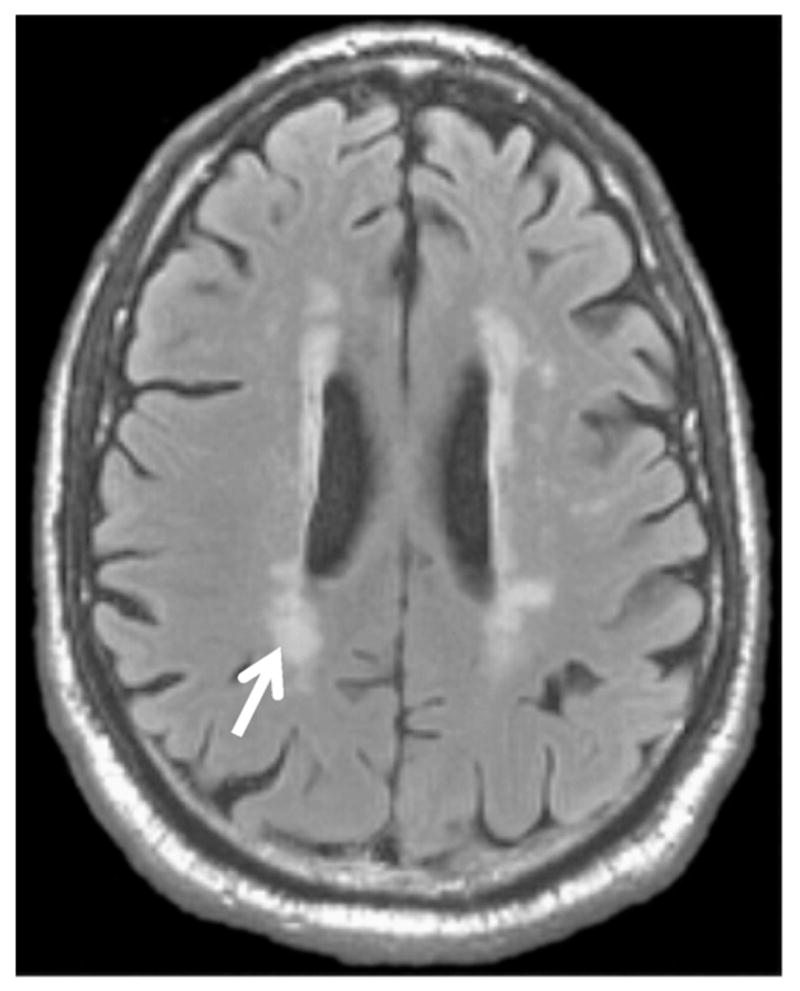
There are no neuroimaging findings entirely specific to diabetes per se, i.e. effects of hyperglycemia. Shown is an axial FLAIR image from a 67-year old diabetic man. It shows typical periventricular hyperintensities (“WMLs”) seen with diabetes (arrow). These lesions are compounded by other factors: age, hypertension, hypercholesterolemia, and homocysteinemia. In a meta-analysis of MRI findings linked to diabetes, population studies found an odds ratio of ~2 for MRI-detected WMLs in diabetes.
Studies performed since the meta-analysis of Van Harten (2006) have further explored the correlation of radiographical changes with the severity of cognitive dysfunction in diabetics [90,93,96–100]. The results of some of these studies are shown in Table 1. Note that the importance and specificity of WMLs appears to be more accentuated in these later studies. Thus, neuroimaging studies help to focus the question with some hope of success, by showing that there are specific anatomical substrates for the cognitive decline seen in diabetics: WMLs, LIs, and CA. Of these three, the two least well understood are WMLs and CA. In the future, technology that probes both functional and structural parameters (fMRI) should provide further insights into diabetes-related brain changes.
Table 1.
Recent MRI studies that correlate anatomical changes with cognitive dysfunction
| Ref. | Patients | MRI findings in association with Type 2 diabetes | Notes |
|---|---|---|---|
| [98] | 113 DM2 51 controls |
WMLs, cortical and subcortical atrophy are associated with cognitive decline | Cognitive dysfunction correlated to WMLs and brain atrophy. A1C was 6.9% (moderately well-controlled) |
| [96] | 122 DM2 56 controls |
WMLs, atrophy are associated with cognitive decline; pathology and other factors show interactions | Cognitive dysfunction correlated with WMLs, atrophy, hypertension, hyperinsulinemia, and “vascular events”; statin use was associated with improved WMLs and with improved cognition |
| [90] | 92 DM2 44 controls |
PVH, WMLs, lacunar infarcts, and cerebral atrophy observed; only PVH was associated with “motor slowing” | MRI findings less well associated with cognitive dysfunction in relation to HbA1C and duration of diabetes, which showed stronger correlation with cognitive dysfunction |
| [93] | 40 DM1 40 DM2 |
DM2 patients have more WMLs and cortical atrophy in comparison to DM1 patients with much longer disease duration | DM2 patients had more cognitive dysfunction but also more of other metabolic aspects including more hypertension, lipid disorders, etc. |
| [97] | 122 DM2 56 controls |
WMLs, atrophy, which were not correlated with peripheral neuropathy within given patients | Authors conclude that CNS and PNS pathology of DM2 may be unrelated because of discrepancies within individuals |
| [100] | 95 DM2 | “White matter hyperintensities” in various areas correlated with declines in cognitive domains | Memory and “mental speed” deficits were associated most strongly with white matter hyperintensities in parietal lobe and thalamus |
5. Human cerebral neuropathology of diabetes
Histopathological studies may complement epidemiological, neuropsychological, and neuroradiographical research regarding the effects of diabetes in the human brain. The discovery of a specific anatomical substrate for CNDM2 would provide needed traction for other experimental systems and for developing therapies. Human CNDM2 data will be described in subsections: first, an overview of human studies on lesions described in diabetics’ brains, including a review of the literature concerning specifically whether or not diabetes is linked to AD pathology; second, studies from the UK ADC autopsy cohort include a case–control study on material from the UK ADC Brain Bank to describe our experiences regarding neuropathological findings in the brains of diabetics, with a series of photomicrographs to demonstrate findings in diabetics’ brains including radiographical–pathological correlations; and finally, a summary and conclusions.
6. Peripheral and autonomic nervous system pathology of diabetes
Details about the manifestations of diabetes in the peripheral and autonomic nervous systems are outside the scope of this review. Briefly, peripheral and autonomic neuropathic changes are prevalent, occurring in ~15–25% of diabetic patients [101–107]. Hyperglycemia itself is the main risk factor [103,104]. The often painful “glove and stocking” (long-fiber) sensorimotor polyneuropathy is the most common neuropathic syndrome in Western countries [105,106]. Autonomic neuropathy is selective but affects many systems, and cranial neuropathy (worst in the oculomotor nerve) is also quite prevalent [108,109]. The histopathology for these changes is relatively nonspecific. Nerves show demyelination and remyelination, a dropout of small and large axons, impaired axonal regeneration, Schwann cell dropout, and/or neuritic dystrophy, often with nearby microangiopathy [104,109–117]. The pathological changes may be caused or exacerbated by the polyol sorbital pathways, glycation reactions, oxidative/inflammatory mechanisms, and other pathways [109,110,118–120]. The prevalent but unspecific peripheral nervous system diabetes-related pathology may be relevant biologically to changes in the brain. However, those who have carefully evaluated both in parallel have found the brain pathology far more difficult to discern [85] and/or often disproportionate in degree with the PNS pathology in individual patients [97].
Ultimately, the conclusions from the Pathology of Diabetes, 4th Edition (1966) still holds true: “there is still no agreement on the pathogenesis or basic mechanism of diabetic peripheral neuropathy” (p.273) [121].
7. Histopathology of diabetes in the brain: literature review
7.1. Historical note
Much of the work on diabetes pathology dates from before the advent of the ‘Pubmed Era’. Naturally, these “classical” studies could only localize diabetes-related brain changes using techniques that are somewhat crude by modern standards. However, it is significant that highly observant researchers, superbly trained in anatomic pathology, and in an era with widespread un-controlled diabetes, could find meager evidence of specific changes in the brains of diabetics.
In the Pathology of Diabetes, 4th Edition (1966) [121], the authors designate a chapter to the CNS pathology of diabetes. Here are some relevant excerpts:
“From the brains which we have examined, and from the reports in the literature, there are no changes distinctive of diabetes other than the abnormal glycogen deposits reported in certain cases of diabetic coma…” [121] (p. 279) “…findings of Vonderahe [122] and of Morgan et al. [123] of a reduced number of ganglion cells in the paraventricular regions have not been confirmed…Hagen [124] has described granular inclusions in nerve cells of the hypothalamic region…” [121] (p.280)
Throughout the human body, according to these authors, diabetes affects all blood vessels from the largest to the smallest. However, special focus was merited for arterioles and capillaries:
“Ever since the distinguished studies of Bell [125–127] it has been recognized that arteriolosclerosis is likely to be more severe and extensive in the diabetic, even in the absence of hypertension.” [121] (p.313) “The capillaries have frequently been regarded as the site of ‘diabetic microangiopathy’, largely because of the importance attributed to the nodular glomerular lesion of Kimmelsteil and Wilson as the one specific lesion of diabetes and the popularity of the common, though nonspecific micro-aneurysms of the retinal capillaries.” [121] (p.317)
In the specific context of brain disease, “minor changes are difficult to evaluate since a number follow vascular damage and vascular change is very frequent in the diabetic…” [121] (p.280). Forty-two years after this final edition of The Pathology of Diabetes, pathologists still only appreciate two relatively specific diabetes-related lesions outside the pancreas–Kimmelstiel–Wilson nodules in the kidney and diabetic retinopathy (Fig. 2).
Fig. 2.
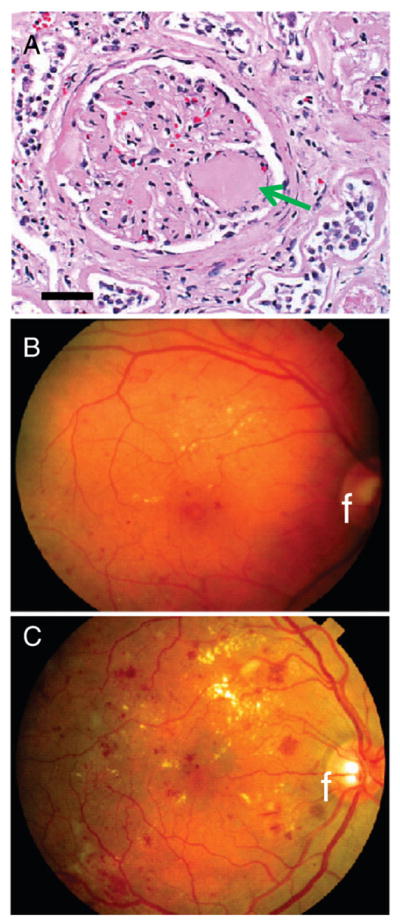
Universally recognized diabetes-related histopathology outside the brain is referent to vascular complications in the kidney and eye. Because of prior studies, these lesions are “pathognomonic” in the sense of indicating diabetes in a patient independent of whether or not the clinical history is well-documented. Diabetic nephropathy involves glomerular vascular lesions termed Kimmelstiel–Wilson nodules (arrow in A). Thin-walled blood vessels in the roughly-spherical glomerulus comprise the anatomical substrate for plasma filtration. With diabetes over a decade in duration, a nodular glomerulosclerosis may develop heralded by albuminuria that can lead to kidney failure. Diabetic retinopathy (B and C) is evaluated using fundus microscopy (fundus indicated by an “f”). The pathology can be parsed into two subtypes: non-proliferative (B) and proliferative (C) retinopathy. Non-proliferative changes (B) are less severe with small capillary micro-aneurysms, dot-type hemorrhages, and microinfarcts (“cotton-wool” spots). This change is associated with gradual but generalized visual dysfunction. Proliferative changes (C) involve neovascularization, fibrosis, and hemorrhages, which can be extensive. Sudden vision loss can occur with vitreous hemorrhage and/or retinal detachment.
7.2. Recent work on cerebral neuropathology of DM2
With regard to more recent ‘conventional’ cerebral histopathological studies, the scientific literature on diabetes/hyperglycemia in human brains has been nearly mute for several decades. A survey of four popular, comprehensive clinical atlases on neuropathology [85, 128–130] provides little information about diabetes-related pathology. Only the Textbook in Neuropathology, 3rd Ed (Davis and Robertson Eds, 1997) [85] has a section dedicated to describing the histopathology of diabetic encephalopathy. This section begins: “Most clinicians regard the cerebral manifestations in diabetes to be due to cerebrovascular disease.” [85] (p.590).
The same neuropathology atlas [85] goes on to enumerate several other histopathological findings associated with diabetic encephalopathy: thickening of cerebral cortical capillary basement membrane; possible abnormality in blood-brain barrier; diffuse degeneration of “ganglion cells” and nerve fibers throughout the brain (attributed to Reske-Nielsen and Lundbaek [86]) or diffuse degeneration of cortical neurons similar to those seen in anoxia and ischemia (attributed to Olsson et al. [131]), with poor correlation to hypertension or uremia. Since these are the only authors with a relatively recent summary of the human neuropathology of diabetic encephalopathy, it is worth quoting their conclusions–.
“Although it is conceivable that the primary ganglion cell abnormalities in diabetic encephalopathy may be related to microangiopathy and increased vascular permeability, many aspects in its pathogenesis are still unknown.” [85] (p.591)
In addition to neuropathology atlases, there have been individual studies about the pathology in diabetics’ brains. Some autopsy series have included evaluations of cerebral pathology in relation to diabetes [86,132–138]; however, there have been few such studies published during the past several decades. Hypothesis-based studies have sought to confirm in human tissues features seen in animal or other models [139–141]. Various findings have been reported, none definitive, except that there is a positive association between diabetes and various strokes [88,132,142–144]. Of the issues pertinent to brain pathology in diabetes that have received most attention, two are conspicuous: the role of cerebrovascular diseases in diabetics, and the pathogenetic connection to AD.
Relatively few recent studies have described in detail the cerebrovascular histopathology linked to diabetes. The scarcity of recent studies stands in contrast to the impressive epidemiological and neuroimaging evidence indicating that diabetic brain dysfunction is mediated at least partly via cerebrovascular disease [88,90,98,132,142–145] (and see below). The lack of human histopathological studies is also remarkable since there are few animal models of diabetes-linked atheromatous brain infarcts [76]. Furthermore, there is cause to re-examine the older autopsy series, because in one study more than a third of patients with cerebral infarcts had an elevated glycosylated hemoglobin although they were not known previously to be diabetics [146,147]. There have been few large human autopsy series performed since that of Aronson (1973) [136]. This study included 4802 non-diabetics and 677 diabetics from consecutive, complete autopsies performed at Kings County Hospital in Providence, RI. In this study, the frequency of encephalomalacia –softening of the brain, often with rarefaction of white matter – was far more frequent in the diabetics. The author concluded that this pathology reflected small-vessel disease that was “presumably not lethal and frequently subclinical, which is distinctly greater” in diabetics [136]. The correlation of diabetes with small-vessel disease is important and has been repeatedly described [90,96,99,148,149]. This type of pathology is associated with clinical manifestations in sharp contrast to the “stroke” syndromes that are characterized by acute, catastrophic neurological deficits that often culminate in severe disability or death. Instead, the neurological deficits referent to small-vessel disease can be subtle but progressive over time [150,151]. The exact mechanisms that underlie small-vessel disease are not well understood.
Although diabetes is most specifically linked to small-vessel disease, it should be underscored that a connection is also firmly established between diabetes and other subtypes of cerebrovascular disease [6,132,145,152–157]. This pertains to large vessel atherosclerosis, lacunar infarcts, thromboembolic stroke, hemorrhagic stroke, and aneurismal subarachnoid infarcts [15,158–167], all of which can produce a spectrum of clinical syndromes. The relative risk for clinically detectable stroke overall in diabetics is in the range of 1.7 to 5.5 (see review in ref. [146]). This is a devastating disease with an incidence of ~760,000/year in the U.S. [168–170], and it is estimated that 11 million more Americans have clinically “silent” strokes per year [169]. Any additional hypotheses about the effects of DM2 on cognition must include the much higher cerebrovascular risk in diabetics as a scientific fact. If additional types of pathological or clinical manifestations are to be suggested, then diabetes-related cerebrovascular disease is also a strong potential experimental confound.
In contrast to the literature concerning cerebrovascular pathology related to diabetes, the association of DM2 with AD pathology is more controversial but has attracted intense scientific attention [171]. This topic is challenging to address from a neutral perspective because the results of the studies are mixed and sometimes seem mutually contradictory. We describe below some of the data and hypotheses –both “pro” and “con”– pertaining to the possibility that AD is pathogenetically linked to DM2.
7.2.1. Data/hypotheses that suggest that AD is linked specifically to DM2
Numerous epidemiological and clinical–pathological studies have reported an increased risk in DM2 patients for developing AD, possibly in connection with ApoE allele 4 [16,154,172–180].
Neuroimaging studies show shrinkage of mesial temporal structures (hippocampus and amygdala) in DM2 patients linked to loss of cognition; these are also areas affected by AD [91,181–183].
The diabetic pancreas contains amyloid substance, similar histologically to that found in AD brain [184,185].
There are inter-related pathways linked to the metabolic syndrome and to dyslipidemia (including cholesterol transport and ApoE alleles) that may credibly affect both DM2 and AD [15,183,186–189].
PET neuroimaging studies have shown brain glucose regulatory deficits even in young adults at risk for developing AD decades later (finding not linked to DM2 per se) [190–193].
AD is linked to hyperglycemia by the hypothesized importance of insulin/IGF-1 regulated pathways, RAGE, PPAR-gamma, and other advanced glycation end-products in AD brain [48,49,66,79,139,140, 182,194–197].
DM2 may potentiate in the brain AD-stimulating pathways that are pertinent to dysfunction in blood-brain barrier, reactive oxygen species, proteases, and leptin or other metabolism-regulatory molecules [48,65,67,69,70,198–201].
Survival bias is an important potential confound that may lead to artificially decreased diagnoses of DM2 and AD [202,203]. Persons with DM2 are at increased risk to die of cardiovascular causes and this risk renders them less likely to die of AD, all other things being equal. This effect may bias studies away from recognizing a positive link between DM2 and AD (this does not explain why DM2 patients are found consistently to have higher risk for stroke, however).
A number of rodent models including therapy-relevant strategies show linkage between DM2-related effects on the rodent brain and AD-relevant pathways [65,66,69,199,204,205].
7.2.2. Data/hypotheses that suggest that AD is not linked specifically to DM2
Numerous epidemiological and clinical–pathological studies have reported that risk for decreased cognition in DM2 patients is not mediated through AD, but through cerebrovascular disease instead [13,14,47,153,206–216].
Confounds related to the metabolic syndrome (hypertension, dyslipidemia, smoking, obesity, genetic factors, and inflammatory mechanisms) also favor brain infarctions, so DM2 may be a strong surrogate for stroke risk and/or low socioeconomic status [8,9,142,143,152,217–220].
Most published neuroimaging findings in DM2 are apparently referent to cerebrovascular disease [90,95–99,144,221,222].
Whatever the mechanism of DM2-related cognitive decline, the presence of that additive dysfunction in someone developing AD would “lower the threshold” to detection, independent of a specific contribution by DM2. This phenomenon was previously demonstrated in AD pathology [223]. This may help explain epidemiological data linking DM2 to AD.
If hyperglycemia and/or poor glycemic control induces AD, then longer-lived juvenile-onset diabetes might be expected to lead to AD, and there is no evidence for this. Several studies have reported that juvenile-onset diabetes patients do not show hippocampal atrophy [224,225].
Whereas advanced glycation products are increased in AD brains, there is not compelling evidence for these markers being elevated in the brains of human DM2 patients [196].
To date, no therapies have been described in humans connected to the hypothesis of DM2–AD link that have worked independently of stroke risk.
In a microenvironment with abundant neuronal death and secondary changes such as the AD brain, there are intuitively good reasons for there to be associations, irrespective of DM2, with deficits in blood-brain barrier, reactive oxygen species, advanced glycation end-products and other inflammatory molecules, and other biochemical perturbations.
Findings in cell culture and rodent models must be scrutinized critically before relevance to the aged human brain – much less to AD pathology – is accepted.
8. The UK ADC experience: a retrospective case–control study from an autopsy convenience sample stratified by identified diagnosis of diabetes, and photomicrographs showing histopathology from select diabetic and non-diabetic cases with clinical–radiographical correlation
8.1. Rationale
8.1.1. Database analyses
Analysis of data about volunteers in a UK ADC research cohort may provide evidence relevant to the controversy about the hypothetical associations between DM2 and cerebrovascular or AD pathology. Our autopsy series includes a group of longitudinally followed individuals who were recruited while non-demented and followed for years. The UK ADC database can be queried to indicate the clinical and pathological indices are associated with DM2 before the advent of severe, debilitating dementia. We hypothesized that pathology in patients with DM2 would reflect their differential vulnerabilities to cerebrovascular disease and/or to AD-type pathology.
8.1.2. Photomicrographs from selected diabetics’ brains
Few recent studies have described histopathological features in aged diabetics’ brains. Some pathological–radiographical studies exist on small-vessel brain disease [226–235], but are not related to diabetes per se. We performed this preliminary experiment in the context of the UK ADC older adult research cohort. These studies include participants who underwent premortem MRI scans to enable radiographical–pathological correlation. This strategy may provide the basis for developing novel hypotheses about mechanism(s) of diabetic encephalopathy.
8.2. Methods: patients, assessments, neuropathology, and analyses
Research protocols were approved by the UK IRB. Details of inclusion criteria and recruitment have been described previously [223]. Patients who had come to autopsy from the UK normal volunteer cohorts were the basis for the study (total N=234 patients), with some patients excluded for a variety of factors including tumors, large contusions, missing clinical data, etc., as described previously (N=95 excluded) [223]. These patients were relatively intact cognitively, having been recruited as non-demented persons and followed for years. Their pathology (if any) was presumed to represent the earlier phases of cognitive decline. Only patients who had come to autopsy with either “Yes” (N=50) or “No” (N=89) in the UK ADC database for a diagnosis of diabetes were used (total N=139). These diagnoses were derived from medical examinations during life, which included blood evaluations, as well as from extensive evaluations of medical charts as part of the UK ADC database management. Demographic characteristics and particular clinical indices from these groups are shown in Table 2. All initially normal individuals were contacted at 6-month intervals, had detailed mental status testing, and had neurological and physical examinations at least annually. Mental status testing and neuropathological assessments were described in detail previously [223]. Simple means were obtained from each group for the clinical and pathological indices shown (Table 3). Significance was tested via Student’s t test (unpaired, two-tailed).
Table 2.
UK ADC: demographics and clinical indices
| Diabetics | Non-diabetics | p-value | |
|---|---|---|---|
| Demographics | |||
| N | 50 | 89 | – |
| Age at death, yrs (mean±SD) | 84.7±8.0 | 88.2±6.7 | <0.011 |
| Sex (%F) | 54 | 63 | NS2 |
| Formal education, yrs (mean±SD) | 15.6±2.6 | 16.0±2.0 | NS1 |
| ApoE alleles (2/3/4), % | 4/84/12 | 11/76/13 | NS3 |
| Last MMSE score (mean±SD) | 26.4±5.1 | 24.4±7.4 | NS1 |
| Interval between last evaluation and death, yrs (mean±SD) | 0.64 | 0.96 | NS1 |
| Clinical parameters | |||
| Depression, % | 20 | 20 | NS2 |
| CABG, % | 14 | 7 | NS3 |
| Peripheral vascular disease, % | 26 | 15 | NS2 |
| TIA, % | 14 | 8 | NS2 |
| Hypertension, % | 64 | 55 | NS2 |
| Daily intake of drugs/meds (mean±SD) | 12.2±7.7 | 19.8±8.5 | <0.00011 |
The demographic and clinical characteristics between diabetics (N=50) and non-diabetics (N=89) in the BRAiNS program at the UK ADC. This group, which was recruited from non-demented individuals, has been described previously [245]. The demographic and clinical indices are similar between diabetics and non-diabetics. The clinical parameters were dichotomous (0 or 1) except for “Number of drugs” (range: 2–39). Definitions: ApoE = Apolipoprotein E; MMSE = Mini-mental status examination (0–30 scale); CABG = coronary artery bypass graft operation; TIA = history of transient ischemic attack(s).
Statistical tests:
Two-tailed Student’s t test
Chi-square
Fisher’s exact test.
Table 3.
UK ADC: pathological indices
| Diabetics | Non-diabetics | p-value | |
|---|---|---|---|
| Pathological parameters, non-Alzheimer’s type | |||
| Lacunar infarcts, % | 14 | 7 | NS2 |
| Micro-infarcts, % | 52 | 30 | 0.012 |
| Large infarcts, % | 2 | 10 | NS2 |
| Hemorrhagic infarcts, % | 8 | 4 | NS2 |
| Any infarcts, % | 56 | 37 | 0.032 |
| Hippocampal sclerosis, % | 9 | 6 | NS2 |
| Argyrophilic grains, % | 22 | 26 | NS2 |
| Lewy bodies in isocortex, % | 6 | 11 | NS2 |
| Brain wgt, g (mean±SD) | 1196±134 | 1193±139 | NS1 |
| Pathological parameters, Alzheimer’s type | |||
| Braak stage (median, range) | 2 (0–6) | 3 (0–6) | NS3 |
| CERAD score (median, range) | 0 (0–3) | 2 (0–3) | 0.023 |
| NIARI score (median, range) | 0 (0–3) | 1 (0–3) | 0.033 |
| Probable or definite AD, % | 24 | 22 | NS2 |
| NFT counts (mean±SD) | |||
| Temporal lobe | 1.9±4.62 | 3.4±8.8 | NS1 |
| Frontal lobe | 0.6±1.9 | 1.3±3.4 | NS1 |
| Parietal lobe | 0.8±2.3 | 1.8±4.8 | NS1 |
| Hippocampal CA1 | 9.5±15.2 | 12.1±21.4 | NS1 |
| Subiculum | 10.9±16.8 | 25.3±40.0 | 0.0041 |
| Neuritic plaque counts (mean±SD) | |||
| Temporal lobe | 3.9±5.9 | 6.4±7.4 | 0.041 |
| Frontal lobe | 4.6±6.9 | 6.6±7.3 | NS1 |
| Parietal lobe | 5.3±7.6 | 7.5±8.3 | NS1 |
| Hippocampal CA1 | 1.5±3.0 | 1.1±2.4 | NS1 |
| Subiculum | 1.9±3.9 | 1.9±3.6 | NS1 |
Pathological indices stratified by diabetics (N=50) and non-diabetics (N=89) in the BRAiNS program at the UK ADC. Note that small infarcts tended to be present more often in diabetics, but AD-related pathology tended to be slightly more abundant in non-diabetics.
Statistical tests:
Two-tailed Student’s t test
Chi-square
Wilcoxon Rank Sum.
A sample of convenience was selected for photomicroscopy:
Diabetes Case 1 (male) died at the age of 88 with history of DM2. Last MMSE score was 26 out of a possible 30 (mild cognitive decline).
Diabetes Case 2 (male) died at the age of 87 with history of DM2. Last MMSE score was 21 out of a possible 30 (moderate-to-severe cognitive decline). MRI was obtained within 4 months of patient’s death.
Diabetes Case 3 (female) died at the age of 81 with history of DM2. Last MMSE score was 29. MRI was obtained within a year before patient’s death.
Diabetes Case 4 (female) died at age 82 with history of DM2 and diagnosed clinically with AD. Last MMSE score was 19 (severe cognitive decline). MRI was obtained 5 years before patient’s death.
Diabetes Case 5 (male) died at age 75 with history of DM2. Last MMSE score was 28 (non-demented). MRI was obtained four years before patient’s death.
Diabetic Case 6 (female) died at age 72 years with history of poorly controlled DM2, last MMSE score 29 (non-demented).
Control Case 1 (male) died at age 78 years with no history of diabetes, last MMSE score of 28 (non-demented).
Control Case 2 (male) died at age 99 years no history of diabetes but with a history of hypertension, last MMSE score of 26 (mild cognitive impairment).
8.3. Results
This retrospective case–control study from an autopsy convenience sample includes patients that are relatively well matched with regard to levels of formal education and ApoE alleles (Table 2). However, the diabetics died at a slightly younger age (84 years versus 88 years, p<0.02 by two-tailed Student’s t test), and tended to have a slightly higher final MMSE scores (26 versus 24, p<0.1 by two-tailed Student’s t test). In terms of pathological parameters, the diabetics tended to have more small infarcts but a slightly less degree of AD-type lesions (Table 3). AD lesions (NFTs and neuritic plaques) are directly quantified by counting or defined according to Consortium to Establish a Registry for AD staging and Braak staging, which are based on consensus criteria for staging AD pathological severity [236,237] (Table 3).
Photomicrographs portray a spectrum of changes in these older persons with differing cognitive changes. The photomicrographs show hematoxylin and eosin (H and E)-stained sections with a special focus on changes in blood vessels and white matter. Histology of cerebral blood vessels and the surrounding Virchow–Robin spaces are shown in Fig. 3 (from Control Case 1 and Diabetes Case 1). Fig. 4 depicts a patient (Diabetes Case 2) with an MRI obtained just prior to a significant decline in cognition as reflected in a drop in MMSE scores (down to 21 near death). In this patient, there were moderate changes referent to Alzheimer’s pathology (Braak stage 3), but the predominant pathological changes were related to the cerebral vasculature. In Diabetes Case 3 (Fig. 5), there were also cerebrovascular disease changes including frank infarctions in the frontal cortices bilaterally. There were small infarcts in the temporal lobe including the hippocampal formation, where there was also minimal Alzheimer’s-type pathology. By contrast, in Diabetes Case 4 (Fig. 6), there was both dementia clinically as well as advanced AD pathologically. Still, even in this case, notable small-vessel cerebrovascular disease was present. Diabetes Case 5 (Fig. 7) had no dementia, no Alzheimer-type pathology, but had subtle white matter pathology including some periventricular enhancement near the basal ganglia. The photomicrograph shows the corresponding region that contained expanded Virchow–Robin spaces and many corpora amylacea. In Diabetes Case 6 (Fig. 8), a patient with poorly controlled DM2 and mild cognitive decline, there were many corpora amylacea within the cornu ammonis of the hippocampal formation, as well as near the inferior horn of the lateral ventricle.
Fig. 3.
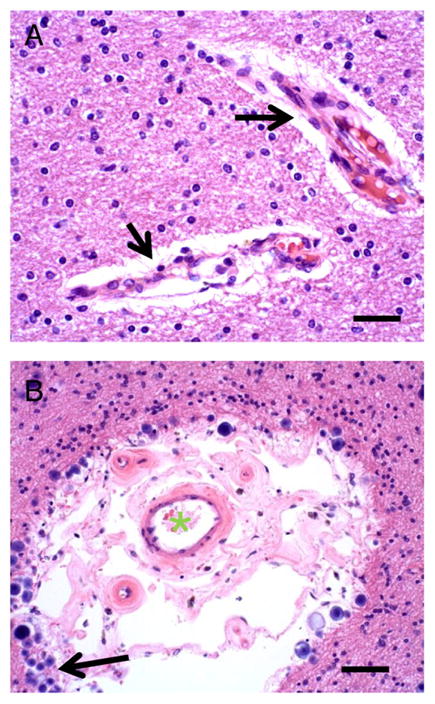
Control Case 1 (A) shows normal appearance of small arteries in white matter (arrows). Surrounding the blood vessels, partly as an artifact of fixation, is a space without cells or parenchyma (Virchow–Robin space). Scale bar=150 μm. (B) in some cases, such as this person (Diabetes Case 1, an 88-year old male with mild cognitive impairment), the Virchow–Robin space is enlarged. The lumen of the blood vessel is shown with a green “*”. Note that in the Virchow–Robin space are extra blood vessels, and the tissue surrounding has many corpora amylacea (arrow). Scale bar=100 μm.
Fig. 4.

Diabetes Case 2–87-year old male with dementia and predominantly cerebrovascular disease by pathology. A shows a chart that depicts the MMSE scores by this patient. Note that the MRI was obtained prior to a significant decrease in the patient’s MMSE score (final score =21). B and C show MRI scans that show hydrocephalus ex vacuo and extensive pathological white matter enhancement. The red box in C shows the area depicted in photomicrographs D and E. D shows a small blood vessel in the white matter of visual cortex away from the ventricle. This vascular profile shows expansion of the Virchow–Robin space with organizing cellular material that includes new small blood vessels (arrow). E shows the area immediately subjacent to the ventricle with frank necrosis (vertical arrow) and calcification (horizontal arrow). Scale bars=150 μm in D and 300 μm in E.
Fig. 5.

Diabetes Case 3–81-year old female with mild cognitive impairment and with predominantly cerebrovascular pathology (final MMSE score=29). (A and B) MRIs show the infarctions in the frontal cortices (arrow in A) and the subtle hippocampal atrophy (B). Histopathology confirmed the presence of frontal cortex infarcts (not shown). C and D show hippocampal histopathology. In this patient, there were blood vessel profiles with expanded Virchow–Robin spaces with many corpora amylacea (C) and there were small infarcts and areas of white matter rarefaction in the fimbria fornix (D), however, Alzheimer’s-type pathology in CA1 of the hippocampus was mild (not shown). Scale bar=150 μm in C, 250 μm in D.
Fig. 6.

Diabetes Case 4–82-year old female diabetic patient with dementia and Alzheimer’s disease diagnosis during life (last MMSE score=19). A and B show the final MRI that was obtained 4 years prior to the patient’s demise. This already showed hippocampal atrophy (arrow in A) but also some periventricular white matter lesions (such as in arrow in B). Photomicrographs show the histopathological features from the red boxes. C depicts a section from CA1 field of the hippocampus stained with the Gallyas silver impregnation technique and shows severe involvement by Alzheimer’s-type neuritic plaques (arrow) and many NFTs. This patient had Braak stage 6 and satisfied CERAD criteria for “Definite Alzheimer’s disease” by pathology. In addition to the AD pathology, there was also some cerebrovascular disease including areas with rarefaction of white matter. D is a section from the left parietal lobe (box in B) which shows an expanded Virchow–Robin space with organized cellular and acellular material. In an 82-year old patient such as this, some degree of concomitant pathology is the rule and not the exception. Scale bars=150 μm in C, 100 μm in D.
Fig. 7.

Diabetes Case 5–75-year old male diabetic patient with no dementia and with subtle changes on MRI (last MMSE score 29). A shows the MRI with mild periventricular white matter changes including some enhancement near in the subependymal basal ganglia. B depicts a photomicrograph from the same area, which includes pathology surrounding medium-sized blood vessels with expanded Virchow–Robin spaces. Surrounding the vessels are many corpora amylacea and gliosis. Scale bar=150 μm.
Fig. 8.
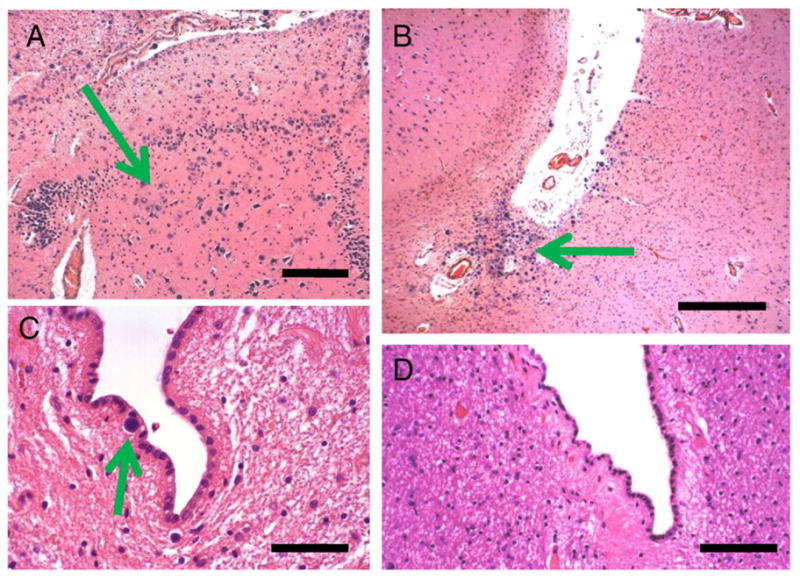
Diabetes Case 6–72-year old female patient with poorly controlled diabetes (two confirmed readings in excess of 150 mg/dl) and mild cognitive decline. In this case, the hippocampal formation showed many corpora amylacea including in the cornu ammonis subfields. These are shown in the CA4/dentate gyrus area, where there is some effacement of the normal cytoarchitecture in association with the presence of many corpora amylacea. B shows the crux of the inferior horn of the lateral ventricle, which also contained many corpora amylacea. By contrast, in Control Cases 1 and 2, there were few (arrow in C) or no corpora amylacea there. Scale bars: 300 μm in A and B, 150 μm in C and D.
8.4. Discussion
This study focused on persons who were recruited without cognitive impairment and followed longitudinally in a research clinic. This is an important strength of the study because long-term medical and neuropsychological studies could be performed on each patient. It also allowed us to minimize various recruitment biases related to already-demented subjects. However, there are several caveats that are germane to these data. Most importantly, the degree of glycemic control was not thoroughly documented by an endocrinologist in all patients. This is only a study of the association of a clinical diagnosis of diabetes, rather than an association that is related to glycemic control per se. The importance of the age difference (average age at death 84 for diabetics, 88 for non-diabetics, p<0.02) may constitute an important confound to this analysis. However, note that the cerebrovascular pathology, despite the age difference, was more severe in the diabetics. There are other trends, including a trend to increased hypertension, in the diabetics that may be contributory to the different prevalence of cerebrovascular disease. In summary, results from our database are in agreement with prior studies indicating that DM2 is associated with increased risk for cerebrovascular disease, and yet there is no positive association between the diagnosis of DM2 and the development of AD pathology [13,14,47,153,206–216].
We also performed a study of a subset of patients including pathological–radiographical correlation. This preliminary, descriptive study underscores our impression from the research literature: cerebrovascular pathology, including extensive small-vessel disease, is an important component of CNDM2. This type of pathology can exist in relative isolation, or together with other diseases such as AD. It is hoped that the pathological–radiographical correlation will help some clinicians to “visualize” at a cellular and sub-cellular level the radiographical changes that have been recorded in the brains of DM2 patients.
In this limited sample, there were relatively many corpora amylacea in the brains of the diabetic patients. By contrast, in the non-diabetics (Control Case 1 and Case 2) there were few or no corpora amylacea in this location. The presence of corporal amylacea in the brains of older patients is generally considered a nonspecific sign of tissue damage or cell loss. However, since these lesions are indeed aberrant, ubiquitinated intracellular deposits of glycated material [238,239], it is possible that their presence in the brains of diabetics may have specific and pathogenetic implications.
These results are neither intended, nor statistically powered, to make comparative assessments. They offer a preliminary, and purely descriptive, portrayal of some of the histopathological features in a limited subset of patients with DM2. However, we hope that these photomicrographs may help demonstrate the potential value of evaluating the histopathological features in human brains with well-documented antemortem characteristics.
9. Summary and conclusions
In the absence of a known pathognomonic anatomic substrate for cognitive dysfunction in diabetics, the question arises–perhaps there are no definitive histopathological changes in diabetic brains? Many metabolic disorders induce mental changes, or delirium, out of proportion to known neuropathological changes [240,241]. Such may be induced by fluxes in blood levels of insulin, glucose, and other metabolic parameters in DM2. Other diseases such as hypoxia can produce brain atrophy and cell death in the absence of a pathognomonic change [85,128]. Hence, delirium or a disease with entirely nonspecific pathology may partly contribute to diabetic encephalopathy.
With the above caveat about the specificity of CNDM2, several associations between DM2 and brain pathology appear to exist (Fig. 9). Neuropsychological studies have indicated that there is enduring cognitive deficits in DM2 patients. There is compelling evidence from neuroimaging studies in humans that there are changes in brain parenchyma that are present disproportionately in the brains of diabetics. The specificity of these changes cannot be reliably ruled out because relatively few studies have tackled the histopathology of diabetes in the brain over the past few decades. This is unfortunate because the changing times have produced new tools available to neuropathologists. There is also increased prevalence of DM2 in Western populations [1] and it would be helpful to evaluate brains in the context of current trends in comorbidities and treatments.
Fig. 9.

Research has provided insights into diabetes-related cognitive dysfunction. However, the specifics are unclear about how the chemical perturbations of diabetes correlate to the hypothesized anatomic substrates associated with “diabetic encephalopathy”. In turn, the contribution to cognitive changes from the pathological lesions is poorly understood. The dashed arrow and question mark at the bottom indicate the possibility that metabolic perturbations in diabetes may produce cognitive changes in the absence of detectable anatomic pathology.
As with many other past studies, we are unable to supply new and definitive answers about CNDM2 based on our illustrative cases and analyses. It is somewhat surprising that there have been so few studies specifically addressing the neuropathology of small-vessel disease in DM2. Moreover, a few questions may be worthy of being addressed. These questions derive from a synthesis of the known scientific literature and the preliminary and descriptive studies that are presented above.
Are corpora amylacea really benign and/or nonspecific in DM2 brains? Perhaps these structures, comprising glycated material [130,239], may cause or reflect a more specific disease process than previously thought. Corpora amylacea have previously been noted in the context of brain pathology [238,242–244], but not correlated to the diagnosis of diabetes per se.
Are there specific markers for microangiopathy in DM2?
Is there a specific process involving Virchow–Robin spaces in DM2?
Why do the results of studies about the relationship between AD and DM2 vary so much?
Future studies are needed to address these and the other many outstanding questions regarding CNDM2. This field is increasingly topical as the number of DM2 patients increase, and as the average age and longevities of Western populations increase. Many experimental systems should be brought to bear in studying this widespread disease, including the direct evaluation of human brain tissue.
Acknowledgments
We are deeply grateful to all of the participants in our longitudinal aging study and to the patients with Alzheimer’s disease in our Alzheimer’s Disease Center’s research clinic. We thank Richard Kryscio, PhD and Marta Mendiondo, PhD for data management and statistical support, Ela Patel, Ann Tudor, Paula Thomason, Dr. Huaichen Liu, and Sonya Anderson, for technical support, and Gregory Cooper, MD, PhD, Nancy Stiles, MD, and Allison Caban-Holt, PhD, for clinical evaluations.
Footnotes
This study was supported by grants 5-P30-AG028383 and K08 NS050110 from the National Institutes of Health, Bethesda, MD, and a grant from the Healy Family Foundation.
Publisher's Disclaimer: This article appeared in a journal published by Elsevier. The attached copy is furnished to the author for internal non-commercial research and education use, including for instruction at the authors institution and sharing with colleagues.
Other uses, including reproduction and distribution, or selling or licensing copies, or posting to personal, institutional or third party websites are prohibited.
In most cases authors are permitted to post their version of the article (e.g. in Word or Tex form) to their personal website or institutional repository. Authors requiring further information regarding Elsevier’s archiving and manuscript policies are encouraged to visit:
References
- 1.Adeghate E, Schattner P, Dunn E. An update on the etiology and epidemiology of diabetes mellitus. Ann N Y Acad Sci. 2006;1084:1–29. doi: 10.1196/annals.1372.029. [DOI] [PubMed] [Google Scholar]
- 2.James WP. The epidemiology of obesity: the size of the problem. J Intern Med. 2008;263:336–352. doi: 10.1111/j.1365-2796.2008.01922.x. [DOI] [PubMed] [Google Scholar]
- 3.Alrefai H, Allababidi H, Levy S, Levy J. The endocrine system in diabetes mellitus. Endocrine. 2002;18:105–119. doi: 10.1385/ENDO:18:2:105. [DOI] [PubMed] [Google Scholar]
- 4.Mooradian AD. Pathophysiology of central nervous system complications in diabetes mellitus. Clin Neurosci. 1997;4:322–326. [PubMed] [Google Scholar]
- 5.Biessels GJ, Deary IJ, Ryan CM. Cognition and diabetes: a lifespan perspective. Lancet Neurol. 2008;7:184–190. doi: 10.1016/S1474-4422(08)70021-8. [DOI] [PubMed] [Google Scholar]
- 6.Wolf PA, Kannel WB, Dawber TR. Prospective investigations: the Framingham study and the epidemiology of stroke. Adv Neurol. 1978;19:107–120. [PubMed] [Google Scholar]
- 7.Dursun B, Dursun E, Capraz I, Ozben T, Apaydin A, Suleymanlar G. Are uremia, diabetes, and atherosclerosis linked with impaired antioxidant mechanisms? J Investig Med. 2008;56:545–552. doi: 10.2310/JIM.0b013e3181641ce3. [DOI] [PubMed] [Google Scholar]
- 8.Nagpal J, Bhartia A. Cardiovascular risk profile of subjects with known diabetes from the middle- and high-income group population of Delhi: the DEDICOM survey. Diabet Med. 2008;25:27–36. doi: 10.1111/j.1464-5491.2007.02307.x. [DOI] [PubMed] [Google Scholar]
- 9.Voeks JH, McClure LA, Go RC, Prineas RJ, Cushman M, Kissela BM, Roseman JM. Regional differences in diabetes as a possible contributor to the geographic disparity in stroke mortality: the REasons for Geographic And Racial Differences in Stroke Study. Stroke. 2008;39(6):1675–1680. doi: 10.1161/STROKEAHA.107.507053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ding EL, Powe NR, Manson JE, Sherber NS, Braunstein JB. Sex differences in perceived risks, distrust, and willingness to participate in clinical trials: a randomized study of cardiovascular prevention trials. Arch Intern Med. 2007;167:905–912. doi: 10.1001/archinte.167.9.905. [DOI] [PubMed] [Google Scholar]
- 11.Herpertz S, Albus C, Kielmann R, Hagemann-Patt H, Lichtblau K, Kohle K, Mann K, Senf W. Comorbidity of diabetes mellitus and eating disorders: a follow-up study. J Psychosom Res. 2001;51:673–678. doi: 10.1016/s0022-3999(01)00246-x. [DOI] [PubMed] [Google Scholar]
- 12.Morgenstern LB, Steffen-Batey L, Smith MA, Moye LA. Barriers to acute stroke therapy and stroke prevention in Mexican Americans. Stroke. 2001;32:1360–1364. doi: 10.1161/01.str.32.6.1360. [DOI] [PubMed] [Google Scholar]
- 13.Akomolafe A, Beiser A, Meigs JB, Au R, Green RC, Farrer LA, Wolf PA, Seshadri S. Diabetes mellitus and risk of developing Alzheimer disease: results from the Framingham Study. Arch Neurol. 2006;63:1551–1555. doi: 10.1001/archneur.63.11.1551. [DOI] [PubMed] [Google Scholar]
- 14.Beeri MS, Silverman JM, Davis KL, Marin D, Grossman HZ, Schmeidler J, Purohit DP, Perl DP, Davidson M, Mohs RC, Haroutunian V. Type 2 diabetes is negatively associated with Alzheimer’s disease neuropathology. J Gerontol A Biol Sci Med Sci. 2005;60:471–475. doi: 10.1093/gerona/60.4.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blair CK, Folsom AR, Knopman DS, Bray MS, Mosley TH, Boerwinkle E. APOE genotype and cognitive decline in a middle-aged cohort. Neurology. 2005;64:268–276. doi: 10.1212/01.WNL.0000149643.91367.8A. [DOI] [PubMed] [Google Scholar]
- 16.Peila R, Rodriguez BL, Launer LJ. Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: the Honolulu-Asia Aging Study. Diabetes. 2002;51:1256–1262. doi: 10.2337/diabetes.51.4.1256. [DOI] [PubMed] [Google Scholar]
- 17.Stewart R, Liolitsa D. Type 2 diabetes mellitus, cognitive impairment and dementia. Diabet Med. 1999;16:93–112. doi: 10.1046/j.1464-5491.1999.00027.x. [DOI] [PubMed] [Google Scholar]
- 18.Bober E, Buyukgebiz A. Hypoglycemia and its effects on the brain in children with type 1 diabetes mellitus. Pediatr Endocrinol Rev. 2005;2:378–382. [PubMed] [Google Scholar]
- 19.McNay EC. The impact of recurrent hypoglycemia on cognitive function in aging. Neurobiol Aging. 2005;26(Suppl 1):76–79. doi: 10.1016/j.neurobiolaging.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 20.Warren RE, Frier BM. Hypoglycaemia and cognitive function. Diabetes Obes Metab. 2005;7:493–503. doi: 10.1111/j.1463-1326.2004.00421.x. [DOI] [PubMed] [Google Scholar]
- 21.Frier BM. Morbidity of hypoglycemia in type 1 diabetes. Diabetes Res Clin Pract. 2004;65(Suppl 1):S47–52. doi: 10.1016/j.diabres.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 22.Cryer PE. Symptoms of hypoglycemia, thresholds for their occurrence, and hypoglycemia unawareness. Endocrinol Metab Clin North Am. 1999;28:495–500. v–vi. doi: 10.1016/s0889-8529(05)70084-0. [DOI] [PubMed] [Google Scholar]
- 23.Auer RN. Hypoglycemic brain damage. Metab Brain Dis. 2004;19:169–175. doi: 10.1023/b:mebr.0000043967.78763.5b. [DOI] [PubMed] [Google Scholar]
- 24.Auer RN, Siesjo BK. Hypoglycaemia: brain neurochemistry and neuropathology. Baillieres Clin Endocrinol Metab. 1993;7:611–625. doi: 10.1016/s0950-351x(05)80210-1. [DOI] [PubMed] [Google Scholar]
- 25.de Courten-Myers GM, Xi G, Hwang JH, Dunn RS, Mills AS, Holland SK, Wagner KR, Myers RE. Hypoglycemic brain injury: potentiation from respiratory depression and injury aggravation from hyperglycemic treatment overshoots. J Cereb Blood Flow Metab. 2000;20:82–92. doi: 10.1097/00004647-200001000-00012. [DOI] [PubMed] [Google Scholar]
- 26.Sarafidis PA. Thiazolidinediones and diabetic nephropathy: need for a closer examination? J Cardiometab Syndr. 2007;2:297–301. doi: 10.1111/j.1559-4564.2007.07834.x. [DOI] [PubMed] [Google Scholar]
- 27.Tibaldi J. Actions of insulin beyond glycemic control: a perspective on insulin detemir. Adv Ther. 2007;24:868–882. doi: 10.1007/BF02849980. [DOI] [PubMed] [Google Scholar]
- 28.Blaschke F, Spanheimer R, Khan M, Law RE. Vascular effects of TZDs: new implications. Vascul Pharmacol. 2006;45:3–18. doi: 10.1016/j.vph.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 29.Khan AH, Pessin JE. Insulin regulation of glucose uptake: a complex interplay of intracellular signalling pathways. Diabetologia. 2002;45:1475–1483. doi: 10.1007/s00125-002-0974-7. [DOI] [PubMed] [Google Scholar]
- 30.Horecker BL. The biochemistry of sugars. Int Z Vitam Ernahrungsforsch Beih. 1976;15:1–21. [PubMed] [Google Scholar]
- 31.Cerami A, Vlassara H, Brownlee M. Glucose and aging. Sci Am. 1987;256:90–96. doi: 10.1038/scientificamerican0587-90. [DOI] [PubMed] [Google Scholar]
- 32.Vlassara H, Brownlee M, Cerami A. Nonenzymatic glycosylation: role in the pathogenesis of diabetic complications. Clin Chem. 1986;32:B37–41. [PubMed] [Google Scholar]
- 33.Schousboe A, Bak LK, Sickmann HM, Sonnewald U, Waagepetersen HS. Energy substrates to support glutamatergic and GABAergic synaptic function: role of glycogen, glucose and lactate. Neurotox Res. 2007;12:263–268. doi: 10.1007/BF03033909. [DOI] [PubMed] [Google Scholar]
- 34.Iadecola C, Nedergaard M. Glial regulation of the cerebral microvasculature. Nat Neurosci. 2007;10:1369–1376. doi: 10.1038/nn2003. [DOI] [PubMed] [Google Scholar]
- 35.Bolanos JP, Herrero-Mendez A, Fernandez-Fernandez S, Almeida A. Linking glycolysis with oxidative stress in neural cells: a regulatory role for nitric oxide. Biochem Soc Trans. 2007;35:1224–1227. doi: 10.1042/BST0351224. [DOI] [PubMed] [Google Scholar]
- 36.McKenna MC. The glutamate–glutamine cycle is not stoichiometric: fates of glutamate in brain. J Neurosci Res. 2007;85:3347–3358. doi: 10.1002/jnr.21444. [DOI] [PubMed] [Google Scholar]
- 37.Nehlig A, Coles JA. Cellular pathways of energy metabolism in the brain: is glucose used by neurons or astrocytes? Glia. 2007;55:1238–1250. doi: 10.1002/glia.20376. [DOI] [PubMed] [Google Scholar]
- 38.Sima AA, Kamiya H. Is C-peptide replacement the missing link for successful treatment of neurological complications in type 1 diabetes? Curr Drug Targets. 2008;9:37–46. doi: 10.2174/138945008783431745. [DOI] [PubMed] [Google Scholar]
- 39.Sima AA, Li ZG. The effect of C-peptide on cognitive dysfunction and hippocampal apoptosis in type 1 diabetic rats. Diabetes. 2005;54:1497–1505. doi: 10.2337/diabetes.54.5.1497. [DOI] [PubMed] [Google Scholar]
- 40.Li ZG, Sima AA. C-peptide and central nervous system complications in diabetes. Exp Diabesity Res. 2004;5:79–90. doi: 10.1080/15438600490424550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jezova D, Vigas M, Sadlon J. C-peptide-like material in rat brain: response to fasting and glucose ingestion. Endocrinol Exp. 1985;19:261–266. [PubMed] [Google Scholar]
- 42.Dorn A, Rinne A, Bernstein HG, Hahn HJ, Ziegler M. Insulin and C-peptide in human brain neurons (insulin/C-peptide/brain peptides/immunohistochemistry/radioimmunoassay) J Hirnforsch. 1983;24:495–499. [PubMed] [Google Scholar]
- 43.Brands AM, Kessels RP, de Haan EH, Kappelle LJ, Biessels GJ. Cerebral dysfunction in type 1 diabetes: effects of insulin, vascular risk factors and blood-glucose levels. Eur J Pharmacol. 2004;490:159–168. doi: 10.1016/j.ejphar.2004.02.053. [DOI] [PubMed] [Google Scholar]
- 44.Guisado R, Arieff AI. Neurologic manifestations of diabetic comas: correlation with biochemical alterations in the brain. Metabolism. 1975;24:665–679. doi: 10.1016/0026-0495(75)90146-8. [DOI] [PubMed] [Google Scholar]
- 45.Harati Y. Diabetes and the nervous system. Endocrinol Metab Clin North Am. 1996;25:325–359. doi: 10.1016/s0889-8529(05)70327-3. [DOI] [PubMed] [Google Scholar]
- 46.Kodl CT, Seaquist ER. Cognitive dysfunction and diabetes mellitus. Endocr Rev. 2008;29(4):494–511. doi: 10.1210/er.2007-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Panza F, Solfrizzi V, Colacicco AM, D’Introno A, Capurso C, Palasciano R, Todarello O, Capurso S, Pellicani V, Capurso A. Cerebrovascular disease in the elderly: lipoprotein metabolism and cognitive decline. Aging Clin Exp Res. 2006;18:144–148. doi: 10.1007/BF03327430. [DOI] [PubMed] [Google Scholar]
- 48.Qiu WQ, Folstein MF. Insulin, insulin-degrading enzyme and amyloid-beta peptide in Alzheimer’s disease: review and hypothesis. Neurobiol Aging. 2006;27:190–198. doi: 10.1016/j.neurobiolaging.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 49.Takeuchi M, Yamagishi S. Possible involvement of advanced glycation end-products (AGEs) in the pathogenesis of Alzheimer’s disease. Curr Pharm Des. 2008;14:973–978. doi: 10.2174/138161208784139693. [DOI] [PubMed] [Google Scholar]
- 50.Ginsberg MD, Prado R, Dietrich WD, Busto R, Watson BD. Hyperglycemia reduces the extent of cerebral infarction in rats. Stroke. 1987;18:570–574. doi: 10.1161/01.str.18.3.570. [DOI] [PubMed] [Google Scholar]
- 51.McCall AL. In: Cerebral Microvascular Transport and Metabolism: Implications for Diabetes. Ruderman N, Williamson J, Brownlee M, editors. Oxford University Press; New York: 1992. pp. 59–106. [Google Scholar]
- 52.Prado R, Ginsberg MD, Dietrich WD, Watson BD, Busto R. Hyperglycemia increases infarct size in collaterally perfused but not end-arterial vascular territories. J Cereb Blood Flow Metab. 1988;8:186–192. doi: 10.1038/jcbfm.1988.48. [DOI] [PubMed] [Google Scholar]
- 53.Smith ML, von Hanwehr R, Siesjo BK. Changes in extra- and intracellular pH in the brain during and following ischemia in hyperglycemic and in moderately hypoglycemic rats. J Cereb Blood Flow Metab. 1986;6:574–583. doi: 10.1038/jcbfm.1986.104. [DOI] [PubMed] [Google Scholar]
- 54.Zasslow MA, Pearl RG, Shuer LM, Steinberg GK, Lieberson RE, Larson CP., Jr Hyperglycemia decreases acute neuronal ischemic changes after middle cerebral artery occlusion in cats. Stroke. 1989;20:519–523. doi: 10.1161/01.str.20.4.519. [DOI] [PubMed] [Google Scholar]
- 55.Berger K, Ajani UA, Kase CS, Gaziano JM, Buring JE, Glynn RJ, Hennekens CH. Light-to-moderate alcohol consumption and risk of stroke among U.S. male physicians. N Engl J Med. 1999;341:1557–1564. doi: 10.1056/NEJM199911183412101. [DOI] [PubMed] [Google Scholar]
- 56.Ferreira MP, Willoughby D. Alcohol consumption the good the bad and the indifferent. Appl Physiol Nutr Metab. 2008;33:12–20. doi: 10.1139/H07-175. [DOI] [PubMed] [Google Scholar]
- 57.Sesso HD. Alcohol and cardiovascular health: recent findings. Am J Cardiovasc Drugs. 2001;1:167–172. doi: 10.2165/00129784-200101030-00002. [DOI] [PubMed] [Google Scholar]
- 58.Gronbaek M. Alcohol, type of alcohol, and all-cause and coronary heart disease mortality. Ann N Y Acad Sci. 2002;957:16–20. doi: 10.1111/j.1749-6632.2002.tb02902.x. [DOI] [PubMed] [Google Scholar]
- 59.Shaper AG, Wannamethee SG. The J-shaped curve and changes in drinking habit. Novartis Found Symp. 1998;216:173–188. doi: 10.1002/9780470515549.ch11. discussion 188–192. [DOI] [PubMed] [Google Scholar]
- 60.Gaziano JM, Buring JE. Alcohol intake, lipids and risks of myocardial infarction. Novartis Found Symp. 1998;216:86–95. doi: 10.1002/9780470515549.ch7. discussion 95–110. [DOI] [PubMed] [Google Scholar]
- 61.Poikolainen K. Alcohol and mortality: a review. J Clin Epidemiol. 1995;48:455–465. doi: 10.1016/0895-4356(94)00174-o. [DOI] [PubMed] [Google Scholar]
- 62.Rosenfeld L. Insulin: discovery and controversy. Clin Chem. 2002;48:2270–2288. [PubMed] [Google Scholar]
- 63.Murray I. The search for insulin. Scott Med J. 1969;14:286–293. doi: 10.1177/003693306901400807. [DOI] [PubMed] [Google Scholar]
- 64.Biessels GJ, Gispen WH. The impact of diabetes on cognition: what can be learned from rodent models? Neurobiol Aging. 2005;26(Suppl 1):36–41. doi: 10.1016/j.neurobiolaging.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 65.Hamel E, Nicolakakis N, Aboulkassim T, Ongali B, Tong XK. Oxidative stress and cerebrovascular dysfunction in mouse models of Alzheimer’s disease. Exp Physiol. 2008;93:116–120. doi: 10.1113/expphysiol.2007.038729. [DOI] [PubMed] [Google Scholar]
- 66.Ho L, Qin W, Pompl PN, Xiang Z, Wang J, Zhao Z, Peng Y, Cambareri G, Rocher A, Mobbs CV, Hof PR, Pasinetti GM. Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer’s disease. FASEB J. 2004;18:902–904. doi: 10.1096/fj.03-0978fje. [DOI] [PubMed] [Google Scholar]
- 67.Mattson MP, Duan W, Guo Z. Meal size and frequency affect neuronal plasticity and vulnerability to disease: cellular and molecular mechanisms. J Neurochem. 2003;84:417–431. doi: 10.1046/j.1471-4159.2003.01586.x. [DOI] [PubMed] [Google Scholar]
- 68.Piotrowski P. Are experimental models useful for analysis of pathogenesis of changes in central nervous system in human diabetes? Folia Neuropathol. 2003;41:167–174. [PubMed] [Google Scholar]
- 69.Planel E, Tatebayashi Y, Miyasaka T, Liu L, Wang L, Herman M, Yu WH, Luchsinger JA, Wadzinski B, Duff KE, Takashima A. Insulin dysfunction induces in vivo tau hyperphosphorylation through distinct mechanisms. J Neurosci. 2007;27:13635–13648. doi: 10.1523/JNEUROSCI.3949-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sadowski M, Pankiewicz J, Scholtzova H, Li YS, Quartermain D, Duff K, Wisniewski T. Links between the pathology of Alzheimer’s disease and vascular dementia. Neurochem Res. 2004;29:1257–1266. doi: 10.1023/b:nere.0000023612.66691.e6. [DOI] [PubMed] [Google Scholar]
- 71.Lu B, Song X, Dong X, Yang Y, Zhang Z, Wen J, Li Y, Zhou L, Zhao N, Zhu X, Hu R. High prevalence of chronic kidney disease in population-based patients diagnosed with type 2 diabetes in downtown Shanghai. J Diabetes Complications. 2008;22:96–103. doi: 10.1016/j.jdiacomp.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 72.Lu B, Wen J, Song XY, Dong XH, Yang YH, Zhang ZY, Zhao NQ, Ye HY, Mou B, Chen FL, Liu Y, Shen Y, Wang XC, Zhou LN, Li YM, Zhu XX, Hu RM. High prevalence of albuminuria in population-based patients diagnosed with type 2 diabetes in the Shanghai downtown. Diabetes Res Clin Pract. 2007;75:184–192. doi: 10.1016/j.diabres.2006.06.024. [DOI] [PubMed] [Google Scholar]
- 73.Coresh J, Astor BC, Greene T, Eknoyan G, Levey AS. Prevalence of chronic kidney disease and decreased kidney function in the adult US population: Third National Health and Nutrition Examination Survey. Am J Kidney Dis. 2003;41:1–12. doi: 10.1053/ajkd.2003.50007. [DOI] [PubMed] [Google Scholar]
- 74.Breyer MD, Bottinger E, Brosius FC, Coffman TM, Fogo A, Harris RC, Heilig CW, Sharma K. Diabetic nephropathy: of mice and men. Adv Chronic Kidney Dis. 2005;12:128–145. doi: 10.1053/j.ackd.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 75.Breyer MD, Bottinger E, Brosius FC, III, Coffman TM, Harris RC, Heilig CW, Sharma K. Mouse models of diabetic nephropathy. J Am Soc Nephrol. 2005;16:27–45. doi: 10.1681/ASN.2004080648. [DOI] [PubMed] [Google Scholar]
- 76.Cefalu WT. Animal models of type 2 diabetes: clinical presentation and pathophysiological relevance to the human condition. ILAR J. 2006;47:186–198. doi: 10.1093/ilar.47.3.186. [DOI] [PubMed] [Google Scholar]
- 77.Salkovic-Petrisic M, Hoyer S. Central insulin resistance as a trigger for sporadic Alzheimer-like pathology: an experimental approach. J Neural Transm Suppl. 2007:217–233. doi: 10.1007/978-3-211-73574-9_28. [DOI] [PubMed] [Google Scholar]
- 78.Hoyer S, Lannert H. Long-term abnormalities in brain glucose/energy metabolism after inhibition of the neuronal insulin receptor: implication of tau-protein. J Neural Transm Suppl. 2007:195–202. doi: 10.1007/978-3-211-73574-9_25. [DOI] [PubMed] [Google Scholar]
- 79.Grunblatt E, Salkovic-Petrisic M, Osmanovic J, Riederer P, Hoyer S. Brain insulin system dysfunction in streptozotocin intracerebroventricularly treated rats generates hyperphosphorylated tau protein. J Neurochem. 2007;101:757–770. doi: 10.1111/j.1471-4159.2006.04368.x. [DOI] [PubMed] [Google Scholar]
- 80.de la Monte SM, Tong M, Lester-Coll N, Plater M, Jr, Wands JR. Therapeutic rescue of neurodegeneration in experimental type 3 diabetes: relevance to Alzheimer’s disease. J Alzheimers Dis. 2006;10:89–109. doi: 10.3233/jad-2006-10113. [DOI] [PubMed] [Google Scholar]
- 81.Lester-Coll N, Rivera EJ, Soscia SJ, Doiron K, Wands JR, de la Monte SM. Intracerebral streptozotocin model of type 3 diabetes: relevance to sporadic Alzheimer’s disease. J Alzheimers Dis. 2006;9:13–33. doi: 10.3233/jad-2006-9102. [DOI] [PubMed] [Google Scholar]
- 82.Itagaki S, Nishida E, Lee MJ, Doi K. Histopathology of subacute renal lesions in mice induced by streptozotocin. Exp Toxicol Pathol. 1995;47:485–491. doi: 10.1016/s0940-2993(11)80332-5. [DOI] [PubMed] [Google Scholar]
- 83.Farr AG, Mannschreck JW, Anderson SK. Expression of Ia antigens by murine kidney epithelium after exposure to streptozotocin. Am J Pathol. 1987;126:561–568. [PMC free article] [PubMed] [Google Scholar]
- 84.Schmezer P, Eckert C, Liegibel UM. Tissue-specific induction of mutations by streptozotocin in vivo. Mutat Res. 1994;307:495–499. doi: 10.1016/0027-5107(94)90260-7. [DOI] [PubMed] [Google Scholar]
- 85.Davis R, Robertson D, editors. Textbook of Neuropathology. Williams and Wilkins; Philadelphia: 1997. [Google Scholar]
- 86.Reske-Nielsen E, Lundbaek K. Diabetic encephalopathy. Diffuse and focal lesions of the brain in long-term diabetes. Acta Neurol Scand Suppl. 1963;39(Suppl 4):273–290. [PubMed] [Google Scholar]
- 87.Strachan MW, Deary IJ, Ewing FM, Frier BM. Is type II diabetes associated with an increased risk of cognitive dysfunction? A critical review of published studies. Diabetes Care. 1997;20:438–445. doi: 10.2337/diacare.20.3.438. [DOI] [PubMed] [Google Scholar]
- 88.Biessels GJ, Staekenborg S, Brunner E, Brayne C, Scheltens P. Risk of dementia in diabetes mellitus: a systematic review. Lancet Neurol. 2006;5:64–74. doi: 10.1016/S1474-4422(05)70284-2. [DOI] [PubMed] [Google Scholar]
- 89.Kloppenborg RP, van den Berg E, Kappelle LJ, Biessels GJ. Diabetes and other vascular risk factors for dementia: Which factor matters most? A systematic review. Eur J Pharmacol. 2008;585:97–108. doi: 10.1016/j.ejphar.2008.02.049. [DOI] [PubMed] [Google Scholar]
- 90.van Harten B, Oosterman J, Muslimovic D, van Loon BJ, Scheltens P, Weinstein HC. Cognitive impairment and MRI correlates in the elderly patients with type 2 diabetes mellitus. Age Ageing. 2007;36:164–170. doi: 10.1093/ageing/afl180. [DOI] [PubMed] [Google Scholar]
- 91.Gold SM, Dziobek I, Sweat V, Tirsi A, Rogers K, Bruehl H, Tsui W, Richardson S, Javier E, Convit A. Hippocampal damage and memory impairments as possible early brain complications of type 2 diabetes. Diabetologia. 2007;50:711–719. doi: 10.1007/s00125-007-0602-7. [DOI] [PubMed] [Google Scholar]
- 92.Brands AM, Biessels GJ, de Haan EH, Kappelle LJ, Kessels RP. The effects of type 1 diabetes on cognitive performance: a meta-analysis. Diabetes Care. 2005;28:726–735. doi: 10.2337/diacare.28.3.726. [DOI] [PubMed] [Google Scholar]
- 93.Brands AM, Van den Berg E, Manschot SM, Biessels GJ, Kappelle LJ, De Haan EH, Kessels RP. A detailed profile of cognitive dysfunction and its relation to psychological distress in patients with type 2 diabetes mellitus. J Int Neuropsychol Soc. 2007;13:288–297. doi: 10.1017/S1355617707070312. [DOI] [PubMed] [Google Scholar]
- 94.Jacobson AM, Musen G, Ryan CM, Silvers N, Cleary P, Waberski B, Burwood A, Weinger K, Bayless M, Dahms W, Harth J. Long-term effect of diabetes and its treatment on cognitive function. N Engl J Med. 2007;356:1842–1852. doi: 10.1056/NEJMoa066397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.van Harten B, de Leeuw FE, Weinstein HC, Scheltens P, Biessels GJ. Brain imaging in patients with diabetes: a systematic review. Diabetes Care. 2006;29:2539–2548. doi: 10.2337/dc06-1637. [DOI] [PubMed] [Google Scholar]
- 96.Manschot SM, Biessels GJ, de Valk H, Algra A, Rutten GE, van der Grond J, Kappelle LJ. Metabolic and vascular determinants of impaired cognitive performance and abnormalities on brain magnetic resonance imaging in patients with type 2 diabetes. Diabetologia. 2007;50:2388–2397. doi: 10.1007/s00125-007-0792-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Manschot SM, Biessels GJ, Rutten GE, Kessels RC, Gispen WH, Kappelle LJ. Peripheral and central neurologic complications in type 2 diabetes mellitus: no association in individual patients. J Neurol Sci. 2008;264:157–162. doi: 10.1016/j.jns.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 98.Manschot SM, Brands AM, van der Grond J, Kessels RP, Algra A, Kappelle LJ, Biessels GJ. Brain magnetic resonance imaging correlates of impaired cognition in patients with type 2 diabetes. Diabetes. 2006;55:1106–1113. doi: 10.2337/diabetes.55.04.06.db05-1323. [DOI] [PubMed] [Google Scholar]
- 99.van Harten B, Oosterman JM, Potter van Loon BJ, Scheltens P, Weinstein HC. Brain lesions on MRI in elderly patients with type 2 diabetes mellitus. Eur Neurol. 2007;57:70–74. doi: 10.1159/000098054. [DOI] [PubMed] [Google Scholar]
- 100.Akisaki T, Sakurai T, Takata T, Umegaki H, Araki A, Mizuno S, Tanaka S, Ohashi Y, Iguchi A, Yokono K, Ito H. Cognitive dysfunction associates with white matter hyperintensities and subcortical atrophy on magnetic resonance imaging of the elderly diabetes mellitus Japanese elderly diabetes intervention trial (J-EDIT) Diabetes Metab Res Rev. 2006;22:376–384. doi: 10.1002/dmrr.632. [DOI] [PubMed] [Google Scholar]
- 101.Benbow SJ, Chan AW, Bowsher D, MacFarlane IA, Williams G. A prospective study of painful symptoms, small-fibre function and peripheral vascular disease in chronic painful diabetic neuropathy. Diabet Med. 1994;11:17–21. doi: 10.1111/j.1464-5491.1994.tb00223.x. [DOI] [PubMed] [Google Scholar]
- 102.Das AK, Vijayaraghavan MV. Diabetic peripheral neuropathy. J Assoc Physicians India. 1991;39:760–763. [PubMed] [Google Scholar]
- 103.Dyck PJ, Windebank A, Yasuda H, Service FJ, Rizza R, Zimmerman B. Diabetic neuropathy. Adv Exp Med Biol. 1985;189:305–320. doi: 10.1007/978-1-4757-1850-8_17. [DOI] [PubMed] [Google Scholar]
- 104.Greene DA, Sima AF, Pfeifer MA, Albers JW. Diabetic neuropathy. Annu Rev Med. 1990;41:303–317. doi: 10.1146/annurev.me.41.020190.001511. [DOI] [PubMed] [Google Scholar]
- 105.Greene DA, Stevens MJ, Feldman EL. Diabetic neuropathy: scope of the syndrome. Am J Med. 1999;107:2S–8S. doi: 10.1016/s0002-9343(99)00007-8. [DOI] [PubMed] [Google Scholar]
- 106.Ward JD. Diabetic neuropathy in NIDDM. Diabetologia. 1996;39:1676–1678. doi: 10.1007/s001250050634. [DOI] [PubMed] [Google Scholar]
- 107.Younger DS, Bronfin L. Overview of diabetic neuropathy. Semin Neurol. 1996;16:107–113. doi: 10.1055/s-2008-1040965. [DOI] [PubMed] [Google Scholar]
- 108.Carroll SL, Byer SJ, Dorsey DA, Watson MA, Schmidt RE. Ganglion-specific patterns of diabetes-modulated gene expression are established in prevertebral and paravertebral sympathetic ganglia prior to the development of neuroaxonal dystrophy. J Neuropathol Exp Neurol. 2004;63:1144–1154. doi: 10.1093/jnen/63.11.1144. [DOI] [PubMed] [Google Scholar]
- 109.Schmidt RE. Neuropathology and pathogenesis of diabetic autonomic neuropathy. Int Rev Neurobiol. 2002;50:257–292. doi: 10.1016/s0074-7742(02)50080-5. [DOI] [PubMed] [Google Scholar]
- 110.Sima AA, Cherian PV. Neuropathology of diabetic neuropathy and its correlations with neurophysiology. Clin Neurosci. 1997;4:359–364. [PubMed] [Google Scholar]
- 111.Britland ST, Young RJ, Sharma AK, Clarke BF. Acute and remitting painful diabetic polyneuropathy: a comparison of peripheral nerve fibre pathology. Pain. 1992;48:361–370. doi: 10.1016/0304-3959(92)90085-P. [DOI] [PubMed] [Google Scholar]
- 112.Malik RA, Tesfaye S, Newrick PG, Walker D, Rajbhandari SM, Siddique I, Sharma AK, Boulton AJ, King RH, Thomas PK, Ward JD. Sural nerve pathology in diabetic patients with minimal but progressive neuropathy. Diabetologia. 2005;48:578–585. doi: 10.1007/s00125-004-1663-5. [DOI] [PubMed] [Google Scholar]
- 113.Dyck PJ, Lais A, Karnes JL, O’Brien P, Rizza R. Fiber loss is primary and multifocal in sural nerves in diabetic polyneuropathy. Ann Neurol. 1986;19:425–439. doi: 10.1002/ana.410190503. [DOI] [PubMed] [Google Scholar]
- 114.Powell HC. Pathology of diabetic neuropathy: new observations, new hypotheses. Lab Invest. 1983;49:515–518. [PubMed] [Google Scholar]
- 115.Vital C, Brechenmacher C, Serise JM, Bellance R, Vital A, Dartigues JF, Boissieras P. Ultrastructural study of peripheral nerve in arteritic diabetic patients. Acta Neuropathol. 1983;61:225–231. doi: 10.1007/BF00691990. [DOI] [PubMed] [Google Scholar]
- 116.Clements RS., Jr Pathogenesis of diabetic neuropathy. N Y State J Med. 1982;82:864–871. [PubMed] [Google Scholar]
- 117.Behse F, Buchthal F, Carlsen F. Nerve biopsy and conduction studies in diabetic neuropathy. J Neurol Neurosurg Psychiatry. 1977;40:1072–1082. doi: 10.1136/jnnp.40.11.1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Cumbie BC, Hermayer KL. Current concepts in targeted therapies for the pathophysiology of diabetic microvascular complications. Vasc Health Risk Manag. 2007;3:823–832. [PMC free article] [PubMed] [Google Scholar]
- 119.Camera A, Hopps E, Caimi G. Diabetic microangiopathy: physiopathological, clinical and therapeutic aspects. Minerva Endocrinol. 2007;32:209–229. [PubMed] [Google Scholar]
- 120.Yagihashi S, Yamagishi S, Wada R. Pathology and pathogenetic mechanisms of diabetic neuropathy: correlation with clinical signs and symptoms. Diabetes Res Clin Pract. 2007;77(Suppl 1):S184–189. doi: 10.1016/j.diabres.2007.01.054. [DOI] [PubMed] [Google Scholar]
- 121.Warren S, LeCompte PM, Legg MA, editors. The Pathology of Diabetes Mellitus. Lea and Febiger; Philadelphia: 1966. [Google Scholar]
- 122.Vonderahe AR. CNS pathology of diabetes. Arch Int Med. 1937;60:694. [Google Scholar]
- 123.Morgan LO, Vonderahe AR, Malone EF. CNS pathology of diabetes mellitus. J Nerv and MEnt Dis. 1937;85:125. [Google Scholar]
- 124.Hagen E. Hypothalamic pathology in diabetes. Deut Ztschr f Nervenheilk. 1957;177:73. [Google Scholar]
- 125.Bell ET. Angiopathic changes in diabetes. Am J Clin Path. 1957;28:27. [Google Scholar]
- 126.Bell ET. Angiopathic changes in diabetes. Diabetes. 1953;2:376. [Google Scholar]
- 127.Bell ET. Angiopathic changes in diabetes. Arch Pathol. 1952;53:444. [Google Scholar]
- 128.Garcia J, editor. Neuropathology – the Diagnostic Approach. Mosby: Philadelphia; 1997. [Google Scholar]
- 129.Graham D, Lantos P, editors. Greenfield’s Neuropathology. Hodder Arnold; 2002. [Google Scholar]
- 130.Ellison D, Love S, Chimelli L, Harding B, Lowe J, Roberts G, Vinters H, editors. Neuropathology. Mosby; Philadelphia: 2000. [Google Scholar]
- 131.Olsson Y. Pathoanatomical study of the central and peripheral nervous systems in diabetes of early onset and long duration. Pathol Eur. 1968;3:62. [PubMed] [Google Scholar]
- 132.Kameyama M, Fushimi H, Udaka F. Diabetes mellitus and cerebral vascular disease. Diabetes Res Clin Pract. 1994;24(Suppl):S205–208. doi: 10.1016/0168-8227(94)90250-x. [DOI] [PubMed] [Google Scholar]
- 133.Howard CF., Jr Diabetes mellitus: relationships of nonhuman primates and other animal models to human forms of diabetes. Adv Vet Sci Comp Med. 1984;28:115–149. doi: 10.1016/b978-0-12-039228-5.50010-3. [DOI] [PubMed] [Google Scholar]
- 134.Laidler P, Pieterse AS, Pounder DJ. Diabetes mellitus and hypopituitarism. Forensic Sci Int. 1981;18:169–174. doi: 10.1016/0379-0738(81)90156-0. [DOI] [PubMed] [Google Scholar]
- 135.Timperley WR, Preston FE, Ward JD. Cerebral intravascular coagulation in diabetic ketoacidosis. Lancet. 1974;1:952–956. doi: 10.1016/s0140-6736(74)91261-6. [DOI] [PubMed] [Google Scholar]
- 136.Aronson SM. Intracranial vascular lesions in patients with diabetes mellitus. J Neuropathol Exp Neurol. 1973;32:183–196. doi: 10.1097/00005072-197304000-00001. [DOI] [PubMed] [Google Scholar]
- 137.Fraley DS, Totten RS. An autopsy study of endocrine organ changes in diabetes mellitus. Metabolism. 1968;17:896–900. doi: 10.1016/0026-0495(68)90155-8. [DOI] [PubMed] [Google Scholar]
- 138.Gerstl B, Tavaststjerna MG, Hayman RB, Smith JK, Eng LF. Lipid studies of white matter and thalamus of human brains. J Neurochem. 1963;10:889–902. doi: 10.1111/j.1471-4159.1963.tb11916.x. [DOI] [PubMed] [Google Scholar]
- 139.Schmitt HP. epsilon-Glycation, APP and Abeta in ageing and Alzheimer disease: a hypothesis. Med Hypotheses. 2006;66:898–906. doi: 10.1016/j.mehy.2005.11.016. [DOI] [PubMed] [Google Scholar]
- 140.Southern L, Williams J, Esiri MM. Immunohistochemical study of N-epsilon-carboxymethyl lysine (CML) in human brain: relation to vascular dementia. BMC Neurol. 2007;7:35. doi: 10.1186/1471-2377-7-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.van Deutekom AW, Niessen HW, Schalkwijk CG, Heine RJ, Simsek S. Increased Nepsilon-(carboxymethyl)-lysine levels in cerebral blood vessels of diabetic patients and in a (streptozotocin-treated) rat model of diabetes mellitus. Eur J Endocrinol. 2008;158:655–660. doi: 10.1530/EJE-08-0024. [DOI] [PubMed] [Google Scholar]
- 142.Meetoo D, McGovern P, Safadi R. An epidemiological overview of diabetes across the world. Br J Nurs. 2007;16:1002–1007. doi: 10.12968/bjon.2007.16.16.27079. [DOI] [PubMed] [Google Scholar]
- 143.Roman GC. Vascular dementia: distinguishing characteristics, treatment, and prevention. J Am Geriatr Soc. 2003;51:S296–304. doi: 10.1046/j.1532-5415.5155.x. [DOI] [PubMed] [Google Scholar]
- 144.Roman GC. Facts, myths, and controversies in vascular dementia. J Neurol Sci. 2004;226:49–52. doi: 10.1016/j.jns.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 145.Cooper R, Sempos C, Hsieh SC, Kovar MG. Slowdown in the decline of stroke mortality in the United States, 1978–1986. Stroke. 1990;21:1274–1279. doi: 10.1161/01.str.21.9.1274. [DOI] [PubMed] [Google Scholar]
- 146.Gorelick PB, editor. Atlas of Cerebrovascular Disease. Philadelphia: 1996. [Google Scholar]
- 147.Riddle MC, Hart J. Hyperglycemia, recognized and unrecognized, as a risk factor for stroke and transient ischemic attacks. Stroke. 1982;13:356–359. doi: 10.1161/01.str.13.3.356. [DOI] [PubMed] [Google Scholar]
- 148.Arboix A, Altes E, Garcia-Eroles L, Massons J. Clinical study of lacunar infarcts in non-hypertensive patients. J Stroke Cerebrovasc Dis. 2003;12:232–236. doi: 10.1016/j.jstrokecerebrovasdis.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 149.Gouw AA, van der Flier WM, Fazekas F, van Straaten EC, Pantoni L, Poggesi A, Inzitari D, Erkinjuntti T, Wahlund LO, Waldemar G, Schmidt R, Scheltens P, Barkhof F. Progression of white matter hyperintensities and incidence of new lacunes over a 3-year period: the Leukoaraiosis and Disability study. Stroke. 2008;39:1414–1420. doi: 10.1161/STROKEAHA.107.498535. [DOI] [PubMed] [Google Scholar]
- 150.Llewellyn DJ, Lang IA, Xie J, Huppert FA, Melzer D, Langa KM. Framingham Stroke Risk Profile and poor cognitive function: a population-based study. BMC Neurol. 2008;8:12. doi: 10.1186/1471-2377-8-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Au R, Massaro JM, Wolf PA, Young ME, Beiser A, Seshadri S, D’Agostino RB, DeCarli C. Association of white matter hyperintensity volume with decreased cognitive functioning: the Framingham Heart Study. Arch Neurol. 2006;63:246–250. doi: 10.1001/archneur.63.2.246. [DOI] [PubMed] [Google Scholar]
- 152.Boston PF, Dennis MS, Jagger C. Factors associated with vascular dementia in an elderly community population. Int J Geriatr Psychiatry. 1999;14:761–766. doi: 10.1002/(sici)1099-1166(199909)14:9<761::aid-gps14>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 153.Curb JD, Rodriguez BL, Abbott RD, Petrovitch H, Ross GW, Masaki KH, Foley D, Blanchette PL, Harris T, Chen R, White LR. Longitudinal association of vascular and Alzheimer’s dementias, diabetes, and glucose tolerance. Neurology. 1999;52:971–975. doi: 10.1212/wnl.52.5.971. [DOI] [PubMed] [Google Scholar]
- 154.Luchsinger JA, Tang MX, Stern Y, Shea S, Mayeux R. Diabetes mellitus and risk of Alzheimer’s disease and dementia with stroke in a multiethnic cohort. Am J Epidemiol. 2001;154:635–641. doi: 10.1093/aje/154.7.635. [DOI] [PubMed] [Google Scholar]
- 155.Brand FN, Abbott RD, Kannel WB. Diabetes, intermittent claudication, and risk of cardiovascular events. The Framingham Study. Diabetes. 1989;38:504–509. doi: 10.2337/diab.38.4.504. [DOI] [PubMed] [Google Scholar]
- 156.Hiller R, Sperduto RD, Podgor MJ, Ferris FL, 3rd, Wilson PW. Diabetic retinopathy and cardiovascular disease in type II diabetics. The Framingham Heart Study and the Framingham Eye Study. Am J Epidemiol. 1988;128:402–409. doi: 10.1093/oxfordjournals.aje.a114980. [DOI] [PubMed] [Google Scholar]
- 157.Kannel WB, McGee DL. Diabetes and glucose tolerance as risk factors for cardiovascular disease: the Framingham study. Diabetes Care. 1979;2:120–126. doi: 10.2337/diacare.2.2.120. [DOI] [PubMed] [Google Scholar]
- 158.Suwanwela NC, Chutinetr A. Risk factors for atherosclerosis of cervicocerebral arteries: intracranial versus extracranial. Neuroepidemiology. 2003;22:37–40. doi: 10.1159/000067112. [DOI] [PubMed] [Google Scholar]
- 159.Folsom AR, Rasmussen ML, Chambless LE, Howard G, Cooper LS, Schmidt MI, Heiss G. Prospective associations of fasting insulin, body fat distribution, and diabetes with risk of ischemic stroke. The Atherosclerosis Risk in Communities (ARIC) Study Investigators. Diabetes Care. 1999;22:1077–1083. doi: 10.2337/diacare.22.7.1077. [DOI] [PubMed] [Google Scholar]
- 160.Kuusisto J, Mykkanen L, Pyorala K, Laakso M. Non-insulin-dependent diabetes and its metabolic control are important predictors of stroke in elderly subjects. Stroke. 1994;25:1157–1164. doi: 10.1161/01.str.25.6.1157. [DOI] [PubMed] [Google Scholar]
- 161.Rodriguez BL, D’Agostino R, Abbott RD, Kagan A, Burchfiel CM, Yano K, Ross GW, Silbershatz H, Higgins MW, Popper J, Wolf PA, Curb JD. Risk of hospitalized stroke in men enrolled in the Honolulu Heart Program and the Framingham Study: a comparison of incidence and risk factor effects. Stroke. 2002;33:230–236. doi: 10.1161/hs0102.101081. [DOI] [PubMed] [Google Scholar]
- 162.Koren-Morag N, Goldbourt U, Tanne D. Relation between the metabolic syndrome and ischemic stroke or transient ischemic attack: a prospective cohort study in patients with atherosclerotic cardiovascular disease. Stroke. 2005;36:1366–1371. doi: 10.1161/01.STR.0000169945.75911.33. [DOI] [PubMed] [Google Scholar]
- 163.Mohan V, Gokulakrishnan K, Sandeep S, Srivastava BK, Ravikumar R, Deepa R. Intimal media thickness, glucose intolerance and metabolic syndrome in Asian Indians–the Chennai Urban Rural Epidemiology Study (CURES-22) Diabet Med. 2006;23:845–850. doi: 10.1111/j.1464-5491.2006.01898.x. [DOI] [PubMed] [Google Scholar]
- 164.Mohan V, Ravikumar R, Shanthi Rani S, Deepa R. Intimal medial thickness of the carotid artery in South Indian diabetic and non-diabetic subjects: the Chennai Urban Population Study (CUPS) Diabetologia. 2000;43:494–499. doi: 10.1007/s001250051334. [DOI] [PubMed] [Google Scholar]
- 165.Feldmann E, Broderick JP, Kernan WN, Viscoli CM, Brass LM, Brott T, Morgenstern LB, Wilterdink JL, Horwitz RI. Major risk factors for intracerebral hemorrhage in the young are modifiable. Stroke. 2005;36:1881–1885. doi: 10.1161/01.STR.0000177480.62341.6b. [DOI] [PubMed] [Google Scholar]
- 166.Gu YX, Chen XC, Song DL, Leng B, Zhao F. Risk factors for intracranial aneurysm in a Chinese ethnic population. Chin Med J (Engl) 2006;119:1359–1364. [PubMed] [Google Scholar]
- 167.Uehara T, Tabuchi M, Mori E. Risk factors for occlusive lesions of intracranial arteries in stroke-free Japanese. Eur J Neurol. 2005;12:218–222. doi: 10.1111/j.1468-1331.2004.00959.x. [DOI] [PubMed] [Google Scholar]
- 168.Prevalence of stroke–United States. MMWR Morb Mortal Wkly Rep. 2007. Vol. 56. 2005. pp. 469–474. [PubMed] [Google Scholar]
- 169.Leary MC, Saver JL. Annual incidence of first silent stroke in the United States: a preliminary estimate. Cerebrovasc Dis. 2003;16:280–285. doi: 10.1159/000071128. [DOI] [PubMed] [Google Scholar]
- 170.Fang J, Alderman MH. Trend of stroke hospitalization, United States, 1988–1997. Stroke. 2001;32:2221–2226. doi: 10.1161/hs1001.096193. [DOI] [PubMed] [Google Scholar]
- 171.Haan MN. Therapy insight: type 2 diabetes mellitus and the risk of late-onset Alzheimer’s disease. Nat Clin Pract Neurol. 2006;2:159–166. doi: 10.1038/ncpneuro0124. [DOI] [PubMed] [Google Scholar]
- 172.Irie F, Fitzpatrick AL, Lopez OL, Kuller LH, Peila R, Newman AB, Launer LJ. Enhanced risk for Alzheimer disease in persons with type 2 diabetes and APOE epsilon4: the Cardiovascular Health Study Cognition Study. Arch Neurol. 2008;65:89–93. doi: 10.1001/archneurol.2007.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Profenno LA, Faraone SV. Diabetes and overweight associate with non-APOE4 genotype in an Alzheimer’s disease population. Am J Med Genet B Neuropsychiatr Genet. 2008;147B(6):822–829. doi: 10.1002/ajmg.b.30694. [DOI] [PubMed] [Google Scholar]
- 174.Xu W, Qiu C, Winblad B, Fratiglioni L. The effect of borderline diabetes on the risk of dementia and Alzheimer’s disease. Diabetes. 2007;56:211–216. doi: 10.2337/db06-0879. [DOI] [PubMed] [Google Scholar]
- 175.Hayden KM, Zandi PP, Lyketsos CG, Khachaturian AS, Bastian LA, Charoonruk G, Tschanz JT, Norton MC, Pieper CF, Munger RG, Breitner JC, Welsh-Bohmer KA. Vascular risk factors for incident Alzheimer disease and vascular dementia: the Cache County study. Alzheimer Dis Assoc Disord. 2006;20:93–100. doi: 10.1097/01.wad.0000213814.43047.86. [DOI] [PubMed] [Google Scholar]
- 176.Luchsinger JA, Reitz C, Honig LS, Tang MX, Shea S, Mayeux R. Aggregation of vascular risk factors and risk of incident Alzheimer disease. Neurology. 2005;65:545–551. doi: 10.1212/01.wnl.0000172914.08967.dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Borenstein AR, Wu Y, Mortimer JA, Schellenberg GD, McCormick WC, Bowen JD, McCurry S, Larson EB. Developmental and vascular risk factors for Alzheimer’s disease. Neurobiol Aging. 2005;26:325–334. doi: 10.1016/j.neurobiolaging.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 178.Janson J, Laedtke T, Parisi JE, O’Brien P, Petersen RC, Butler PC. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes. 2004;53:474–481. doi: 10.2337/diabetes.53.2.474. [DOI] [PubMed] [Google Scholar]
- 179.Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology. 1999;53:1937–1942. doi: 10.1212/wnl.53.9.1937. [DOI] [PubMed] [Google Scholar]
- 180.Leibson CL, Rocca WA, Hanson VA, Cha R, Kokmen E, O’Brien PC, Palumbo PJ. The risk of dementia among persons with diabetes mellitus: a population–based cohort study. Ann N Y Acad Sci. 1997;826:422–427. doi: 10.1111/j.1749-6632.1997.tb48496.x. [DOI] [PubMed] [Google Scholar]
- 181.Biessels GJ, De Leeuw FE, Lindeboom J, Barkhof F, Scheltens P. Increased cortical atrophy in patients with Alzheimer’s disease and type 2 diabetes mellitus. J Neurol Neurosurg Psychiatry. 2006;77:304–307. doi: 10.1136/jnnp.2005.069583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.den Heijer T, Vermeer SE, van Dijk EJ, Prins ND, Koudstaal PJ, Hofman A, Breteler MM. Type 2 diabetes and atrophy of medial temporal lobe structures on brain MRI. Diabetologia. 2003;46:1604–1610. doi: 10.1007/s00125-003-1235-0. [DOI] [PubMed] [Google Scholar]
- 183.Knopman DS, Mosley TH, Catellier DJ, Sharrett AR. Cardiovascular risk factors and cerebral atrophy in a middle-aged cohort. Neurology. 2005;65:876–881. doi: 10.1212/01.wnl.0000176074.09733.a8. [DOI] [PubMed] [Google Scholar]
- 184.Nicolls MR. The clinical and biological relationship between Type II diabetes mellitus and Alzheimer’s disease. Curr Alzheimer Res. 2004;1:47–54. doi: 10.2174/1567205043480555. [DOI] [PubMed] [Google Scholar]
- 185.Miklossy J, Taddei K, Martins R, Escher G, Kraftsik R, Pillevuit O, Lepori D, Campiche M. Alzheimer disease: curly fibers and tangles in organs other than brain. J Neuropathol Exp Neurol. 1999;58:803–814. doi: 10.1097/00005072-199908000-00003. [DOI] [PubMed] [Google Scholar]
- 186.Mielke MM, Rosenberg PB, Tschanz J, Cook L, Corcoran C, Hayden KM, Norton M, Rabins PV, Green RC, Welsh-Bohmer KA, Breitner JC, Munger R, Lyketsos CG. Vascular factors predict rate of progression in Alzheimer disease. Neurology. 2007;69:1850–1858. doi: 10.1212/01.wnl.0000279520.59792.fe. [DOI] [PubMed] [Google Scholar]
- 187.Luchsinger JA, Mayeux R. Adiposity and Alzheimer’s disease. Curr Alzheimer Res. 2007;4:127–134. doi: 10.2174/156720507780362100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Scarmeas N, Stern Y, Mayeux R, Luchsinger JA. Mediterranean diet, Alzheimer disease, and vascular mediation. Arch Neurol. 2006;63:1709–1717. doi: 10.1001/archneur.63.12.noc60109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Silvestrini M, Pasqualetti P, Baruffaldi R, Bartolini M, Handouk Y, Matteis M, Moffa F, Provinciali L, Vernieri F. Cerebrovascular reactivity and cognitive decline in patients with Alzheimer disease. Stroke. 2006;37:1010–1015. doi: 10.1161/01.STR.0000206439.62025.97. [DOI] [PubMed] [Google Scholar]
- 190.Scarmeas N, Habeck CG, Hilton J, Anderson KE, Flynn J, Park A, Stern Y. APOE related alterations in cerebral activation even at college age. J Neurol Neurosurg Psychiatry. 2005;76:1440–1444. doi: 10.1136/jnnp.2004.053645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Scarmeas N, Zarahn E, Anderson KE, Hilton J, Flynn J, Van Heertum RL, Sackeim HA, Stern Y. Cognitive reserve modulates functional brain responses during memory tasks: a PET study in healthy young and elderly subjects. Neuroimage. 2003;19:1215–1227. doi: 10.1016/s1053-8119(03)00074-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, Saunders AM, Hardy J. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proc Natl Acad Sci U S A. 2004;101:284–289. doi: 10.1073/pnas.2635903100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Caselli RJ, Graff-Radford NR, Reiman EM, Weaver A, Osborne D, Lucas J, Uecker A, Thibodeau SN. Preclinical memory decline in cognitively normal apolipoprotein E-epsilon4 homozygotes. Neurology. 1999;53:201–207. doi: 10.1212/wnl.53.1.201. [DOI] [PubMed] [Google Scholar]
- 194.Yan SD, Stern D, Schmidt AM. What’s the RAGE? The receptor for advanced glycation end products (RAGE) and the dark side of glucose. Eur J Clin Invest. 1997;27:179–181. doi: 10.1046/j.1365-2362.1996.00072.x. [DOI] [PubMed] [Google Scholar]
- 195.Thornalley PJ. Cell activation by glycated proteins. AGE receptors, receptor recognition factors and functional classification of AGEs. Cell Mol Biol (Noisy-legrand) 1998;44:1013–1023. [PubMed] [Google Scholar]
- 196.Sasaki N, Toki S, Chowei H, Saito T, Nakano N, Hayashi Y, Takeuchi M, Makita Z. Immunohistochemical distribution of the receptor for advanced glycation end products in neurons and astrocytes in Alzheimer’s disease. Brain Res. 2001;888:256–262. doi: 10.1016/s0006-8993(00)03075-4. [DOI] [PubMed] [Google Scholar]
- 197.Luth HJ, Ogunlade V, Kuhla B, Kientsch-Engel R, Stahl P, Webster J, Arendt T, Munch G. Age- and stage-dependent accumulation of advanced glycation end products in intracellular deposits in normal and Alzheimer’s disease brains. Cereb Cortex. 2005;15:211–220. doi: 10.1093/cercor/bhh123. [DOI] [PubMed] [Google Scholar]
- 198.Ronnemaa E, Zethelius B, Sundelof J, Sundstrom J, Degerman-Gunnarsson M, Berne C, Lannfelt L, Kilander L. Impaired insulin secretion increases the risk of Alzheimer disease. Neurology. 2008 doi: 10.1212/01.wnl.0000310646.32212.3a. [DOI] [PubMed] [Google Scholar]
- 199.Burdo JR, Chen Q, Calcutt NA, Schubert D. The pathological interaction between diabetes and presymptomatic Alzheimer’s disease. Neurobiol Aging. 2008 doi: 10.1016/j.neurobiolaging.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 200.Luchsinger JA, Tang MX, Shea S, Mayeux R. Hyperinsulinemia and risk of Alzheimer disease. Neurology. 2004;63:1187–1192. doi: 10.1212/01.wnl.0000140292.04932.87. [DOI] [PubMed] [Google Scholar]
- 201.Liolitsa D, Powell J, Lovestone S. Genetic variability in the insulin signalling pathway may contribute to the risk of late onset Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2002;73:261–266. doi: 10.1136/jnnp.73.3.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Kurland BF, Heagerty PJ. Directly parameterized regression conditioning on being alive: analysis of longitudinal data truncated by deaths. Biostatistics. 2005;6:241–258. doi: 10.1093/biostatistics/kxi006. [DOI] [PubMed] [Google Scholar]
- 203.Tyas SL, Salazar JC, Snowdon DA, Desrosiers MF, Riley KP, Mendiondo MS, Kryscio RJ. Transitions to mild cognitive impairments, dementia, and death: findings from the Nun Study. Am J Epidemiol. 2007;165:1231–1238. doi: 10.1093/aje/kwm085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Cao D, Lu H, Lewis TL, Li L. Intake of sucrose-sweetened water induces insulin resistance and exacerbates memory deficits and amyloidosis in a transgenic mouse model of Alzheimer disease. J Biol Chem. 2007;282:36275–36282. doi: 10.1074/jbc.M703561200. [DOI] [PubMed] [Google Scholar]
- 205.Munch G, Apelt J, Rosemarie Kientsch E, Stahl P, Luth HJ, Schliebs R. Advanced glycation endproducts and pro-inflammatory cytokines in transgenic Tg2576 mice with amyloid plaque pathology. J Neurochem. 2003;86:283–289. doi: 10.1046/j.1471-4159.2003.01837.x. [DOI] [PubMed] [Google Scholar]
- 206.Heitner J, Dickson D. Diabetics do not have increased Alzheimer-type pathology compared with age-matched control subjects. A retrospective postmortem immunocytochemical and histofluorescent study. Neurology. 1997;49:1306–1311. doi: 10.1212/wnl.49.5.1306. [DOI] [PubMed] [Google Scholar]
- 207.Sanderson M, Wang J, Davis DR, Lane MJ, Cornman CB, Fadden MK. Co-morbidity associated with dementia. Am J Alzheimers Dis Other Demen. 2002;17:73–78. doi: 10.1177/153331750201700210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Hassing LB, Johansson B, Nilsson SE, Berg S, Pedersen NL, Gatz M, McClearn G. Diabetes mellitus is a risk factor for vascular dementia, but not for Alzheimer’s disease: a population-based study of the oldest old. Int Psychogeriatr. 2002;14:239–248. doi: 10.1017/s104161020200844x. [DOI] [PubMed] [Google Scholar]
- 209.Chiang CJ, Yip PK, Wu SC, Lu CS, Liou CW, Liu HC, Liu CK, Chu CH, Hwang CS, Sung SF, Hsu YD, Chen CC, Liu SI, Yan SH, Fong CS, Chang SF, You SL, Chen CJ. Midlife risk factors for subtypes of dementia: a nested case–control study in Taiwan. Am J Geriatr Psychiatry. 2007;15:762–771. doi: 10.1097/JGP.0b013e318050c98f. [DOI] [PubMed] [Google Scholar]
- 210.Arvanitakis Z, Schneider JA, Wilson RS, Li Y, Arnold SE, Wang Z, Bennett DA. Diabetes is related to cerebral infarction but not to AD pathology in older persons. Neurology. 2006;67:1960–1965. doi: 10.1212/01.wnl.0000247053.45483.4e. [DOI] [PubMed] [Google Scholar]
- 211.Yamada M. Risk factors for cerebral amyloid angiopathy in the elderly. Ann N Y Acad Sci. 2002;977:37–44. doi: 10.1111/j.1749-6632.2002.tb04797.x. [DOI] [PubMed] [Google Scholar]
- 212.MacKnight C, Rockwood K, Awalt E, McDowell I. Diabetes mellitus and the risk of dementia, Alzheimer’s disease and vascular cognitive impairment in the Canadian Study of Health and Aging. Dement Geriatr Cogn Disord. 2002;14:77–83. doi: 10.1159/000064928. [DOI] [PubMed] [Google Scholar]
- 213.Bruce DG, Harrington N, Davis WA, Davis TM. Dementia and its associations in type 2 diabetes mellitus: the Fremantle Diabetes Study. Diabetes Res Clin Pract. 2001;53:165–172. doi: 10.1016/s0168-8227(01)00266-2. [DOI] [PubMed] [Google Scholar]
- 214.Rastas S, Pirttila T, Mattila K, Verkkoniemi A, Juva K, Niinisto L, Lansimies E, Sulkava R. Vascular risk factors and dementia in the general population aged <85 years. Prospective population-based study. Neurobiol Aging. 2008 doi: 10.1016/j.neurobiolaging.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 215.Biessels GJ, Koffeman A, Scheltens P. Diabetes and cognitive impairment. Clinical diagnosis and brain imaging in patients attending a memory clinic. J Neurol. 2006;253:477–482. doi: 10.1007/s00415-005-0036-4. [DOI] [PubMed] [Google Scholar]
- 216.Fuh JL, Wang SJ, Hwu CM, Lu SR. Glucose tolerance status and cognitive impairment in early middle-aged women. Diabet Med. 2007;24:788–791. doi: 10.1111/j.1464-5491.2007.02170.x. [DOI] [PubMed] [Google Scholar]
- 217.Xu WL, Qiu CX, Wahlin A, Winblad B, Fratiglioni L. Diabetes mellitus and risk of dementia in the Kungsholmen project: a 6-year follow-up study. Neurology. 2004;63:1181–1186. doi: 10.1212/01.wnl.0000140291.86406.d1. [DOI] [PubMed] [Google Scholar]
- 218.Schneider B, Martin S, Heinemann L, Lodwig V, Kolb H. Interrelations between diabetes therapy, self-monitoring of blood glucose, blood glucose and non-fatal or fatal endpoints in patients with type 2 diabetes/results of a longitudinal cohort study (ROSSO 5) Arzneimittelforschung. 2007;57:762–769. doi: 10.1055/s-0031-1296677. [DOI] [PubMed] [Google Scholar]
- 219.McLean G, Guthrie B, Sutton M. Differences in the quality of primary medical care for CVD and diabetes across the NHS: evidence from the quality and outcomes framework. BMC Health Serv Res. 2007;7:74. doi: 10.1186/1472-6963-7-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Mollah AS, Rahman SW, Das KK, Hassanuzzaman M. Characteristics of patients admitted with stroke. Mymensingh Med J. 2007;16:20–24. doi: 10.3329/mmj.v16i1.242. [DOI] [PubMed] [Google Scholar]
- 221.Rosendorff C, Beeri MS, Silverman JM. Cardiovascular risk factors for Alzheimer’s disease. Am J Geriatr Cardiol. 2007;16:143–149. doi: 10.1111/j.1076-7460.2007.06696.x. [DOI] [PubMed] [Google Scholar]
- 222.Jongen C, van der Grond J, Kappelle LJ, Biessels GJ, Viergever MA, Pluim JP. Automated measurement of brain and white matter lesion volume in type 2 diabetes mellitus. Diabetologia. 2007;50:1509–1516. doi: 10.1007/s00125-007-0688-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Nelson PT, Jicha GA, Schmitt FA, Liu H, Davis DG, Mendiondo MS, Abner EL, Markesbery WR. Clinicopathologic correlations in a large Alzheimer disease center autopsy cohort: neuritic plaques and neurofibrillary tangles “do count” when staging disease severity. J Neuropathol Exp Neurol. 2007;66:1136–1146. doi: 10.1097/nen.0b013e31815c5efb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Lobnig BM, Kromeke O, Optenhostert-Porst C, Wolf OT. Hippocampal volume and cognitive performance in long-standing Type 1 diabetic patients without macrovascular complications. Diabet Med. 2006;23:32–39. doi: 10.1111/j.1464-5491.2005.01716.x. [DOI] [PubMed] [Google Scholar]
- 225.Brands AM, Kessels RP, Hoogma RP, Henselmans JM, van der Beek Boter JW, Kappelle LJ, de Haan EH, Biessels GJ. Cognitive performance, psychological well-being, and brain magnetic resonance imaging in older patients with type 1 diabetes. Diabetes. 2006;55:1800–1806. doi: 10.2337/db05-1226. [DOI] [PubMed] [Google Scholar]
- 226.Braffman BH, Zimmerman RA, Trojanowski JQ, Gonatas NK, Hickey WF, Schlaepfer WW. MR Brain pathologic correlation with gross and histopathology. 2. Hyperintense white-matter foci in the elderly. AJR Am J Roentgenol. 1988;151:559–566. doi: 10.2214/ajr.151.3.559. [DOI] [PubMed] [Google Scholar]
- 227.Raffman BH, Zimmerman RA, Trojanowski JQ, Gonatas NK, Hickey WF, Schlaepfer WW. MRBrain Pathologic correlation with gross and histopathology. 1. Lacunar infarction and Virchow–Robin spaces. AJR Am J Roentgenol. 1988;151:551–558. doi: 10.2214/ajr.151.3.551. [DOI] [PubMed] [Google Scholar]
- 228.Chimowitz MI, Estes ML, Furlan AJ, Awad IA. Further observations on the pathology of subcortical lesions identified on magnetic resonance imaging. Arch Neurol. 1992;49:747–752. doi: 10.1001/archneur.1992.00530310095018. [DOI] [PubMed] [Google Scholar]
- 229.Fazekas F, Kleinert R, Offenbacher H, Schmidt R, Kleinert G, Payer F, Radner H, Lechner H. Pathologic correlates of incidental MRI white matter signal hyperintensities. Neurology. 1993;43:1683–1689. doi: 10.1212/wnl.43.9.1683. [DOI] [PubMed] [Google Scholar]
- 230.Golomb J, Kluger A, Gianutsos J, Ferris SH, de Leon MJ, George AE. Nonspecific leukoencephalopathy associated with aging. Neuroimaging Clin N Am. 1995;5:33–44. [PubMed] [Google Scholar]
- 231.Johnson KA, Davis KR, Buonanno FS, Brady TJ, Rosen TJ, Growdon JH. Comparison of magnetic resonance and roentgen ray computed tomography in dementia. Arch Neurol. 1987;44:1075–1080. doi: 10.1001/archneur.1987.00520220071020. [DOI] [PubMed] [Google Scholar]
- 232.Leifer D, Buonanno FS, Richardson EP., Jr Clinicopathologic correlations of cranial magnetic resonance imaging of periventricular white matter. Neurology. 1990;40:911–918. doi: 10.1212/wnl.40.6.911. [DOI] [PubMed] [Google Scholar]
- 233.Matsusue E, Sugihara S, Fujii S, Kinoshita T, Ohama E, Ogawa T. Wallerian degeneration of the corticospinal tracts: postmortem MR-pathologic correlations. Acta Radiol. 2007;48:690–694. doi: 10.1080/02841850701342112. [DOI] [PubMed] [Google Scholar]
- 234.Matsusue E, Sugihara S, Fujii S, Ohama E, Kinoshita T, Ogawa T. White matter changes in elderly people: MR-pathologic correlations. Magn Reson Med Sci. 2006;5:99–104. doi: 10.2463/mrms.5.99. [DOI] [PubMed] [Google Scholar]
- 235.Pullicino PM, Miller LL, Alexandrov AV, Ostrow PT. Infraputaminal ‘lacunes’. Clinical and pathological correlations. Stroke. 1995;26:1598–1602. doi: 10.1161/01.str.26.9.1598. [DOI] [PubMed] [Google Scholar]
- 236.Gearing M, Mirra SS, Hedreen JC, Sumi SM, Hansen LA, Heyman A. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part X. Neuropathology confirmation of the clinical diagnosis of Alzheimer’s disease. Neurology. 1995;45:461–466. doi: 10.1212/wnl.45.3.461. [DOI] [PubMed] [Google Scholar]
- 237.Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 238.Cisse S, Perry G, Lacoste-Royal G, Cabana T, Gauvreau D. Immunochemical identification of ubiquitin and heat-shock proteins in corpora amylacea from normal aged and Alzheimer’s disease brains. Acta Neuropathol. 1993;85:233–240. doi: 10.1007/BF00227716. [DOI] [PubMed] [Google Scholar]
- 239.Keller JN. Age-related neuropathology, cognitive decline, and Alzheimer’s disease. Ageing Res Rev. 2006;5:1–13. doi: 10.1016/j.arr.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 240.Kirshner HS. Delirium: a focused review. Curr Neurol Neurosci Rep. 2007;7:479–482. doi: 10.1007/s11910-007-0074-7. [DOI] [PubMed] [Google Scholar]
- 241.Skenazy JA, Bigler ED. Neuropsychological findings in diabetes mellitus. J Clin Psychol. 1984;40:246–258. doi: 10.1002/1097-4679(198401)40:1<246::aid-jclp2270400148>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 242.Botez G, Rami A. Immunoreactivity for Bcl-2 and C-Jun/AP1 in hippocampal corpora amylacea after ischaemia in humans. Neuropathol Appl Neurobiol. 2001;27:474–480. doi: 10.1046/j.1365-2990.2001.00362.x. [DOI] [PubMed] [Google Scholar]
- 243.Mann DM, Bonshek RE, Marcyniuk B, Stoddart RW, Torgerson E. Saccharides of senile plaques and neurofibrillary tangles in Alzheimer’s disease. Neurosci Lett. 1988;85:277–282. doi: 10.1016/0304-3940(88)90365-5. [DOI] [PubMed] [Google Scholar]
- 244.Singhrao SK, Neal JW, Newman GR. Corpora amylacea could be an indicator of neurodegeneration. Neuropathol Appl Neurobiol. 1993;19:269–276. doi: 10.1111/j.1365-2990.1993.tb00437.x. [DOI] [PubMed] [Google Scholar]
- 245.Schmitt FA, Wetherby MM, Wekstein DR, Dearth CM, Markesbery WR. Brain donation in normal aging: procedures, motivations, and donor characteristics from the Biologically Resilient Adults in Neurological Studies (BRAiNS) Project. Gerontologist. 2001;41:716–722. doi: 10.1093/geront/41.6.716. [DOI] [PubMed] [Google Scholar]